
Abstract
Fractionated radiation therapy (RT) leads to adaptive changes in the tumor microenvironment that may limit the generation of an antitumor immune response. We demonstrated that fractionated RT led to increased tumor cell expression of programmed cell death ligand 1 (PD-L1) in response to CD8+ T cell production of interferon gamma. Our data reveal that the efficacy of fractionated RT can be significantly improved through the generation of durable systemic immune responses when combined with concurrent, but not sequential, blockade of the PD-1/PD-L1 pathway.
Radiation therapy (RT) is delivered to approximately 50–60% of all cancer patients and is a component of curative treatment in approximately 40% of cancer patients. Several studies demonstrate that, in addition to its direct cytoreductive effect, RT-induced cell death can be immunogenic. Treatment with RT leads to the release of damage-associated molecular patterns (DAMPs) by cancer cells that can facilitate the recruitment and activation of antigen presenting cells (APC), such as dendritic cells (DCs), and priming of tumor antigen-specific T cells (reviewed in ref. 1Citation1). The contribution of the anticancer immune response to the clinical efficacy of RT is under investigation; however, generation of systemic immunity and the “abscopal effect” are rare in clinical practice. Preclinical data demonstrate that whereas vaccination of mice with irradiated tumor cells can lead to potent T-cell priming, in situ vaccination of established experimental tumors with RT alone is rarely able to induce durable systemic antitumor immune responses.Citation2,3 This lack of a durable immune response to RT in established tumors is thought to be a consequence of an immunosuppressive tumor microenvironment that may limit the magnitude and durability of the anticancer immune response.Citation4 This immune failure may contribute to disease recurrence, which is the leading cause of death following RT.
To date, the majority of preclinical studies have investigated the effect of a large single dose of RT on the generation of systemic immune responses. Despite substantial improvements in image-guided stereotactic body radiation therapy (SBRT), which permits the delivery of larger doses of RT over a much shorter time period (hypofractionation), and in the administration of a single large ablative dose, the majority of RT in the clinic is delivered in multiple daily fractions of approximately 1.8–2 Gy. However, relatively few studies have addressed the immunogenicity of fractionated RT with this lower dose (reviewed in Citation5), therefore this was the focus of our study.
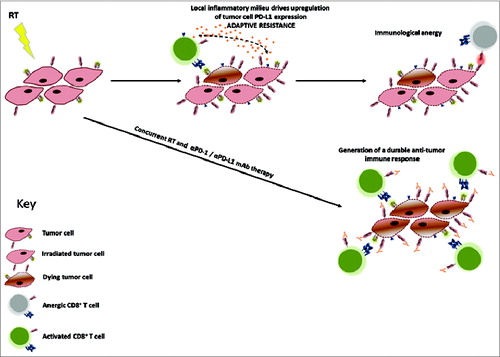
We conducted phenotypic analysis of the tumor microenvironment following treatment with fractionated RT to identify drivers of immunosuppression. Our data revealed that local fractionated RT to the tumor led to upregulation of programed cell death ligand (PD-L)-1 on cancer cells in vivo. Through binding its cognate receptor PD-1 expressed on lymphocytes, PD-L1 is involved in the maintenance of peripheral tolerance and modulation of acute inflammatory responses through inhibition of T-cell receptor (TCR) signal transduction, leading to apoptosis of antigen-specific T-cells.Citation6,7 Our studies revealed that PD-L1 expression was induced on tumor cells in vivo, but not in vitro, following RT. Given this disparity, we evaluated the contribution of the local tumor microenvironment to the expression of PD-L1 by tumor cells. We determined that RT led to CD8+ T-cell production of interferon gamma (IFNγ), which was responsible for the upregulation of tumor cell PD-L1 expression. These data reveal that tumor cells can adapt to the microenvironmental changes following RT to circumvent T-cell immunity, potentially contributing to treatment failure. Moreover, treatment with RT also led to increased expression of PD-L1 on cells with a myeloid-derived suppressor cell (MDSC) phenotype (CD11b+GR1Hi), further demonstrating the potential of blockade of the PD-1/PD-L1 axis to improve the effectiveness of RT. Using syngeneic models of melanoma, colorectal cancer, and triple negative breast cancer, we demonstrated that the efficacy of RT can be enhanced when delivered in combination with monoclonal antibody (mAb) targeted against PD-1 or PD-L1.Citation8 The efficacy of combination therapy was dependent on the activity of CD8+ T cells, and long-term (>100 days) surviving mice were protected against disease recurrence by the generation of a tumor antigen-specific memory immune response. In a separate preclinical study, Deng and colleagues demonstrated that high-dose RT (12 or 20 Gy) led to increased expression of PD-L1 on the surface of both tumor cells and DCs, and that anti-PD-L1 treatment improved the efficacy of RT.Citation9 Together, these studies rationalize blockade of the PD-1/PD-L1 axis to improve outcomes in patients receiving SBRT or low-dose fractionated RT (see ).
Figure 1. Tumor cell adaptive resistance following radiation therapy. Radiation therapy (RT) leads to upregulation of programmed death ligand 1 (PD-L1) expression on tumor cells through the activation of tumor resident ανδ infiltrating CD8+ T cells and local production of interferon gamma (IFNγ). This adaptive upregulation of PD-L1 leads to T-cell anergy through binding of T-cell–expressed PD-1. Blockade of either PD-1 or PD-L1 using monoclonal antibody administered concurrently with RT inhibits this immunosuppressive pathway, augmenting the generation of a durable and effective antitumor immune response.
A critical question to support translation of this therapeutic approach is which combination schedule should be adopted for maximal immunogenicity. To address this issue we tested 3 different regimens: (1) concurrent RT and αPD-L1 mAb starting at the beginning of the RT cycle, (2) concurrent RT and αPD-L1 mAb starting at the end of the RT cycle, and (3) sequential therapy with αPD-L1 mAb administered 7 d after the completion of RT. We showed that concurrent therapy was optimal for generating effective antitumor immunity and long-term tumor control. In contrast, the efficacy of sequential therapy was not significantly different to that of RT alone. Measurement of the kinetics of tumor cell PD-L1 expression revealed an acute increase following RT that persisted for at least 7 d after completion of the RT cycle. In addition, we showed that the expression of PD-1 on tumor-infiltrating T cells, which has been shown to identify the tumor antigen-specific T-cell repertoire,Citation10 is elevated immediately after RT. Taken together, these data suggest that blockade of the PD-1/PD-L1 pathway is optimal when synchronized with the delivery of fractionated RT to prevent the induction of immunological anergy. These observations may be important for clinical translation as a sequential dosing schedule is often the de facto regimen adopted where there are uncertainties about the tolerability of a novel combination.
In summary, we have demonstrated that fractionated RT can prime antitumor CD8+ T-cell responses, which may be attenuated by signaling through the PD-1/PD-L1 axis. Inhibition of this pathway augments the generation of durable effective antitumor responses and improves survival. However, scheduling of the combination may be important for optimal outcome and our findings suggest that concurrent blockade of the PD-1/PD-L1 axis is required to improve the efficacy of fractionated RT. This study provides a strong rationale for clinical translation and has important implications for clinical trial design.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31:51-72; PMID:23157435; http://dx.doi.org/10.1146/annurev-immunol-032712-100008
- Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005; 174:7516-23; PMID:15944250; http://dx.doi.org/10.4049/jimmunol.174.12.7516
- Dovedi SJ, Melis MH, Wilkinson RW, Adlard AL, Stratford IJ, Honeychurch J, Illidge TM. Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood 2013; 121:251-9; PMID:23086756; http://dx.doi.org/10.1182/blood-2012-05-432393
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263-74; PMID:15776005; http://dx.doi.org/10.1038/nrc1586
- Demaria S, Formenti SC. Radiation as an immunological adjuvant: current evidence on dose and fractionation. Front Oncol 2012; 2:153; PMID:23112958; http://dx.doi.org/10.3389/fonc.2012.00153
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8:793-800; PMID:12091876; http://dx.doi.org/10.1038/nm0902-1039c
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027-34; PMID:11015443; http://dx.doi.org/10.1084/jem.192.7.1027
- Dovedi SJ, Adlard AL, Lipowska-Bhalla G, Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, Stratford IJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res 2014; 74:5458-68; PMID:25274032; http://dx.doi.org/10.1158/0008-5472.CAN-14-1258
- Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014; 124:687-95; PMID:24382348; http://dx.doi.org/10.1172/JCI67313
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124:2246-59; PMID:24667641; http://dx.doi.org/10.1172/JCI73639