
Abstract
Impairment of natural killer (NK) cell activity is an important mechanism of tumor immunoevasion. We have previously shown that expression of granulin-epithelin precursor (GEP) in hepatocellular carcinoma (HCC) cells rendered the cells resistant to NK cell immunosurveillance. Here, we examined whether targeting GEP could rescue NK activity in HCC patients. The current study demonstrated that quantities and activities of NK cells were significantly lower in HCC patients compared with healthy individuals, and were negatively correlated with GEP levels in HCC cells. NK cells demonstrated enhanced expression of the stimulatory receptors natural-killer group 2, member D (NKG2D) and CD69, increased secretion of IFN-γ and perforin, and cytotoxicity against HCC cells upon GEP suppression. Opposite phenotypes of NK cells were observed when GEP was overexpressed in HCC cells. Importantly, GEP blockage by monoclonal antibody A23 restored NK activity in HCC patients and sensitized HCC cells to NK cytotoxicity. Furthermore, A23 induced NK-mediated antibody-dependent cell-mediated cytotoxicity against HCC. In summary, the activity of NK cells in HCC was impaired by GEP expression, which could be rescued by GEP antibody. This study provides new insight for treatments targeting GEP to boost NK activity in HCC patients.
Introduction
Tumors have developed diverse mechanisms to generate a suppressive network and evade the host's immune response. Impairment of natural killer (NK) activity is an important mechanism of tumor immunoevasion. NK cells represent a major component of innate immunity—they can exert direct cellular cytotoxicity on tumor cells without prior sensitization and secrete immunostimulatory cytokines such as the typical T helper type 1 (Th1) cytokine interferon (IFN)-γ, which controls both local tumor growth and metastasis.Citation1,2 An epidemiologic survey showed that low peripheral blood NK cell activity was associated with increased cancer risk.Citation3 In addition, NK intratumoral infiltration was reported to be associated with better disease prognosis in various cancers.Citation4-6
In hepatocellular carcinoma (HCC), high NK cell number and activity are associated with early stages and improved patient survival.Citation7,8 Increased frequencies of NK cells expressing higher levels of stimulatory NK receptors, together with higher functional activity such as IFN-γ production and cytotoxicity, were shown to have a favorable role in controlling HCC.Citation9 However, NK cells from HCC patients were frequently found to be defective in lytic function and cytokine secretion,Citation10-12 although the underlying mechanism is not clear.
Granulin-epithelin precursor (GEP) is a pluripotent growth factor that regulates fetal development, tissue repair, and tumorigenesis in various cancers.Citation13 Our group has demonstrated overexpression and a tumorigenic role of GEP in HCC.Citation14-17 GEP blockage by antibody inhibits the growth of established HCCCitation18,19 and sensitizes HCC to chemotherapeutic agents.Citation18,19 Moreover, GEP is a hepatic oncofetal protein that regulates hepatic cancer stem cell (CSC) propertiesCitation20 and renders HCC cells resistant to NK cytotoxic activity.Citation21 Importantly, we showed that GEP promotes the production of soluble MHC class I chain-related gene A (MICA) (sMICA), which in turn downregulates the surface expression of stimulatory receptor natural-killer group 2, member D (NKG2D) on NK cells.Citation21 Since NK activity was reported to be compromised in HCC patients, we hypothesized that GEP overexpression might be associated with impaired NK function. In this study, we further investigated whether targeting GEP could rescue NK activity in HCC patients and sensitize the aggressive HCC cells to NK immunosurveillance.
Results
Reduced NK cell quantity and activity in HCC patients
To characterize the phenotype of NK cells in HCC, we compared the quantity of NK cells between HCC patients and healthy individuals. Two major subsets of peripheral blood NK cells have been identified in humans on the basis of surface expression of CD56 and CD16.Citation22 The CD56+CD16+ NK subset, which constitutes approximately 90–95% of peripheral blood NK cells, has high cytotoxic activity through the production of a high level of cytotoxic granular proteins such as perforin whereas the CD56+CD16− NK subset is mainly present in tissues or secondary lymphoid organs. Although weakly cytotoxic, the CD56+CD16− NK subset can produce large amounts of cytokines including IFNγ, tumor necrosis factor (TNF)α, and granulocyte-macrophage colony-stimulating factor (GM-CSF) and is critical for development of Th1 responses.Citation23
In peripheral blood, the percentage of total NK cells (CD56+) in PBMCs was significantly reduced in HCC patients when compared with healthy individuals (). Further characterization showed that the reduction of NK cells in HCC patients was attributed to a reduced CD56+CD16+, but not CD56+CD16− subset (). We then compared NK infiltration in HCC tumors and paired adjacent non-tumor liver tissues. Infiltration of total NK cells (CD56+) was significantly reduced in tumors compared with matching non-tumor liver tissues. Further characterization showed that such reduction was attributed to both CD56+CD16+ and CD56+CD16− NK subsets (). The above findings echo previous reports of reduced number and impaired activity of NK cells in HCC patients.Citation10,11
Figure 1. Reduced natural killer cell quantity and activity in patients with hepatocellular carcinoma. (A) Peripheral blood mononuclear cells (PBMCs) were isolated from healthy individuals and HCC patients and the percentages of total NK (CD56+) cells and CD56+CD16+ and CD56+CD16− subsets were quantified (n ≥ 16). (B) Tumors and paired adjacent non-tumor liver tissues of HCC patients were dissociated and the percentages of total NK (CD56+) cells, CD56+CD16+ and CD56+CD16− subsets were quantified (n = 11). (C) Surface expression of stimulatory receptors NKG2D and CD69 on peripheral blood CD56+ NK cells were measured (n ≥ 15). MFI: mean fluorescence intensity. Peripheral blood CD56+ NK cells were isolated and co-cultured with Hep3B or HepG2 HCC cells at an effector cell:target cell (E:T) ratio of 4:1 for 24 h. IFN-γ and perforin levels in the culture supernatants were examined by enzyme-linked immunosorbent assay (ELISA), and NK cytotoxicity against HCC cells was measured by flow cytometry (n ≥ 5).
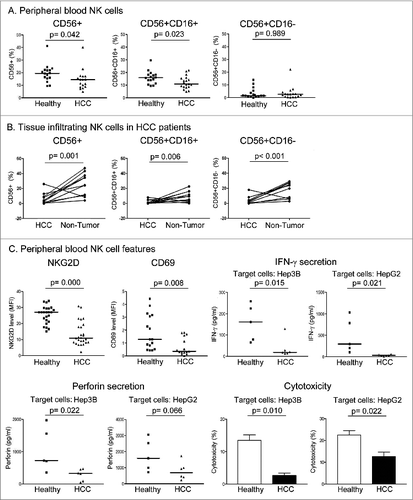
The phenotype of peripheral blood NK cells was then characterized in HCC patients and healthy individuals in terms of surface expression of the stimulatory receptors NKG2D and CD69 (). Both NKG2D and CD69 levels were significantly lower on peripheral NK cells of HCC patients than on those of healthy individuals, implying a lower activity level in peripheral blood NK cells of HCC patients. We then assessed the NK functional properties by co-culturing the NK cells with HCC cell lines Hep3B or HepG2 cells (). Upon co-culture, production of IFN-γ and perforin and cytotoxic activity against Hep3B and HepG2 cells of NK cells from HCC patients were significantly reduced compared with NK cells from healthy individuals.
Correlation of GEP and NK activity in HCC patients
Our group previously demonstrated that GEP rendered HCC cells resistant to NK cytotoxicity,Citation21 although the NK cells used were isolated from healthy individuals. Here, we investigated the association of GEP expression in HCC cells with the number and activity of NK cells in HCC patients. GEP was quantified by flow cytometry in terms of the percentage of GEP+ cells in tumors and paired adjacent non-tumor liver tissues. Consistent with our previous reports, the GEP level was significantly higher in HCC tumors than in non-tumor liver tissues (). GEP level in tumors was not associated with the number of NK cells in peripheral blood of HCC patients (); however, it was negatively correlated with NK cell surface expression of the stimulatory receptors NKG2D and CD69 (), suggesting that tumor GEP level was negatively associated with the activation of peripheral blood NK cells in HCC patients.
Figure 2. GEP level correlated with impaired NK activity and infiltration in patients with hepatocellular carcinoma. (A) GEP levels in tumors and paired adjacent non-tumor liver tissues in HCC. GEP level was measured by flow cytometry and presented as percentage of GEP+ cells (n = 20). (B) Correlation between GEP level and peripheral blood total NK (CD56+) cells, CD56+CD16+, and CD56+CD16− NK subsets of HCC patients, and surface expression of NKG2D and CD69 on peripheral blood CD56+ NK cells (n = 11). (C) Correlation between GEP level and infiltrating total NK cells (CD56+), CD56+CD16+, and CD56+CD16− subsets in HCC tumors (n = 11). (D) GEP expression levels and NK cell (CD56+) infiltration were examined in HCC tissues by immunohistochemical staining. Representative photographs show tumors with high (n = 7) and low (n = 4) GEP expression levels and the corresponding NK frequencies. HCC cases were categorized into 2 groups: high and low levels of GEP expression. The number of infiltrating NK cells per field (200×) was counted and compared between the 2 groups.
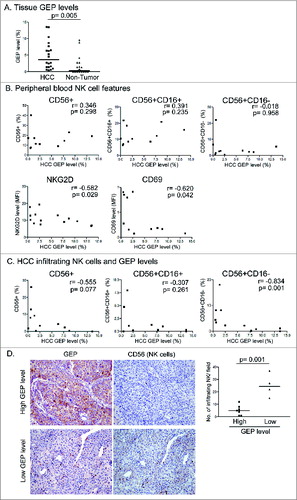
Next, we studied the association of GEP levels with NK infiltration in HCC tumors (). A trend toward a negative correlation between GEP levels and the quantity of infiltrating CD56+ NK cells in tumors was observed although statistical significance was not reached, possibly due in part to the small sample size. Further investigation indicated a significant negative correlation between GEP levels and the number of infiltrating CD56+CD16−, but not CD56+CD16+, NK cells (), suggesting that GEP level was associated with reduced infiltration of the CD56+CD16− NK cell subset in HCC tumors.
To further examine the association of GEP with NK infiltration, GEP level and number of infiltrating NK cells were measured by immunohistochemical (IHC) staining in HCC tumor tissues (). Eleven HCC cases were categorized into 2 groups according to their GEP levels as detected by flow cytometry: high GEP (GEP+ cells: 9.5–20.2%, n = 7) and low GEP (GEP+ cells: 0.6–2.2%, n = 4). The GEP levels of these 2 groups were further analyzed by IHC staining as described previously,Citation21 and confirmed as strong and weak GEP expression, respectively. Comparison of the number of infiltrating CD56+ NK cells between the 2 groups showed that the number of infiltrating NK cells was significantly reduced in the high-GEP expression group, providing further evidence for a negative correlation between GEP expression and NK infiltration in HCC tumors.
Immunoevasive ability of GEP-expressing cells in HCC
To better characterize the immunoevasive properties of GEP-expressing cells, GEPhigh and GEPlow subpopulations were isolated from Hep3B and HepG2 cells and their phenotypes and interactions with NK cells from HCC patients were examined. The purity of the GEPhigh and GEPlow subpopulations was approximately 80–90%, as revealed by post-sorting analysis (). We have previously demonstrated that GEP bestowed HCC cells with the ability to evade immunosurveillance of healthy NK cells through the NKG2D-MICA pathway.Citation21 NKG2D is a major stimulatory receptor on NK cells, and its activation results in cytokine secretion and cytotoxicity.Citation24 Human ligands of NKG2D belong to 2 families, the MHC class I-related chains A and B (MICA/B) and the unique long 16 (UL16)-binding protein (ULBP) family. Reduced expression of MICA and ULBP1 has been reported in HCC patients and this reduction was associated with poor clinical outcome.Citation10,25 We found that GEPhigh cells expressed significantly lower levels of MICA and ULBP1 than GEPlow subpopulations in both Hep3B and HepG2 cells (), suggesting that GEPhigh cells expressed a lower level of stimulatory ligands for NK cell activity. Moreover, GEPhigh cells produced a significantly higher level of sMICA than GEPlow cells (). This is consistent with our previous findings that GEP downregulates surface MICA expression by promoting its cleavage and enhances the secretion of sMICA.Citation21
Figure 3 (See previous page). Immunoevasion ability of GEP-expressing cells in HCC. (A) GEP expression in unsorted Hep3B and HepG2 cells and sorted GEPhigh and GEPlow subpopulations. Results are expressed as percentage of GEP+ cells and mean fluorescence intensity (MFI, mean ± SD). (B) Surface expression of stimulatory NKG2D ligands MICA and ULBP1 on unsorted HCC cells and sorted subpopulations. (C) sMICA secretion levels of unsorted HCC cells and sorted subpopulations after culture for 24 h. Unsorted, sorted GEPhigh, and GEPlow subpopulations were co-cultured with peripheral blood NK cells of HCC patients at an effector cell:target cell ratio of 4:1 for 24 h. Surface expression of (D) NKG2D and (E) CD69 on NK cells, and (F) cytotoxicity against HCC cells were assessed.
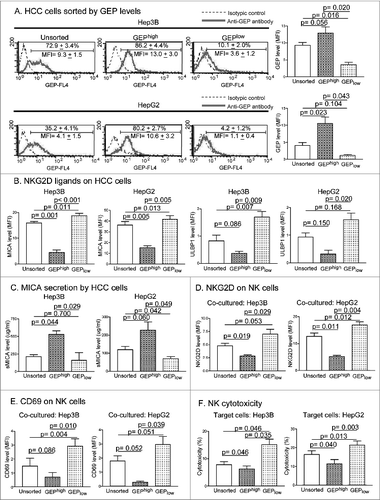
HCC cells and the sorted subpopulations were then co-cultured with NK cells from HCC patients. NK activity upon co-culture was assessed in terms of NKG2D and CD69 expression. Surface expression of NKG2D and CD69 on NK cells co-cultured with GEPhigh HCC cells was significantly lower compared with cells co-cultured with GEPlow HCC cells (). Furthermore, NK cytotoxicity against GEPhigh HCC cells was significantly lower than that against GEPlow HCC cells (), confirming that GEPhigh HCC cells possess a selective advantage to evade NK cytotoxicity in HCC patients.
GEP expression modulated NK activity in HCC patients
To elucidate the role of GEP in modulating NK activity in HCC patients, we established stable transfectants with suppression of GEP using shRNA (sh) in Hep3B cells, a HCC cell line with high endogenous GEP levels, and stable transfectants that overexpress GEP using GEP full-length cDNA (FL) in HepG2, a HCC cell line with low endogenous GEP levels. shRNA negative control and vector control were included as controls (CTL) for transfection in Hep3B and HepG2, respectively. Results from flow cytometric analysis showed that GEP protein levels were significantly downregulated in Hep3B and upregulated in HepG2 cells after transfection (). Secretory GEP levels also showed significant alterations. GEP transfectants were then co-cultured with NK cells from HCC patients. Upon co-culture with GEP-suppressed Hep3B cells the surface expression of NKG2D and CD69 on NK cells was significantly increased compared with that on cells co-cultured with control cells, while the opposite was observed when NK cells were co-cultured with GEP-overexpressing HepG2 cells (). In addition, production of IFN-γ and perforin was significantly increased when NK cells were co-cultured with GEP-suppressed Hep3B cells, but decreased when GEP was overexpressed in HepG2 cells (). The effect of GEP on NK activity was further confirmed by the significantly enhanced cytotoxicity against GEP-suppressed HCC cells and reduced cytotoxicity upon GEP overexpression ().
Figure 4. GEP expression modulated natural killer cell activity in HCC patients. (A) Modulation of GEP in HCC cell lines by transfection. Cellular and secretory levels of GEP in Hep3B (high endogenous GEP) cells were significantly suppressed by shRNA whereas significant overexpression of GEP was induced in HepG2 cells (low endogenous GEP) by transfection of full-length cDNA construct. (B) HCC cells and their GEP transfectants were co-cultured with peripheral blood NK cells from HCC patients at an effector cell:target cell ratio of 4:1 for 24 h. NK cell surface expression of NKG2D and CD69, production of IFN-γ and perforin, and cytotoxicity against HCC cells were assessed. P: parental cells; CTL: shRNA negative control for Hep3B or vector control for HepG2; sh: GEP shRNA; FL: GEP full-length cDNA.
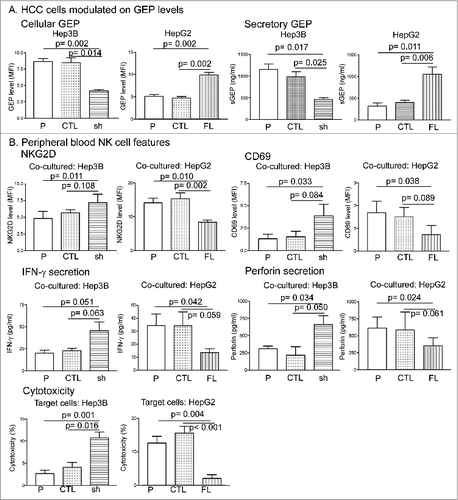
It is noteworthy that upon GEP suppression in HCC cells, perforin production and cytotoxicity of NK cells from patients were restored to levels comparable to those of healthy NK cells (). This implies that the impaired activities of the patients' NK cells can be rescued by suppressing GEP in HCC cells.
Anti-GEP antibody A23 restored NK activity in HCC patients
To investigate the therapeutic potential of GEP, HCC cells were treated with the anti-GEP monoclonal antibody A23 and the effect on activity of NK cells from the patients was assessed. HCC cells were treated with 50 μg/mL A23 or isotype control (mouse IgG1κ) for 24 h prior to co-culture with NK cells. Upon A23 treatment, endogenous GEP levels in both Hep3B and HepG2 cells were significantly reduced compared with those of control cells (). When co-cultured with A23-treated HCC cells, surface expression of NKG2D and CD69 on NK cells was significantly higher than in controls (), suggesting that NK cells were activated upon in vitro A23 treatment of HCC cells. Moreover, production of IFN-γ and perforin was significantly increased when NK cells of patients were co-cultured with A23-treated HCC cells (). Importantly, cytotoxicity of patients' NK cells against HCC cells was augmented upon A23 treatment (), confirming that NK activity was restored by targeting GEP with antibody. The above result echoes that of GEP suppression by transfection, in which perforin production and cytotoxicity of patients' NK cells could be restored to levels comparable to those of healthy NK cells ().
Figure 5. Anti-GEP antibody A23 restored natural killer activity in HCC patients. HCC cells were treated with anti-GEP antibody (A23), mouse IgG isotype (IgG) (50 μg/mL) or without antibody (CTL) in serum-starved condition (1% FBS) for 24 h. (A) GEP levels in HCC cells after A23 treatment. (B) HCC cells were co-cultured with NK cells at an effector cell:target cell (E:T) ratio of 4:1 for 24 h. NK cell surface expression of NKG2D and CD69 and production of IFN-γ and perforin were measured. HCC cells were treated with or without A23 or IgG for 24 h prior to co-culture with NK cells treated with or without IL-12 (50 ng/mL) at an effector cell:target cell ratio of 4:1 for 24 h and NK cytotoxicity was assessed.
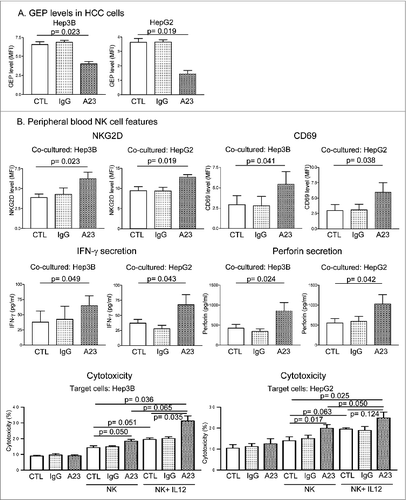
Since A23 could sensitize HCC cells to NK cytotoxicity, it was postulated that an additional antitumor effect might result if the patient's NK cells are pre-activated before A23 treatment of HCC. Therefore, NK cells were treated with NK-activating Th1 cytokine IL-12 prior to co-culture with HCC cells with or without A23 treatment. An additional cytotoxic effect of NK cells was observed upon combination of IL-12 treatment of NK cells and A23 treatment of HCC cells (). The result suggests that combination therapy with anti-GEP monoclonal antibody and immunotherapy targeting NK cell activation might further improve the antitumor effect against HCC.
Anti-GEP antibody A23 elicited antibody-dependent cell-mediated cytotoxicity (ADCC) mediated by NK cells
To further characterize the immunomodulatory mechanism of A23, ADCC induced by A23 was assessed. ADCC occurs when antibodies bind to antigen on tumor cells and the antibody Fc domains engage Fc receptors on the surface of immune effector cells.Citation26 HCC cells were stained with or without anti-GEP antibody A23 or mouse isotype control antibody for 30 min and the antibody-labeled HCC cells were then co-cultured with healthy PBMCs. A23, but not isotype control, significantly induced ADCC of human PBMCs against both Hep3B and HepG2 cells in a dose-dependent manner ().
Figure 6. Anti-GEP antibody A23 elicited ADCC mediated by natural killer cells. (A) HCC cells were incubated with or without anti-GEP antibody A23 (A23) or mouse isotype control (IgG) at the indicated antibody concentrations for 30 min prior to co-culture with or without healthy PBMCs at an effector cell:target cell (E:T) ratio of 25:1 for 5 h. Cytotoxicity against HCC cells was measured. *P < 0.05, **P < 0.01, compared with control cells without antibody. (B) HCC cells were incubated with or without anti-GEP antibody A23 at the indicated concentrations for 30 min prior to co-culture with healthy NK cell-depleted PBMCs at an E:T ratio of 25:1 for 5 h. (C) CD56+ NK cells were isolated from healthy PBMCs and cultured with HCC cells at the indicated E:T ratio for 5 h. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control cells without antibody at the corresponding E:T ratio. (D) CD56+ NK cells were isolated from PBMCs of HCC patients and co-cultured with HCC cells labeled with or without A23 or IgG at an E:T ratio of 4:1 for 5 h. *P < 0.05, **P < 0.01 compared with control cells without antibody.
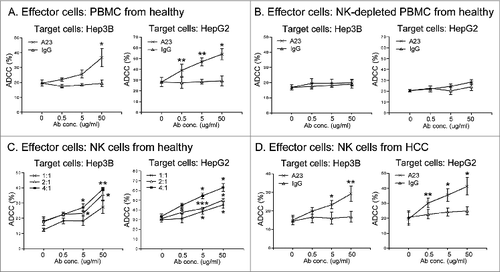
Next, we further elucidated whether NK cells were responsible for the ADCC activity of A23 in humans. When NK cells were depleted from PBMCs the A23-mediated ADCC in both cell lines was markedly abolished (), suggesting that the ADCC effect was at least partially mediated by NK cells. A23-labeled HCC cells were then co-cultured with healthy NK cells at different effector cell:target cell (E:T) ratios, and A23-mediated ADCC against HCC cells increased as the E:T ratio increased (). These data confirmed that NK cells also play an important role as effector cells for A23-induced ADCC in humans.
To validate the ADCC effect of A23 in HCC patients, A23-treated HCC cells were co-cultured with patients' NK cells at an E:T ratio of 4:1. A23, but not isotype control, significantly induced ADCC of patients' NK cells against both Hep3B and HepG2 cells in a dose-dependent manner (), indicating that A23 could also elicit ADCC mediated by NK cells in HCC patients.
Discussion
Impairment of NK activity has been identified in various human cancers including HCC,Citation10,11 but the underlying mechanism remains unclear. In this study, we showed that NK cells in HCC patients were defective in their expression of stimulatory receptors, cytokine secretion, and cytotoxicity. We also provided evidence for the potential linkage between GEP expression in HCC cells and impaired NK activity. By transfection experiments, we confirmed that high GEP expression in HCC cells significantly impaired NK activity in HCC patients. By targeting GEP with antibody, NK activity could be restored and HCC cells were sensitized to NK cytotoxicity. Since NK immunosurveillance represents a crucial antitumor immune response, investigation of the underlying mechanisms for its impairment in HCC patients would open a new avenue for effective treatment targeting the restoration of NK activity.
NK cell function is regulated by a balance between stimulatory and inhibitory signals mediated by an array of cell surface receptors. Recently, the role of the stimulatory receptor NKG2D and its ligands in the antitumor responses of NK cells has received considerable interest.Citation27 NKG2D is known to protect the host from tumor initiation and its activation results in cytokine secretion and exocytosis of cytotoxic granules.Citation24 NKG2D-deficient mice display impaired immunosurveillance of epithelial and lymphoid malignancies.Citation28,29 To circumvent NK immunosurveillance, tumors have evolved to downregulate the expression of NKG2D ligands on their surface. It is crucial to gain insight into such suppressive processes in order to enhance the immunogenicity of tumor cells by restoring the expression of NKG2D ligands.
We previously demonstrated that GEP conferred HCC cells with the ability to evade immunosurveillance of healthy NK cells through the NKG2D–MICA pathway by promoting shedding of MICA from the HCC cell surface through the action of matrix metalloproteinase (MMP)2 and MMP9.Citation21 Here, we showed that GEP-expressing cells expressed significantly lower levels of surface NKG2D ligands MICA and ULBP1, and produced higher levels of sMICA than their GEP-negative counterparts. Upon interaction with GEP-expressing cells, NKG2D levels on patients' NK cells were significantly downregulated. These findings suggested that GEP might impair the activity of NK cells in HCC patients at least partially through the NKG2D–MICA pathway. Reducing or blocking the proteolytic cleavage of soluble NKG2D ligands represents an interesting biological mechanism of potential interest for targeting NKG2D signaling. Although in vitro inhibition of MMP activity was shown to reduce the shedding of NKG2D ligands, specific inhibitors are needed to avoid toxicity.Citation30 More effort is required to translate the experimental evidence for NKG2D signaling into new therapeutic avenues to stimulate the cell-mediated antitumor immunity.
ADCC is an innate immune effector mechanism by which tumor cell death is evoked when antibodies bind to antigens on tumor cells and the antibody Fc domains engage Fc receptors on the surface of cytotoxic immune cells such as NK cells.Citation31 Here, we demonstrated that the anti-GEP monoclonal antibody A23 could induce NK cell-mediated ADCC against HCC cells. Therefore, the antitumor effect of anti-GEP monoclonal antibody A23 can be further amplified by inducing destruction of the A23-coated HCC cells by NK cells.
In addition to functional impairment of NK cells, GEP levels in HCC tumors were also shown to be negatively correlated with reduced NK infiltration in HCC tumors. A previous study reported that NK cells were predominantly present in tumors of patients with longer survival.Citation8,32 Moreover, NK cell frequency was positively correlated with tumor apoptosis, and negatively correlated with tumor proliferation.Citation8 CXCL10, CCL5, and CCL2 were the main chemokines attracting NK cells into the tumor microenvironment in HCC.Citation6 These chemokines attract NK cells, which produce more IFN-γ upon activation and trigger a paracrine loop further amplifying chemokine production and lymphocyte infiltration.Citation6,33 Tumor cells have been shown to acquire the ability to produce chemokines to alter the tumor microenvironment.Citation34,35 Further investigation into the chemokine expression profiles of GEP-expressing HCC cells is required to delineate the mechanism by which GEP expression is associated with NK infiltration in HCC.
Taken together, our findings revealed that GEP reduces NK activities in HCC patients, in addition to rendering the HCC cells resistant to NK immunosurveillance. Targeting GEP with antibody could restore NK activity in HCC patients and sensitize the HCC cells to NK cytotoxicity. The present study has therefore uncovered the immunostimulatory function of the anti-GEP antibody A23, in addition to its direct antigrowth and chemosensitization effects on HCC cells. In fact, the only FDA-approved targeted agent for HCC, sorafenib, was also shown to enhance NK sensitivity of HCC cellsCitation36 and induce antitumor responses of NK cells in HCC.Citation37 Taken together, these findings implicate targeting of GEP as a novel therapeutic approach to suppressing tumor immunoevasion and restoring NK activity in HCC. This study will provide new insights into the development of novel immune-based therapies for HCC.
Materials and Methods
Clinical specimens
The study protocol was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (HKU/HA HKW IRB). Patients who underwent curative partial hepatectomy for HCC at Queen Mary Hospital, Hong Kong, (n = 42) were recruited to the study with written informed consent. All patients had been diagnosed with primary HCC and confirmed by pathologic examination. Heparinized blood samples were collected from HCC patients before tumor resection to isolate PBMCs and NK cells for characterization and functional assays. Tumor and matched adjacent non-tumor tissue from HCC patients were examined for GEP expression levels, NK cell frequencies, and phenotypes.
HCC cell culture
Human HCC cell lines Hep3B and HepG2 were purchased from American Type Culture Collection (Manassas, VA) and authenticated using short tandem repeat DNA profiles by the company. The cells were expanded and cultured immediately, or cryopreserved in multiple aliquots and used within 6 months of resuscitation as described.Citation16 Stable transfectants for GEP overexpression were established by transfecting GEP full-length cDNA (FL) into HepG2 cells, whereas GEP suppression was performed by transfecting GEP shRNA (sh) into Hep3B cells. Vector control and shRNA negative control were included as controls for transfections in HepG2 and Hep3B cells, respectively. All transfectants were maintained in advanced minimum essential media (AMEM) with 10% FBS and 0.4 mg/mL G418 (Life Technologies). GEP blockage in Hep3B was performed by incubating the cells with or without 50 μg/mL anti-GEP monoclonal antibody A23 (Versitech Ltd, Hong Kong) or mouse IgG isotype control (Sigma-Aldrich) for 24 h. Doses of A23 were previously titrated and 50 µg/mL of A23 alone did not exert any cytotoxic effect on HCC cells.Citation21
Isolation of human NK cells
NK cells were isolated from peripheral blood of HCC patients or buffy coats of healthy individuals. PBMCs were collected by density gradient centrifugation using Ficoll-Paque Plus. NK cells were then sorted from PBMCs by negative selection with anti-CD3 using magnetic cell sorting prior to positive selection with anti-CD56 microbeads according to the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). The isolated NK cells were cultured in RPMI 1640 with 10% FBS and subjected to characterization and functional assays.
Isolation of GEP-expressing cells
Isolation of GEPhigh and GEPlow HCC cells was performed by magnetic cell sorting (Miltenyi Biotec) using anti-GEP monoclonal antibody A23 as previously described.Citation20 Purity of the sorted populations was analyzed by flow cytometry using mouse anti-human GEP antibody (R&D Systems) recognizing different epitopes from the antibody used for cell sorting. Post-sorting analysis consistently indicated purities of >80% with minimal cell death (<10%).
Cytotoxicity assay
Cytotoxicity was determined by dual-color flow cytometry using the HCC cell lines Hep3B or HepG2 as target cells and NK cells as effector cells as previously described.Citation21 For ADCC, HCC cells were stained with or without the indicated concentrations of anti-GEP antibody A23 or mouse IgG isotype antibody for 30 min at 4°C. Labeled target cells were then co-cultured with effector cells for 5 h at the indicated effector cell:target cell (E:T) ratio.
Immunofluorescence staining and flow cytometric analysis
For HCC cell surface expression of ligands for the NK cell activating receptors NKG2D, MICA, and unique long 16 (UL16)-binding protein (ULBP)1, cells were stained with APC-conjugated mouse anti-human MICA (Biolegend), ULBP1 (R&D Systems), or an equal amount of corresponding isotype control (Biolegend). For intracellular expression of GEP, cells were permeabilized with 0.1% saponin and then incubated with FITC-conjugated mouse anti-human GEP antibody (in-house, described previouslyCitation18) or an equal amount of FITC-conjugated mouse IgG isotype (Sigma-Aldrich). For NKG2D and CD69 surface expression on NK cells, cells were stained with APC-conjugated mouse anti-human NKG2D, PE-conjugated mouse anti-human CD69 antibody, or an equal amount of corresponding isotype control (BD Biosciences). Results were expressed as percentage of cells positive for the above markers or mean fluorescence intensity (MFI) of the markers, after subtracting the non-specific background signal (isotype controls), by flow cytometry (FACSCalibur, BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
Levels of soluble GEP (Adipogen Inc., Seoul, Korea), MICA (R&D Systems), IFN-γ (R&D Systems), and perforin (Abcam, Cambridge, UK) in culture supernatants were determined by ELISA according to the manufacturers' instructions.
Immunohistochemical (IHC) staining
IHC staining was performed with the Dako Envision Plus System (Dako) following the manufacturer's instruction with modifications as previously described.Citation21 HCC tumors were stained with mouse anti-human GEP antibody (in-house, described previouslyCitation18) and mouse anti-human CD56 antibody (Dako).
Statistical analyses
All analyses were performed using the statistical software GraphPad Prism Version 3.00 for Windows (GraphPad Software). All in vitro data were expressed as mean values ± standard deviation (SD) from at least 3 independent experiments. Continuous variables were assessed by Spearman correlation and compared between groups by ANOVA (clinical samples) or Student t test (in vitro models). A P value <0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
The University of Hong Kong has filed patent applications for the described works. STC has received Pfizer collaborative research grants. Providers of funding had no role in study design, data collection, analysis, interpretation of the data, writing of the article, or the decision to submit the article for publication. The other authors declare no conflict of interest.
2015ONCOIMM0017R-s01.pdf
Download PDF (17.2 KB)Funding
This study was supported in part by Hong Kong Research Grants Council (GRF 764111, 764112), Health and Medical Research Fund (01121536 and 01121566) and Sun C.Y. Research Foundation for Hepatobiliary and Pancreatic Surgery of the University of Hong Kong.
References
- Levy EM, Roberti MP, Mordoh J. Natural killer cells in human cancer: from biological functions to clinical applications. J Biomed Biotechnol 2011; 2011:676198; PMID:21541191; http://dx.doi.org/10.1155/2011/676198
- Caligiuri MA. Human natural killer cells. Blood 2008; 112:461-9; PMID:18650461; http://dx.doi.org/10.1182/blood-2007-09-077438
- Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 2000; 356:1795-9; PMID:11117911; http://dx.doi.org/10.1016/S0140-6736(00)03231-1
- Eckl J, Buchner A, Prinz PU, Riesenberg R, Siegert SI, Kammerer R, Nelson PJ, Noessner E. Transcript signature predicts tissue NK cell content and defines renal cell carcinoma subgroups independent of TNM staging. J Mol Med (Berl) 2011; 90:55-66; PMID:21870102; http://dx.doi.org/10.1007/s00109-011-0806-7
- Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, Koch M, Weitz J, Kloor M, Zoernig I, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res 2011; 17:678-89; PMID:21325295; http://dx.doi.org/10.1158/1078-0432.CCR-10-2173
- Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, Weber A, Slankamenac K, Poon RT, Yang H, et al. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut 2012; 61:427-38; PMID:21930732; http://dx.doi.org/10.1136/gutjnl-2011-300509
- Taketomi A, Shimada M, Shirabe K, Kajiyama K, Gion T, Sugimachi K. Natural killer cell activity in patients with hepatocellular carcinoma: a new prognostic indicator after hepatectomy. Cancer 1998; 83:58-63; PMID:9655293; http://dx.doi.org/10.1002/(SICI)1097-0142(19980701)83:1<58::AID-CNCR8>3.0.CO;2-A
- Chew V, Tow C, Teo M, Wong HL, Chan J, Gehring A, Loh M, Bolze A, Quek R, Lee VK, et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J Hepatol 2010; 52:370-9; PMID:19720422; http://dx.doi.org/10.1016/j.jhep.2009.07.013
- Zerbini A, Pilli M, Laccabue D, Pelosi G, Molinari A, Negri E, Cerioni S, Fagnoni F, Soliani P, Ferrari C, et al. Radiofrequency thermal ablation for hepatocellular carcinoma stimulates autologous NK-cell response. Gastroenterology 2010; 138:1931-42; PMID:20060829; http://dx.doi.org/10.1053/j.gastro.2009.12.051
- Jinushi M, Takehara T, Tatsumi T, Hiramatsu N, Sakamori R, Yamaguchi S, Hayashi N. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol 2005; 43:1013-20; PMID:16168521; http://dx.doi.org/10.1016/j.jhep.2005.05.026
- Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, Shi M, Zhang H, Yang Y, Wu H, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol 2008; 129:428-37; PMID:18824414; http://dx.doi.org/10.1016/j.clim.2008.08.012
- Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, Lehner F, Manns MP, Greten TF, Korangy F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009; 50:799-807; PMID:19551844; http://dx.doi.org/10.1002/hep.23054
- Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays 2009; 31:1245-54; PMID:19795409; http://dx.doi.org/10.1002/bies.200900086
- Cheung ST, Cheung PF, Cheng KC, Wong NC, Fan ST. Granulin-epithelin precursor and ATP-dependent binding cassette (ABC)B5 regulate liver cancer cell chemoresistance. Gastroenterology 2011; 140:344-55; PMID:20682318; http://dx.doi.org/10.1053/j.gastro.2010.07.049
- Cheung ST, Wong SY, Lee YT, Fan ST. GEP associates with wild-type p53 in hepatocellular carcinoma. Oncol Rep 2006; 15:1507-11; PMID:16685387
- Cheung ST, Wong SY, Leung KL, Chen X, So S, Ng IO, Fan ST. Granulin-epithelin precursor overexpression promotes growth and invasion of hepatocellular carcinoma. Clin Cancer Res 2004; 10:7629-36; PMID:15569995; http://dx.doi.org/10.1158/1078-0432.CCR-04-0960
- Yip CW, Cheung PF, Leung IC, Wong NC, Cheng CK, Fan ST, Cheung ST. Granulin-epithelin precursor interacts with heparan sulfate on liver cancer cells. Carcinogenesis 2014; 35:2485-94; PMID:25115442; http://dx.doi.org/10.1093/carcin/bgu164
- Ho JC, Ip YC, Cheung ST, Lee YT, Chan KF, Wong SY, Fan ST. Granulin-epithelin precursor as a therapeutic target for hepatocellular carcinoma. Hepatology 2008; 47:1524-32; PMID:18393387; http://dx.doi.org/10.1002/hep.22191
- Wong NC, Cheung PF, Yip CW, Chan KF, Ng IO, Fan ST, Cheung ST. Antibody against granulin-epithelin precursor sensitizes hepatocellular carcinoma to chemotherapeutic agents. Mol Cancer Ther 2014; 13:3001-12; PMID:25253787; http://dx.doi.org/10.1158/1535-7163.MCT-14-0012
- Cheung PF, Cheng CK, Wong NC, Ho JC, Yip CW, Lui VC, Cheung AN, Fan ST, Cheung ST. Granulin-epithelin precursor is an oncofetal protein defining hepatic cancer stem cells. PLoS One 2011; 6:e28246; PMID:22194816; http://dx.doi.org/10.1371/journal.pone.0028246
- Cheung PF, Yip CW, Wong NC, Fong DY, Ng LW, Wan AM, Wong CK, Cheung TT, Ng IO, Poon RT, et al. Granulin-epithelin precursor renders hepatocellular carcinoma cells resistant to natural killer cytotoxicity. Cancer Immunol Res 2014; 2:1209-19; PMID:25315249; http://dx.doi.org/10.1158/2326-6066.CIR-14-0096
- Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol 2001; 22:633-40; PMID:11698225; http://dx.doi.org/10.1016/S1471-4906(01)02060-9
- Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, Carson WE, Caligiuri MA. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001; 97:3146-51; PMID:11342442; http://dx.doi.org/10.1182/blood.V97.10.3146
- Hayakawa Y, Kelly JM, Westwood JA, Darcy PK, Diefenbach A, Raulet D, Smyth MJ. Cutting edge: tumor rejection mediated by NKG2D receptor-ligand interaction is dependent upon perforin. J Immunol 2002; 169:5377-81; PMID:12421908; http://dx.doi.org/10.4049/jimmunol.169.10.5377
- Kamimura H, Yamagiwa S, Tsuchiya A, Takamura M, Matsuda Y, Ohkoshi S, Inoue M, Wakai T, Shirai Y, Nomoto M, et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J Hepatol 2012; 56:381-8; PMID:21756848; http://dx.doi.org/10.1016/j.jhep.2011.06.017
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012; 12:278-87; PMID:22437872; http://dx.doi.org/10.1038/nrc3236
- Moretta L, Bottino C, Pende D, Castriconi R, Mingari MC, Moretta A. Surface NK receptors and their ligands on tumor cells. Semin Immunol 2006; 18:151-8; PMID:16730454; http://dx.doi.org/10.1016/j.smim.2006.03.002
- Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med 2005; 202:583-8; PMID:16129707; http://dx.doi.org/10.1084/jem.20050994
- Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008; 28:571-80; PMID:18394936; http://dx.doi.org/10.1016/j.immuni.2008.02.016
- Folgueras AR, Pendas AM, Sanchez LM, Lopez-Otin C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol 2004; 48:411-24; PMID:15349816; http://dx.doi.org/10.1387/ijdb.041811af
- Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Disc 2012; 11:311-U87; PMID:22460124; http://dx.doi.org/10.1038/nrd2909
- Guo CL, Yang HC, Yang XH, Cheng W, Dong TX, Zhu WJ, Xu Z, Zhao L. Associations between infiltrating lymphocyte subsets and hepatocellular carcinoma. Asian Pac J Cancer Prev: APJCP 2012; 13:5909-13; PMID:23317279; http://dx.doi.org/10.7314/APJCP.2012.13.11.5909
- Maghazachi AA. Role of chemokines in the biology of natural killer cells. Curr Topics Microbiol Immunol 2010; 341:37-58; PMID:20369317
- Chow MT, Luster AD. Chemokines in Cancer. Cancer Immunol Res 2014; 2:1125-31; PMID:25480554; http://dx.doi.org/10.1158/2326-6066.CIR-14-0160
- Koizumi K, Hojo S, Akashi T, Yasumoto K, Saiki I. Chemokine receptors in cancer metastasis and cancer cell-derived chemokines in host immune response. Cancer Sci 2007; 98:1652-8; PMID:17894551; http://dx.doi.org/10.1111/j.1349-7006.2007.00606.x
- Kohga K, Takehara T, Tatsumi T, Ishida H, Miyagi T, Hosui A, Hayashi N. Sorafenib inhibits the shedding of major histocompatibility complex class I-related chain A on hepatocellular carcinoma cells by down-regulating a disintegrin and metalloproteinase 9. Hepatology 2010; 51:1264-73; PMID:20099300; http://dx.doi.org/10.1002/hep.23456
- Sprinzl MF, Reisinger F, Puschnik A, Ringelhan M, Ackermann K, Hartmann D, Schiemann M, Weinmann A, Galle PR, Schuchmann M, et al. Sorafenib perpetuates cellular anticancer effector functions by modulating the crosstalk between macrophages and natural killer cells. Hepatology 2013; 57:2358-68; PMID:23424039; http://dx.doi.org/10.1002/hep.26328