
Abstract
Interleukin-10 (IL-10) is a potent anti-inflammatory cytokine that regulates immune responses. IL-10 has also been shown to enhance antitumor CD8+ T-cell responses in tumor models although the underlying mechanisms are not fully understood. In this study, we used a series of genetic mouse models and the mouse plasmacytoma J558 model to investigate this issue. J558 tumors grew significantly faster in IL-10−/− mice than in wild type (WT) mice, but similarly in IL-10−/−Rag2−/− and Rag2−/− mice. Tumors from IL-10−/− mice contained fewer IFN-γ-producing CD8+ and CD4+ T cells than tumors from WT mice. Strikingly, depletion of total CD4+ T cells, but not CD25+ cells, resulted in tumor eradication in IL-10−/− mice. Adoptive transfer studies revealed that CD4+ T cells from IL-10−/− mice exhibited more potent suppression of cytotoxic T lymphocyte (CTL)-mediated tumor rejection than their WT counterparts, and IL-10–deficient tumor-infiltrating CD4+ T cells expressed higher levels of PD-L1 and CTLA-4 inhibitory molecules. Although IL-10–deficient CD8+ T cells are not defective in activation and initial rejection of tumors, adoptive transfer studies using IL-10–deficient P1CTL transgenic T cells that recognize the tumor rejection antigen P1A reveal that IL-10 is required for long-term persistence of CTLs and control of tumor growth. Thus, we have found that IL-10 enhances antitumor CTL responses by inhibiting highly suppressive CD4+ T cells and promoting CTL persistence. These data have important implications for the design of immunotherapy for human cancer.
Introduction
Although spontaneous CD8+ T-cell responses against human cancer have long been observed,Citation1 those responses rarely lead to tumor rejection because of a suppressive network in established tumors that not only inhibits functions of cytotoxic T lymphocytes (CTLs) but also permits tumor progression.Citation2-4 This immune suppressive network involves a number of cellular components such as Foxp3+CD4+CD25+ T regulatory cells (Tregs)Citation5 and myeloid-derived suppressor cells (MDSCs).Citation6,7 More recently, inflammatory CD4+ T cells (Th17) have also been implicated in the tumor microenvironment,Citation8,9 consistent with the reported functions of IL-17 in promoting tumor growth.Citation10-15
Interleukin 10 (IL-10) is an anti-inflammatory cytokine that is produced by a variety of cells including T lymphocytes, B lymphocytes, and cells of the myeloid lineage.Citation16,17 IL-10 reduces the expression of major histocompatibility complex (MHC) and co-stimulatory molecules on antigen-presenting cells and inhibits antigen-specific activation of T cells, and is therefore essential in maintaining T-cell tolerance.Citation16 Although the anti-inflammatory effect of IL-10 is well characterized,Citation18,19 reports on the role of IL-10 in tumor immunity are often controversial and suggest a context-dependent effect.Citation20-22 For example, expression of IL-10 in FoxP3+ Treg cells was shown to inhibit inflammation-driven tumorigenesis and antitumor immunity.Citation23,24 Our recent studies suggest that IL-10 derived from tumor-associated myeloid cells can inhibit CD8+ T-cell activation and effector function in a model devoid of CD4+ T cells.Citation25,26 However, increasing evidence also suggests that IL-10 has a potent effect on the induction of antitumor CTL responses in a number of tumor models. IL-10–deficient mice exhibit reduced antitumor CTL responsesCitation27 and increased numbers of FoxP3+ Treg cells,Citation28 whereas in IL-10 transgenic mice antitumor CTL responses are primed and were shown to be responsible for tumor rejection.Citation27,29 Expression of IL-10 in tumor cells or systemic administration of IL-10 resulted in inhibition of tumor growth Citation30-35 and the induction of CD8+ T-cell responses.Citation27,30,34,35 Administration of IL-10 in combination with antitumor vaccines resulted in significantly enhanced tumor-specific CTL responses and therapeutic effectiveness of the vaccines.Citation36,37
Although it has long been observed that IL-10 can enhance antitumor CTL responses in a number of tumor models, the underlying mechanisms are less clearly understood. IL-10 has been shown to directly stimulate in situ CTL expansion through the IL-10 receptor on CD8+ T cells.Citation35 CTL-produced IL-10 has also been shown to be related to better effector functions.Citation38,39 However, it is unclear whether the poor CD8+ T cell responses observed in IL-10−/− mice was caused by defects of the CD8+ T cell itself or by IL-10 deficiency resulting from dysregulation of other immune cell types. To address these issues, we have generated IL-10 and Rag2 double-deficient mice and IL-10–deficient P1CTL transgenic mice, whose T-cell receptor (TCR) recognizes the classic tumor antigen P1A.Citation40-42 Using these genetic models, we have found that IL-10 enhances antitumor CTL responses by inhibiting the priming of highly suppressive CD4+ T cells and by promoting long-term persistence of CTLs.
Results
Accelerated tumor growth in IL-10−/− mice
To investigate the roles of IL-10 in tumor immunity, we injected J558 tumor cells into wild-type and IL-10-deficient BALB/c mice. As shown in , tumors started to establish in IL-10−/− mice within 1 week and grew progressively thereafter; by 3 weeks after tumor cell injection all mice had large established tumors. In wild-type mice, tumors started to establish at approximately 2 weeks and tumors grew much more slowly such that the tumor volumes were significantly smaller than in IL-10−/− mice within the same time frame. To investigate whether the adaptive immune response was responsible for the slower tumor growth in wild-type mice, we generated Rag2 and IL-10 double-deficient mice (IL-10−/−Rag2−/−). When IL-10−/−Rag2−/− mice and IL-10+/+Rag2−/− mice were challenged with the same J558 tumor cells, we found that J558 tumors had similar tumor establishment and growth rate in both types of mice (). Thus, the adaptive immune response caused the slower tumor growth in wild-type mice.
Figure 1. Tumor growth kinetics in IL-10–deficient mice. Plasmacytoma J558 cells were injected into each mouse subcutaneously at a dose of 5 × 106/mouse. Tumor growth was monitored by measuring tumor size in 2 cross directions. Tumor volume was calculated using the formula: volume = (L × W2)/2, where L = length and W = width. (A) Tumor growth in BALB/c (n = 7) and IL-10−/− BALB/c mice (n = 8). **P < 0.01 by Student t test. (B) Tumor growth in Rag2−/− BALB/c (Rag2 KO, n = 5) and IL-10−/−Rag2−/− (DKO, n = 5) mice. Data shown are representative of 5 (A) and 3 (B) experiments with similar results.
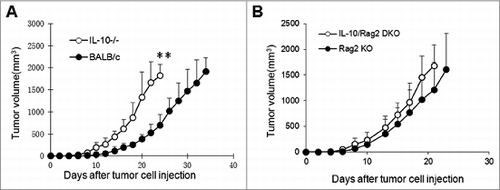
Diminished CD8+ T-cell responses in tumors from IL-10−/− mice
To determine the cellular components that caused the delay in tumor growth in wild-type mice versus IL-10−/− mice, established J558 tumors from wild type and IL-10−/− mice were harvested and single-cell suspensions were prepared. The cells were stained with various fluorescence-conjugated antibodies followed by flow cytometry analysis. As shown in , IL-10−/− and wild-type mice had similar numbers of CD4+ and CD8+ T cells in the spleen. However, the number of CD4+ and CD8+ T cells was significantly reduced in the tumors from IL-10−/− mice compared to tumors from wild-type mice (). In addition, we found that a much higher number of CD8+ T cells produce IFN-γ in wild-type tumors than in IL-10–deficient tumors (). CD4+ T cells in both types of tumors largely failed to produce IFN-γ. Depletion of CD8+ T cells in wild-type mice dramatically enhanced tumor growth (), whereas CD8+ T cell depletion had a limited effect on tumor growth in IL-10−/− mice (). Thus, diminished CD8+ T-cell responses in IL-10−/− mice were responsible for the enhanced growth of J558 tumors.
Figure 2. Diminished antitumor T-cell responses in IL-10−/− mice. (A) Established J558 tumors from WT and IL-10−/− mice were harvested and single-cell suspensions were prepared and stained for CD4 and CD8, followed by flow cytometry analysis. The percentage of CD4+ and CD8+ T lymphocytes in tumors from WT and IL-10−/− mice are summarized (n = 9 per group). Data shown are representative of 3 experiments with similar results. (B) IFN-γ producing CD8+ and CD4+ T cells in tumors from WT and IL-10−/− mice were analyzed and quantified by flow cytometry. The percentage of IFN-γ–producing CD4+ and CD8+ T lymphocytes in tumors from WT and IL-10−/− mice is summarized (n = 7 per group). Data shown are representative of 3 experiments with similar results.
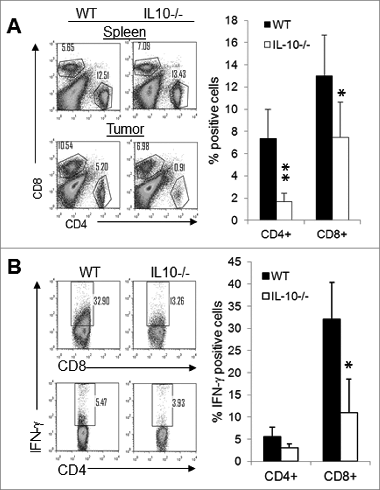
Figure 3. Depletion of CD4+ T cells in IL-10−/− mice facilitates tumor rejection. Four doses of anti-CD8 (53-6.72), anti-CD4 (GK1.5), or control antibodies (2A3 or LTF-2) were injected intraperitoneally into WT (A, C) and IL-10−/− mice (B, D) at a dose of 400 μg/per mouse at 4-day intervals beginning on day 1 after J558 tumor cell injection. Four to 5 mice were used per group. Data shown are representative of 2 to 3 experiments with similar results. **P < 0.01 by Student t test.
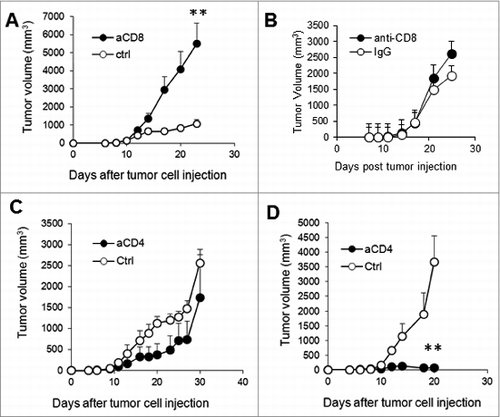
CD4+ T cells in IL-10−/− mice strongly suppress CTL-mediated tumor rejection
Since CD4+ T cells in the IL-10–deficient and sufficient tumors did not produce much IFN-γ, we asked whether these CD4+ T cells were suppressors and, if so, whether depletion of CD4+ T cells in WT and IL-10−/− mice would affect tumor growth. As demonstrated in , depletion of CD4+ T cells did not enhance, but rather slightly reduced, tumor growth in WT mice. Strikingly, depletion of CD4+ T cells in IL-10−/− mice caused nearly complete rejection of J558 tumors (). We detected much higher numbers of CD8+ T cells in tumors from IL-10−/− mice treated with anti-CD4 antibody compared to those treated with control antibody (). To determine whether CD4+ T cells from IL-10–deficient mice are more suppressive than CD4+ T cells from WT mice, we injected IL-10−/−Rag2−/− mice with J558 cells. When tumors were established, we co-injected tumor-bearing mice with IL-10-deficient P1CTL cells and IL-10-deficient or sufficient CD4+ T cells. As shown in , significantly more efficient tumor rejection was observed in mice receiving WT CD4+ T cells than in mice receiving IL-10–deficient CD4+ T cells. Thus, CD4+ T cells from IL-10–deficient mice strongly suppress CTL-mediated tumor rejection. Consistent with their more immunosuppressive functions, we found that tumor-infiltrating CD4+ T cells expressed higher levels of inhibitory molecules including PD-L1 and CTLA-4 ().
Figure 4. IL-10-deficient CD4+ T cells suppress antitumor CTL responses. (A) Depletion of CD4+ T cells in IL-10−/− mice led to CTL infiltration into tumors. Four tumors were analyzed from each group. Data are representative of 2 experiments with similar results. **P < 0.01 by Student t test. (B) J558 cells (5 × 106 cells) were injected subcutaneously into each IL-10−/−Rag2−/− mouse. At 7 days after tumor cell injection, IL-10−/−P1CTL cells (5 × 106) were injected intravenously into each mouse. Each mouse also received 5 × 106 CD4+ T cells from WT (n = 5) or IL-10−/− mice (n = 5) on day 7. Tumor growth was observed. Data shown are representative of 2 experiments with similar results. *P < 0.05 by Student t test. (C) J558 cells (5 × 106 cells) were injected subcutaneously into each IL-10−/− (n = 4) or WT BALB/c (n = 4) mouse. At 2 weeks after tumor cell injection, tumor-infiltrating lymphocytes were stained for CD4 in combination with PD-1, PD-L1 or CTLA-4 followed by flow cytometry analysis. MFI, mean fluorescence intensity; **P < 0.01 by Student t test.
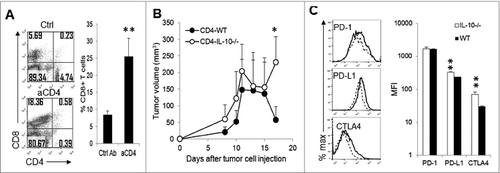
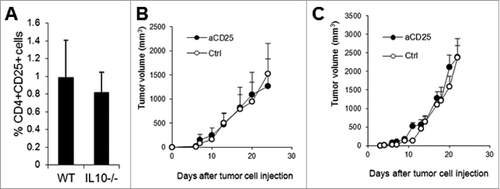
Since Treg cells (CD4+CD25+Foxp3+) from IL-10–deficient mice had been demonstrated to have diminished suppressive activity against T cells,Citation23 and depletion of CD4+ T cells in wild type mice did not result in dramatic tumor rejection (), the severe tumor rejection in CD4-depleted IL-10−/− mice was unlikely to be due to Treg depletion. Indeed, tumors from WT and IL-10−/− mice contained similar numbers of CD4+CD25+ T cells (), and treatment of WT () and IL-10-deficient mice () with anti-CD25 antibody or a control antibody did not affect the growth of J558 tumors.
Figure 5. Anti-CD25 antibody treatment does not affect tumor growth. The percentage of tumor-infiltrating CD4+CD25+ T cells was quantified (n = 7 per group) based on flow cytometry analysis (A). Four doses of anti-CD25 (PC-61.5.3) or control antibody (HRPN) were injected intraperitoneally into WT (B) or IL-10−/− mice (C) at a dose of 400 μg/per mouse at 4-day intervals beginning on day 1 after J558 tumor cell challenge (n = 4 per group). Data shown are representative of 2 experiments with similar results.
Tumors from IL-10−/− mice have an inflammatory environment that promotes Th17 responses
To understand why depletion of CD4+ T cells resulted in dramatic tumor rejection in IL-10–deficient mice, we analyzed the expression of a number of factors in IL-10–deficient and wild-type tumors. As shown in , real-time PCR analysis revealed significantly elevated levels of IL-17 and IL-6 transcripts in IL-10–deficient J558 tumors, whereas TGF-β1 gene expression was not significantly different between IL-10–deficient and wild-type mice. In IL-10–deficient tumors, a larger number of CD4+ T cells produced IL-17, which was therefore present at a significantly higher level than in WT tumors (). Increased expression of IL-6 was detected in tumor lysates from IL-10−/− mice ().
Figure 6. IL-6–Th17 axis in tumors from IL-10−/− and WT mice. (A) Total RNA was isolated from established J558 tumors from IL-10−/− (n = 3) or WT (n = 3) mice, and real-time PCR was performed to detect the expression of different genes. Data shown are representative of 2 experiments with similar results. (B) Intracellular IL-17 staining and flow cytometry analysis of tumor-infiltrating CD4+ T cells. Data in the right panel summarize the percentage of Th17 cells in tumors from IL-10−/−−(n = 13) or WT (n = 10) mice. (C) IL-6 concentration in tumor lysates (n = 3 per group) were determined by ELISA. (D) J558 cells (5 × 106 cells) were injected subcutaneously into IL-10−/− mice. Four doses (day 1, 4, 7, and 10) of anti–IL-6 (MP5-20F3, n = 5) or control antibody (HRPN, n = 5) were injected into IL-10−/−mice at a dose of 400 μg/mouse and tumor size was measured over time. *P < 0.05 by Student t test. (E) J558 cells (5 × 106 cells) were injected subcutaneously into IL-10−/− mice. Five doses (day 1, 4, 7, 10, and 14) of anti–IL-6 plus anti–IL-17A (MP5-20F3+17F3, n = 5) or control antibodies (HRPN plus MOPC-21, n=5) were injected into IL-10−/− mice at a dose of 400 μg/mouse/antibody and tumor size was measured over time.
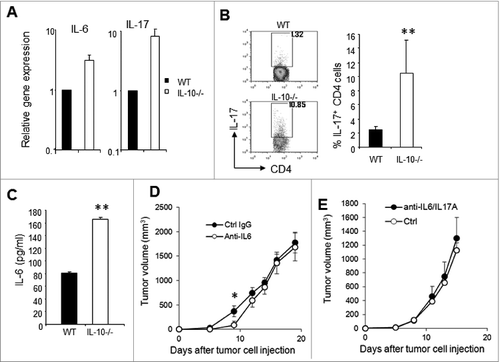
IL-6 is known to drive Th17 differentiationCitation43 and stimulate tumor growth, therefore we tested whether IL-6 neutralization would affect J558 tumor growth in IL-10–deficient mice. As shown in , anti–IL-6 treatment only slightly delayed the growth of J558 tumors in IL-10−/− mice. Moreover, combined anti–IL-6 and anti–IL-17A treatment failed to affect J558 tumor growth in IL-10−/− mice. Thus, increased activity of the IL-6–Th17 axis is not solely responsible for the accelerated tumor growth in IL-10−/− mice.
CTL-derived IL-10 is required for CTL long-term survival
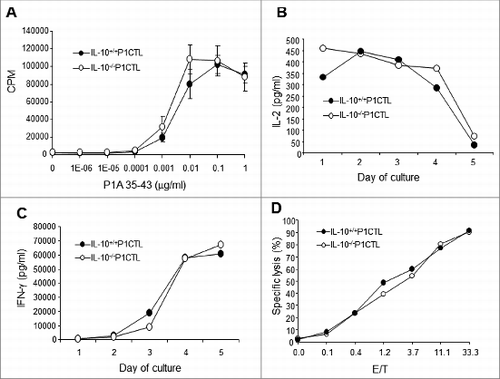
Since CD8+ T cells infiltrated into tumors in CD4+ T cell-depleted IL-10−/− mice and caused tumor rejection (), IL-10–deficient CD8+ T cells appeared to have normal functions. Indeed, as demonstrated in , IL-10–deficient P1CTL cells proliferated similarly to their wild type counterparts in response to P1A antigen stimulation (). Similar amounts of IL-2 () and IFN-γ () were produced after peptide-mediated activation. Moreover, upon activation the IL-10–deficient P1CTLs were equally effective as their wild type counterparts in killing P1A antigen-positive targets ().
Figure 7. IL-10–deficient P1CTL cells have similar activation and effector functions to their wild type counterparts. Splenocytes from IL-10−/−P1CTL and IL-10+/+ P1CTL mice were cultured in the presence of different concentrations of P1A35-43 in U-bottomed 96-well plates or 6-well plates. (A) After 48 h, 3H-tritium was added and the plates were incubated for a further 12 h before harvesting and counting on a scintillation counter. (B, C) Release of IL-2 (B) and IFNγ (C) into the culture supernatants over time was measured by ELISA. (D) P815 cells were used as targets in a 51Cr-release assay. Day 5 stimulated cultured splenocytes of IL-10−/−P1CTL and IL-10+/+P1CTL mice were used as effectors. Data shown are representative of 3–5 experiments with similar results.
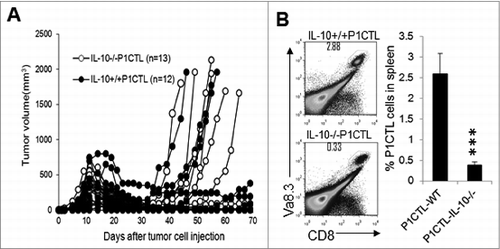
To test whether IL-10–deficient P1CTL cells could effectively destroy established tumors and mediate tumor rejection, we injected J558 tumor cells into Rag2−/− BALB/c mice. When tumors were established (day 9 after tumor cell injection), 5 × 106 CD8+ T cells from either IL-10−/− P1CTL or IL-10+/+ P1CTL mice were injected into each tumor-bearing mouse. As shown in , similar tumor rejection was initially observed. However, mice receiving IL-10−/− P1CTL cells had a greater frequency of tumor recurrence. Our previous studiesCitation44 established that tumor recurrence after P1CTL therapy was due to P1A antigen mutationCitation41,42 or T cell exhaustion.Citation42 Indeed, we found that by 40 days after injection of T cells, mice receiving IL-10−/− P1CTL cell treatment had significantly reduced numbers of P1CTL cells in their spleens compared to mice receiving IL-10+/+ P1CTL cells. Thus, IL-10–deficient CD8+ T cells failed to persist in tumor-bearing mice.
Figure 8. IL-10-deficient CTL cells are defective in long-term persistence. (A) Purified CD8+ cells from IL-10−/− P1CTL or IL-10+/+ P1CTL mice were injected intravenously into Rag2−/−mice with established J558 tumors at a dose of 5 × 106/mouse. Tumor growth was monitored every 2–3 d (B). At 40 d after T cell therapy, mice were sacrificed and P1CTL cells (CD8+Vα8.3+) in spleens were analyzed by flow cytometry and quantified (n = 5 mice/group). ***P < 0.0001 by Student t test. Data shown are representative of 2 experiments with similar results.
Discussion
In this study, we used a series of genetic mouse models and the mouse plasmacytoma model to study the role of endogenous IL-10 in antitumor CTL responses. We have made the following 2 notable observations: First, IL-10–deficient mice exhibited significantly diminished antitumor CTL responses and accelerated tumor growth. However, depletion of CD4+ T cells in IL-10–deficient mice restored CTL response and tumor rejection, suggesting that IL-10–deficient tumor-infiltrating CD4+ T cells are highly immunosuppressive. Second, although initial CTL proliferation, cytokine production, cytotoxicity, and tumor destruction were not affected by IL-10 deficiency, IL-10–deficient CTL cells have intrinsic defects in long-term persistence in tumor-bearing mice.
There are controversial reports in the literature regarding the roles of IL-10 in antitumor immunity.Citation20,21 However, accumulating evidenceCitation30,34,36 supports a role of IL-10 in enhancing CTL responses in tumor-bearing mice. In this study, we found that plasmacytoma J558 tumors grew significantly faster in IL-10−/− mice than in wild-type mice. The similar tumor growth kinetics of J558 tumors in IL-10−/−Rag2−/−−mice supports a role of adaptive immunity in preventing tumor growth. Indeed, we found that high numbers of IFN-γ–producing CD8+ T cells infiltrated into tumors from wild type but not IL-10−/− mice. Depletion of CD8+ T cells dramatically enhanced tumor growth in WT mice but had limited effect on tumor growth in IL-10−/− mice (). Thus, the difference in tumor growth difference between wild type and IL-10−/− mice reflected differences in the CTL response.
The significantly reduced numbers of IFN-γ–producing CD8+ T cells in tumors from IL-10−/− mice suggests that CTL responses were inhibited in the IL-10–deficient tumor microenvironment. Our data indicate that suppression of CTL responses was mediated by tumor-infiltrating CD4+ T cells since depletion of CD4+ T cells restored CTL infiltration into tumors and caused tumor regression in IL-10−/− mice (). An adoptive transfer study confirmed that CD4+ T cells from IL-10−/− mice were more suppressive toward CTL-mediated tumor rejection than CD4+ T cells from WT mice (). Importantly, tumor-infiltrating CD4+ T cells expressed significantly higher levels of PD-L1 and CTLA-4 (), consistent with their immunosuppressive functions. Although increased FoxP3+ Treg responses in tumors from IL-10−/− mice were previously reported,Citation28 other studies suggest that IL-10–deficient Treg cells have diminished suppressive activity.Citation23,45 In our tumor model, Treg numbers were not significantly different between IL-10–deficient and WT tumors. More importantly, anti-CD25 antibody treatment did not cause a difference in tumor growth between WT and IL-10−/− mice (). Thus, the CTL response and tumor rejection induced by CD4 depletion in IL-10−/− mice was unlikely to be mediated by depletion of Treg cells. We found that a significant population of CD4+ T cells in tumors from IL-10−/− mice was of the Th17 phenotype, which was present at significantly higher levels than among CD4+ T cells from WT tumors. This observation is consistent with previous reports that IL-10 signaling can directly inhibit Th17 cellsCitation24,46,47 or indirectly inhibit the production of inflammatory cytokines such as IL-6,Citation48,49 a key differentiation cytokine for Th17 cells.Citation43 Th17 cells have been implicated in suppression of tumor immunity and promotion of tumor growth.Citation10–15 However, our IL-6 and IL-6/IL-17 blockade experiments suggest that inhibition of the IL-6–IL-17 axis does not affect tumor growth (). Thus, it seems likely that the CD4+ T cell-mediated suppression of CTL responses in IL-10–deficient tumors is mediated through the traditional immunosuppressive pathways such as PD-1 and CTLA-4. How IL-10-deficiency leads to upregulation of PD-L1 and CTLA-4 remains to be investigated.
A notable observation in this study is that depletion of CD4+ T cells resulted in more significant tumor rejection in IL-10−/− mice than in wild type mice (). There are 2 possible reasons for the more efficient tumor rejection in IL-10−/− mice in the absence of CD4+ T cells. First, IL-10 is known to inhibit antigen presentation and activation of T cells.Citation16 We have recently shown that IL-10 deficiency also reduced the suppressive activity of myeloid-derived suppressor cells against CD8+ T cells.Citation25 These effects could lead to better activation of CD8+ T cells in the absence of suppressive CD4+ T cells. Second, we have recently shown that IL-10 deficiency results in increased production of cytokines such as TNF-α by tumor-associated myeloid cells and turns these cells into killer cells.Citation25 Together, these effects would result in enhanced tumor eradication.
As some studies have suggested,Citation50,26 the acute functions of CTL cells, including proliferation, production of cytokines, and cytotoxicity () and initial rejection of tumors in vivo () were not affected by IL-10 deficiency. However, we found that adoptively transferred IL-10–deficient P1CTL cells are less efficient at controlling tumor recurrence and fail to persist in tumor-bearing hosts compared to their WT counterparts (). These data suggest that CTL-derived IL-10 is required for long-term CTL persistence. IL-10–producing CD8+ T cells are usually considered to be suppressor cells that downregulate T-cell responses.Citation51 However, some studies have shown that IL-10–producing CTLs are more highly activated and cytolytic than IL-10–deficient CTLs.Citation27,38,52 In our recent study, we found that IL-27–stimulated IL-10–deficient CTLs expressed similar levels of IFN-γ, Granzyme B, and perforin, and exhibited similar levels of cytotoxicity toward target cells compared to their WT counterparts. However, IL-10 deficiency significantly reduced the expression of IL-27–induced survival molecules, including SOCS3, Bcl-2, and Bcl-6, in CTLs.Citation39 Thus, failure of IL-10–deficient P1CTL cells to persist in a tumor-bearing host is probably due to their poor survival potential. The current study has demonstrated the intrinsic defects of IL-10–deficient CTLs in long-term persistence/survival in a tumor-bearing host. This observation is in line with the previous observation that IL-10 is critical for CTL memory.Citation53
In summary, we have identified 2 novel mechanisms by which IL-10 enhances the antitumor CTL responses—by inhibiting the expansion of highly suppressive CD4+ T cells and by promoting CTL long-term survival/persistence in the plasmacytoma J558 tumor model. We believe that these mechanisms apply to many different tumor types in which IL-10 has been shown to play positive roles in promoting antitumor T-cell responses.Citation21 Our results also suggest that amplification of IL-10 signaling may enhance the expansion of CTLs and inhibit regulatory T cells in the tumor microenvironment. Since IL-10 inhibits antigen presentation,Citation54 it is likely that cancer types with pre-existing tumor-infiltrating T cells will respond positively to IL-10 therapy. In this regard, tumor infiltration of CTL and immunoregulatory T cells has been shown to be a feature of many types of human cancer,Citation55,56 for which IL-10 therapy should be applicable.
Materials and Methods
Experimental animals
IL-10–deficient BALB/c mice were purchased from the Jackson Laboratory. Transgenic mice expressing a TCR specific for the tumor rejection antigen H-2Ld:P1A35-43 complex (P1CTL) have been described previously.Citation40 P1CTL TCR transgenic mice were backcrossed with BALB/c mice for at least 15 generations before use in this study. BALB/c mice with a targeted mutation of the Rag2 gene were purchased from Taconic Farms (Germantown, New York, USA). Through breeding P1CTL TCR transgenic mice with Rag2−/− BALB/c mice we have generated Rag2-deficient P1CTL TCR transgenic mice (Rag2−/−P1CTL). IL-10–deficient P1CTL TCR transgenic mice (IL-10−/−P1CTL) were generated by breeding P1CTL TCR transgenic mice with IL-10–deficient BALB/c mice. Through breeding IL-10−/− BALB/c mice with Rag2−/− BALB/c mice we have generated Rag2 and IL-10 double-deficient mice (Rag2−/−IL-10−/−). IL-10 deficiency was confirmed by PCR genotyping using the primers mIL-10.G: 5′-ATAGACTTGC TCTTGCACTA CCAAAG-3′ (forward) and 5′-CTCATGGCTT TCCCTAGGAC TCTCTA-3′ (reverse). All mice were maintained in OSU laboratory animal facilities that were fully accredited by the Institutional Animal Care and Use Committee.
Tumor establishment and T-cell adoptive transfer therapy of mice with established tumors
For tumor establishment in vivo, 5 × 106 J558 cells were injected into each mouse subcutaneously. Tumors were measured for length (L) and width (W) every 2 to 3 d and tumor volumes were calculated as LW2/2.Citation57 For CTL therapy of mice with established tumors, pools of spleen and lymph node cells from P1CTL transgenic mice were incubated with a cocktail of monoclonal Abs (mAbs; anti-CD4 mAb GK1.5, anti-FcR mAb 2.4G2, and anti-CD11c mAb N418). After removal of unbound mAbs, the cells were incubated with anti-IgG coated magnetic beads (Dynal Biotech). The antibody-coated cells were removed using a magnet. Unbound cells consisted of more than 90% CD8+ T cells with no detectable CD4+ T cells. Purified CD8+ T cells (5 × 106/mouse) were injected intravenously (i.v.) into mice bearing established tumors.
In vivo treatment of mice with tumors using antibodies
Monoclonal antibodies to mouse CD4 (GK1.5), CD8 (53-6.72), CD25 (PC-61.5.3), IL-6 (MP5-20F3), IL-17A (17F3) and their relative isotype control antibodies HRPN (rat IgG1), 2A3 (rat IgG2a), LTF2 (rat IgG2b), and MOPC-21 (mouse IgG1) were all purchased from BioXCell (West Lebanon, NH). The treatment protocols for experiments are specifically indicated in the relevant figure legends.
Antibodies and flow cytometry
FITC-, PE-, APC- or Percp-labeled antibodies to CD4, CD8α, CD25, PD-1, PD-L1, CTLA4, Vα8.3, IL-17, IFN-γ, and isotype control antibodies were purchased from BD Biosciences (San Diego, CA). For staining of cell surface markers, cells (splenocytes and single-cell suspensions of tumors) were stained with various antibodies in staining buffer (PBS with 1% FCS) on ice for 30 min, washed with staining buffer, and fixed in 1% paraformaldehyde in PBS. Cells were analyzed on a FACSCalibur flow cytometer. Intracellular cytokine staining procedure as described previously.Citation25 Data were analyzed using Flowjo software (Tree Star, Inc., OR).
Reverse transcription and real-time PCR
Total RNA was isolated from each tumor using the Trizol method (Invitrogen). First strand cDNA of each sample was synthesized using a reverse transcription kit (Invitrogen). Quantitative real-time PCR was performed using an ABI 7900-HT sequence system (PE Applied Biosystems) with the QuantiTect SYBR Green PCR kit (Qiagen) in accordance with the manufacturer's instructions. PCR was performed using previously determined conditionsCitation41 with the following primers to amplify specific genes: mIL-6: 5′-ACTTCACAAG TCGGAGGCTT-3′ (forward) and 5′-TCTGCAAGTG CATCATCGT-3′ (reverse); mIL-17: 5′-CCTCCAGAAT GTGAAGGTCA-3′ (forward) and 5′-CTATCAGGGT CTTCATTGCG-3′; mIFN-γ: 5′-AGCTCTTCCT CATGGCTGTT-3′ (forward) and 5′-TTTGCCAGTT CCTCCAGATA-3′ (reverse). The HPRT gene was simultaneously amplified as the endogenous control using the primers 5′-AGCCTAAGAT GAGCGCAAGT-3′ (forward) and 5′-TTACTAGGCA GATGGCCACA-3′ (reverse). Each sample was assayed in triplicate, and the experiments were repeated twice. The relative amount of mRNA was calculated by plotting the Ct (cycle number) and the average relative expression for each group was determined using the comparative method (2−ΔΔCt).
Cytokine ELISA
ELISA kits for the detection of IL-2, IFN-γ, and IL-6 were obtained from BD Biosciences. Standard procedures were followed to detect the release of cytokines into culture supernatants in a variety of settings as detailed in figure legends for each experiment.
Cytotoxicity assay
Splenocytes from P1CTL TCR transgenic mice were stimulated with P1A peptide (0.1 μg/mL) for 5 d and used as effectors.50 Cr-labeled P815 tumor cells were used as targets. The effector T cells and the targets were incubated together for 6 h, and the percentage of specific lysis was calculated based on the following formula: specific lysis % = 100 × (cpmsample-cpmmedium)/(cpmmax-cpmmedium).
Statistics
The Student t-test was used to compare differences in tumor size between 2 groups. A p value less than 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by a grant from the National Cancer Institute (R01CA138427).
References
- Romero P, Dunbar PR, Valmori D, Pittet M, Ogg GS, Rimoldi D, Chen JL, Lienard D, Cerottini JC, Cerundolo V. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J Exp Med 1998; 188:1641-50; PMID:9802976; http://dx.doi.org/10.1084/jem.188.9.1641
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263-74; PMID:15776005; http://dx.doi.org/10.1038/nrc1586
- Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 2006; 66:5527-36; PMID:16740684; http://dx.doi.org/10.1158/0008-5472.CAN-05-4128
- Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006; 66:605-12; PMID:16423985; http://dx.doi.org/10.1158/0008-5472.CAN-05-4005
- Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer 2010; 127:759-67; PMID:20518016
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499-506; PMID:19342621; http://dx.doi.org/10.4049/jimmunol.0802740
- Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol 2010; 184:1630-41; PMID:20026736; http://dx.doi.org/10.4049/jimmunol.0902813
- Murugaiyan G, Saha B. Protumor vs antitumor functions of IL-17. J Immunol 2009; 183:4169-75; PMID:19767566; http://dx.doi.org/10.4049/jimmunol.0901017
- Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood 2003; 101:2620-7; PMID:12411307; http://dx.doi.org/10.1182/blood-2002-05-1461
- Numasaki M, Watanabe M, Suzuki T, Takahashi H, Nakamura A, McAllister F, Hishinuma T, Goto J, Lotze MT, Kolls JK, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol 2005; 175:6177-89; PMID:16237115; http://dx.doi.org/10.4049/jimmunol.175.9.6177
- Prabhala RH, Pelluru D, Fulciniti M, Prabhala HK, Nanjappa P, Song W, Pai C, Amin S, Tai YT, Richardson PG, et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010; 115:5385-92; PMID:20395418; http://dx.doi.org/10.1182/blood-2009-10-246660
- Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 2009; 206:1457-64; PMID:19564351; http://dx.doi.org/10.1084/jem.20090207
- Tartour E, Fossiez F, Joyeux I, Galinha A, Gey A, Claret E, Sastre-Garau X, Couturier J, Mosseri V, Vives V, et al. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res 1999; 59:3698-704; PMID:10446984
- Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A 2010; 107:5540-4; PMID:20212110; http://dx.doi.org/10.1073/pnas.0912675107
- Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001; 19:683-765; PMID:11244051; http://dx.doi.org/10.1146/annurev.immunol.19.1.683
- Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galan JE, Harhaj E, Flavell RA. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 2006; 25:941-52; PMID:17137799; http://dx.doi.org/10.1016/j.immuni.2006.09.013
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993; 75:263-74; PMID:8402911; http://dx.doi.org/10.1016/0092-8674(93)80068-P
- Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity 2008; 28:468-76; PMID:18400189; http://dx.doi.org/10.1016/j.immuni.2008.03.003
- Vicari AP, Trinchieri G. Interleukin-10 in viral diseases and cancer: exiting the labyrinth? Immunol Rev 2004; 202:223-36; PMID:15546396; http://dx.doi.org/10.1111/j.0105-2896.2004.00216.x
- Mocellin S, Marincola FM, Young HA. Interleukin-10 and the immune response against cancer: a counterpoint. J Leukoc Biol 2005; 78:1043-51; PMID:16204623; http://dx.doi.org/10.1189/jlb.0705358
- Wilke CM, Wei S, Wang L, Kryczek I, Kao J, Zou W. Dual biological effects of the cytokines interleukin-10 and interferon-gamma. Cancer Immunol Immunother 2011; 60:1529-41; PMID:21918895; http://dx.doi.org/10.1007/s00262-011-1104-5
- Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, Tomczak M, Rogers AB, Horwitz BH, Fox JG. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res 2003; 63:6042-50; PMID:14522933
- Stewart CA, Metheny H, Iida N, Smith L, Hanson M, Steinhagen F, Leighty RM, Roers A, Karp CL, Muller W, et al. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J Clin Invest 2013; 123:4859-74; PMID:24216477; http://dx.doi.org/10.1172/JCI65180
- Wang L, Liu JQ, Talebian F, El-Omrani HY, Khattabi M, Yu L, Bai XF. Tumor expression of CD200 inhibits IL-10 production by tumor-associated myeloid cells and prevents tumor immune evasion of CTL therapy. Eur J Immunol 2010; 40:2569-79; PMID:20662098; http://dx.doi.org/10.1002/eji.201040472
- Wang LX, Talebian F, Liu JQ, Khattabi M, Yu L, Bai XF. IL-10 contributes to the suppressive function of tumour-associated myeloid cells and enhances myeloid cell accumulation in tumours. Scand J Immunol 2012; 75:273-81; PMID:22050574
- Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 2011; 20:781-96; PMID:22172723; http://dx.doi.org/10.1016/j.ccr.2011.11.003
- Tanikawa T, Wilke CM, Kryczek I, Chen GY, Kao J, Nunez G, Zou W. Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Res 2012; 72:420-9; PMID:22123924; http://dx.doi.org/10.1158/0008-5472.CAN-10-4627
- Groux H, Cottrez F, Rouleau M, Mauze S, Antonenko S, Hurst S, McNeil T, Bigler M, Roncarolo MG, Coffman RL. A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J Immunol 1999; 162:1723-9; PMID:9973435
- Giovarelli M, Musiani P, Modesti A, Dellabona P, Casorati G, Allione A, Consalvo M, Cavallo F, di Pierro F, De Giovanni C, et al. Local release of IL-10 by transfected mouse mammary adenocarcinoma cells does not suppress but enhances antitumor reaction and elicits a strong cytotoxic lymphocyte and antibody-dependent immune memory. J Immunol 1995; 155:3112-23; PMID:7673726
- Gerard CM, Bruyns C, Delvaux A, Baudson N, Dargent JL, Goldman M, Velu T. Loss of tumorigenicity and increased immunogenicity induced by interleukin-10 gene transfer in B16 melanoma cells. Hum Gene Ther 1996; 7:23-31; PMID:8825865; http://dx.doi.org/10.1089/hum.1996.7.1-23
- Zheng LM, Ojcius DM, Garaud F, Roth C, Maxwell E, Li Z, Rong H, Chen J, Wang XY, Catino JJ, et al. Interleukin-10 inhibits tumor metastasis through an NK cell-dependent mechanism. J Exp Med 1996; 184:579-84; PMID:8760811; http://dx.doi.org/10.1084/jem.184.2.579
- Adris S, Klein S, Jasnis M, Chuluyan E, Ledda M, Bravo A, Carbone C, Chernajovsky Y, Podhajcer O. IL-10 expression by CT26 colon carcinoma cells inhibits their malignant phenotype and induces a T cell-mediated tumor rejection in the context of a systemic Th2 response. Gene Ther 1999; 6:1705-12; PMID:10516719; http://dx.doi.org/10.1038/sj.gt.3301012
- Berman RM, Suzuki T, Tahara H, Robbins PD, Narula SK, Lotze MT. Systemic administration of cellular IL-10 induces an effective, specific, and long-lived immune response against established tumors in mice. J Immunol 1996; 157:231-8; PMID:8683120
- Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, Gorman DM, Oft M. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res 2012; 72:3570-81; PMID:22581824; http://dx.doi.org/10.1158/0008-5472.CAN-12-0721
- Fujii S, Shimizu K, Shimizu T, Lotze MT. Interleukin-10 promotes the maintenance of antitumor CD8(+) T-cell effector function in situ. Blood 2001; 98:2143-51; PMID:11568001; http://dx.doi.org/10.1182/blood.V98.7.2143
- Kaufman HL, Rao JB, Irvine KR, Bronte V, Rosenberg SA, Restifo NP. Interleukin-10 enhances the therapeutic effectiveness of a recombinant poxvirus-based vaccine in an experimental murine tumor model. J Immunother 1999; 22:489-96; PMID:10570747; http://dx.doi.org/10.1097/00002371-199911000-00003
- Trandem K, Zhao J, Fleming E, Perlman S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J Immunol 2011; 186:3642-52; PMID:21317392; http://dx.doi.org/10.4049/jimmunol.1003292
- Liu Z, Liu JQ, Talebian F, Wu LC, Li S, Bai XF. IL-27 enhances the survival of tumor antigen-specific CD8+ T cells and programs them into IL-10-producing, memory precursor-like effector cells. Eur J Immunol 2013; 43:468-79; PMID:23225163; http://dx.doi.org/10.1002/eji.201242930
- Sarma S, Guo Y, Guilloux Y, Lee C, Bai XF, Liu Y. Cytotoxic T lymphocytes to an unmutated tumor rejection antigen P1A: normal development but restrained effector function in vivo. J Exp Med 1999; 189:811-20; PMID:10049945; http://dx.doi.org/10.1084/jem.189.5.811
- Bai XF, Liu J, Li O, Zheng P, Liu Y. Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest 2003; 111:1487-96; PMID:12750398; http://dx.doi.org/10.1172/JCI17656
- Bai XF, Liu JQ, Joshi PS, Wang L, Yin L, Labanowska J, Heerema N, Zheng P, Liu Y. Different Lineages of P1A-Expressing Cancer Cells Use Divergent Modes of Immune Evasion for T-Cell Adoptive Therapy. Cancer Res 2006; 66:8241-9; PMID:16912204; http://dx.doi.org/10.1158/0008-5472.CAN-06-0279
- Kimura A, Kishimoto T. IL-6: Regulator of Treg/Th17 balance. Eur J Immunol 2010; 40:1830-5; PMID: 20583029; http://dx.doi.org/10.1002/eji.201040391
- Liu JQ, Bai XF. Overcoming immune evasion in T cell therapy of cancer: lessons from animal models. Curr Mol Med 2008; 8:68-75; PMID:18289015; http://dx.doi.org/10.2174/156652408783565531
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr., et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008; 28:546-58; PMID:18387831; http://dx.doi.org/10.1016/j.immuni.2008.02.017
- Gu Y, Yang J, Ouyang X, Liu W, Li H, Yang J, Bromberg J, Chen SH, Mayer L, Unkeless JC, et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol 2008; 38:1807-13; PMID:18506885; http://dx.doi.org/10.1002/eji.200838331
- Huber S, Gagliani N, Esplugues E, O'Connor W, Jr., Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011; 34:554-65; PMID:21511184; http://dx.doi.org/10.1016/j.immuni.2011.01.020
- de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 1991; 174:1209-20; PMID:1940799; http://dx.doi.org/10.1084/jem.174.5.1209
- Marshall JS, Leal-Berumen I, Nielsen L, Glibetic M, Jordana M. Interleukin (IL)-10 inhibits long-term IL-6 production but not preformed mediator release from rat peritoneal mast cells. J Clin Invest 1996; 97:1122-8; PMID:8613537; http://dx.doi.org/10.1172/JCI118506
- Dercamp C, Chemin K, Caux C, Trinchieri G, Vicari AP. Distinct and overlapping roles of interleukin-10 and CD25+ regulatory T cells in the inhibition of antitumor CD8 T-cell responses. Cancer Res 2005; 65:8479-86; PMID:16166328; http://dx.doi.org/10.1158/0008-5472.CAN-05-1319
- Noble A, Giorgini A, Leggat JA. Cytokine-induced IL-10-secreting CD8 T cells represent a phenotypically distinct suppressor T-cell lineage. Blood 2006; 107:4475-83; PMID:16467201; http://dx.doi.org/10.1182/blood-2005-10-3994
- Curran MA, Geiger TL, Montalvo W, Kim M, Reiner SL, Al-Shamkhani A, Sun JC, Allison JP. Systemic 4–1BB activation induces a novel T cell phenotype driven by high expression of Eomesodermin. J Exp Med 2013; 210:743-55; PMID:23547098; http://dx.doi.org/10.1084/jem.20121190
- Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity 2011; 35:792-805; PMID:22118527; http://dx.doi.org/10.1016/j.immuni.2011.09.017
- Beissert S, Hosoi J, Grabbe S, Asahina A, Granstein RD. IL-10 inhibits tumor antigen presentation by epidermal antigen-presenting cells. J Immunol 1995; 154:1280-6; PMID:7822797
- Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res 2003; 9:4404-8; PMID:14555512
- Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005; 102:18538-43; PMID:16344461; http://dx.doi.org/10.1073/pnas.0509182102
- Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 1989; 24:148-54; PMID: 2544306; http://dx.doi.org/10.1007/BF00300234