
Abstract
Adoptive immunotherapy with cytotoxic T lymphocytes (CTLs) can result in robust and durable antitumor responses. Tumor-infiltrating CTLs produce IFNγ and mediate antitumor activity, but they simultaneously induce counter-regulatory immunosuppressive mechanisms in the tumor by recruiting monocytic myeloid-derived suppressor cells (MDSCs) that limit their proliferation and effector function. Using a murine model of adoptive immunotherapy for B16 melanoma, we developed a strategy to augment CTL activity by downregulating immunosuppression by MDSCs. Intravenous injection of transgenic pmel-1 CTLs into tumor-bearing mice, resulted in their infiltration into the tumor, but this was accompanied by the accumulation of large numbers of monocytic MDSCs (M-MDSCs). These cells hampered CTL function and reduced their numbers in the tumor. We determined that one mechanism responsible for this immunosuppression was the production of nitric oxide (NO) by MDSCs in the tumor. Therefore, mice were given the NO scavenger carboxy-PTIO (C-PTIO) on the day after CTL transfer. This led to the restoration of impaired proliferative capacity and function of the CTLs, resulting in sustained suppression of tumor growth. Thus, we conclude that CTL therapy can be improved by counter-acting immunosuppression. Targeting NO, one mediator of the immunosuppressive activity of M-MDSCs, may be an appropriate strategy to restore impaired CTL function and improve the efficacy of immunotherapy.
Introduction
In established tumors, complex interactions between the many different cell types and molecules contribute to the creation of an immunosuppressive microenvironment.Citation1,2 Tumor cells express ligands for immunological checkpoint molecules on T cells (e.g. PD-L1, B7-H4) and/or produce factors (e.g. IL-10, TGF-β, galectin-1, gangliosides, PGE2, IDO) which suppress immune responses.Citation3,4 Factors involved in pro-apoptotic pathways which delete T cells (e.g. FasL, TRAIL,), as well as the recruitment of immunosuppressive cells, such as regulatory T cells and MDSC, are also major players in this process.Citation5
The suppressive local milieu of the tumor microenvironment induces a state of functional T cell tolerance which can result in the coexistence of antitumor immunity and growing tumor cells.Citation6 Therefore, overcoming the obstacles represented by multiple suppressive factors is necessary for successful immunotherapy.Citation7 The use of immunomodulatory antibodies specific for the immunoinhibitory receptors CTLA-4 or PD-1, or the PD-L1 ligand, has recently been reported, providing dramatic proof-of-concept that cancer immunotherapy can achieve durable and long-lasting responses in cancer patients.Citation8,9 Adoptive T cell therapy, also a promising and rapidly advancing form of immunotherapy, represent a different strategy to overcome tolerance.Citation10,11 This approach acts at the T cell sensitization stage by generating tumor-specific effector T cells in vitro, where there is no tumor-induced immunosuppressive microenvironment. In this way, ex vivo-expanded-infiltrating-leukocytes (TILs), or T cells that have been genetically engineered to express tumor-specific antigen receptors, can be infused back into the patients. The receptors used in such adoptive T cell immunotherapy may be either T cell receptors (TCR) that recognize tumor-specific antigenic peptides presented by MHC class I molecules, or so-called chimeric antigen receptors (CARs) in which tumor-specific antibody or single chain fragment variable regions of the antibodies are fused with the intracellular signaling machinery of the TCR. These T cells with reactivity thus redirected toward tumor-associated antigens can mediate robust and durable antitumor immune activity.Citation12
Previously, we investigated the cellular immune responses in the tumor induced by adoptively-transferred CTLs.Citation13 We reported that CTLs that had infiltrated the tumor recognized tumor antigen, killed tumor cells, and secreted IFNγ, but that they also recruited a massive number of other tumor-infiltrating cells, the majority of which was CD11b+Gr1intLy6C+ M-MDSCs. The antitumor activity of the tumor-infiltrating CTLs was compromised by these accumulated M-MDSCs in the tumor. The CTL response itself thus provoked a counter-regulatory immunosuppressive mechanism via the recruitment of M-MDSCs. NO and reactive oxygen species (ROS) produced by these MDSCs inhibit the proliferation of the antigen-specific CTL.Citation13 Therefore, strategies for regulating CTL-induced M-MDSCs or factors produced by M-MDSCs in the tumor could be desirable for optimizing adoptive immunotherapy with CTLs.
In the present study, mice received the NO radical scavenger C-PTIO the day after CTL injection, with the aim of eliminating NO produced by CTL-induced M-MDSCs in the tumor. We found that the antitumor activity of CTLs was augmented by the abrogation of NO by C-PTIO.
Results
Adoptively-transferred CTLs lose function in the tumor
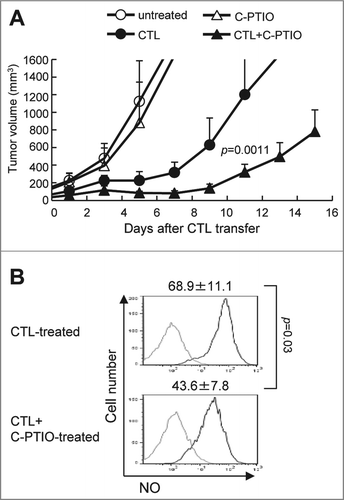
C57BL/6 mice (5 per group) first received a subcutaneous inoculation of B16F10 melanoma cells (1 × 106), and 9 days later, after the tumor had become established, they were given 1 × 107 adoptively-transferred CTLs. As we previously reported,Citation13,14 this treatment suppresses B16 tumor growth, as confirmed here (). Thus, tumor volume already exceeded 300 mm3 by day 4.3 ± 0.9 in untreated mice, but this took 8.4 ± 0.7 d in the CTL-treated mice (p = 0.000058). However, although the adoptively-transferred CTLs caused this substantial delay in tumor growth, it started again around day 7 or 8. Hence, the antitumor activity of a single injection of CTLs was transient. The presence of CTLs in the tumor was found to parallel growth suppression. As shown in , CD8+CD90.1+ CTLs were detected in the tumor but the percentage of these in the TILs gradually decreased from 12.2 ± 2.0% to 7.3 ± 0.6% and further to 2.0 ± 0.6% on day 5, 7 and 17, respectively. This translates to the number of CTLs in the tumor decreasing from 2.3 ± 0.7 × 106 on day 3 to 1.4 ± 0.5 × 105 on day 17 (). As shown in , 25.2 ± 3.4% of CTLs isolated from the tumor produced IFNγ ex vivo without in vitro stimulation on day 3, indicating that they had recognized and responded to antigen in the tumor. When these CTLs were stimulated with gp100 peptide in vitro, 74.2 ± 3.6% produced IFNγ, suggesting that they were fully functional. The percentage of IFNγ producing CTLs in the tumor was found to be 25.2 ± 3.4% on day 3, 6.4 ± 1.4 on day 5, 3.1 ± 0.4 on day 7 and 7.2 ± 3.1% on day 17. Even after gp100 peptide stimulation, only 52.6 ± 1.4%, 25.0 ± 5.0% and 33.8% ± 7.1% of CTLs produced IFNγ on day 5, 7 and 17, respectively. This implies that CTLs could infiltrate in the tumor, recognize tumor cells, and initially exert antitumor activity to suppress tumor growth by a mechanism involving IFNγ, but that they eventually lost this function in the tumor.
Figure 1. Adoptively-transferred CTLs are impaired in the tumor. (A) C57BL/6 mice were injected with B16 melanoma cells (1 × 106), and 9 d later (designated as day 0), tumor-bearing mice (n = 5) received in vitro-activated pmel-1 splenocytes (1 × 107) as CTLs. Tumor volumes were measured every other day. (B) Mice (n = 3) were killed on days 3, 5, 7, and 17 after CTL transfer and infiltration of CTLs into the tumor was analyzed by flow cytometry. The frequency of CTLs in the tumor was determined by quantifying eFluor450−CD45+CD8+CD90.1+ cells. (C) The absolute number of CTLs was calculated as described in the Materials and Methods section and adjusted by the tumor weight (cells/g). (D) IFNγ production by CTLs with or without stimulation with 1 μg/mL hgp100 peptide for 4 h was analyzed on days 3, 5, 7, and 17 after CTL transfer by intracellular staining (n = 3 per group). The experiments were performed independently at least three times with similar results.
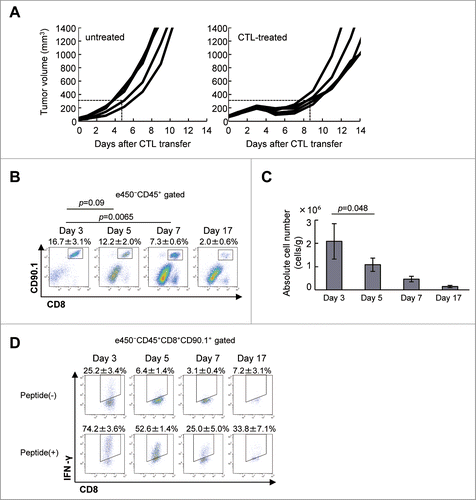
Monocytic MDSCs are recruited into the tumor by adoptively-transferred CTLs
As we reported previously,Citation13 infiltration of CTLs into the tumor resulted in a massive accumulation of tumor-infiltrating cells (), but many of these were not the T cells. Instead, there were large numbers CD11b+Gr1+ MDSCs (), especially CD11b+Gr1intLy6C+ M-MDSC (). Cells with this phenotype have been reported to suppress the function and proliferation of CTLs by releasing NO and ROS.Citation13 The percentages of MDSCs and M-MDSCs in TILs were to some degree comparable between untreated and CTL-treated animals. However, the absolute numbers of these cells were significantly greater in CTL-treated mice (). They displayed a typical MDSC morphology, with a large amount of cytoplasm and an eccentrically-placed kidney bean-shaped nucleus (). CD11b+Gr-1+MDSCs fluoresced green when briefly incubated with the NO indicator DAF-FM (). Therefore, we investigated whether NO production by MDSCs affected the antitumor activity of intratumoral CTLs and if so, whether we could develop strategies to counteract such immunosuppressive activity.
Figure 2 (See previous page). CTL-induced M-MDSCs produce NO. (A) C57BL/6 mice were injected with B16 melanoma cells and 9 days later, tumor-bearing mice (n = 3) were treated as described in . Tumor-infiltrating cells were analyzed on days 0, 5, and 10 after CTL transfer. The absolute number of eFluor450−CD45+ cells at the indicated time points is shown. (B) The eFluor450−CD45+ cells were stained with anti-CD11b and -Gr1 mAbs to detect MDSCs. (C) The absolute number of CD11b+Gr1+ cells at the indicated time points is shown, numbers of cells in each population were calculated as described in the Materials and Methods section and adjusted by the tumor weight (cells/g). (D) Monocytic MDSC were gated as Gr1intLy6C+ cells in the CD11b+Gr1+ population. (E) The absolute numbers of Gr1intLy6C+ monocytic MDSC are shown. (F) CD11b+Gr1+ cells were stained with Diff-quick as described in Materials and Methods; their morphology was assessed using an OLYMPUS BX41 microscope (magnification ×400 left, ×1000 right). (G) Sorted CD11b+Gr1+ cells in F were incubated for 30 min at 37°C with 5 μM DAF-FM (Diaminofluorescein-FM) (SEKISUI MEDICAL) and then stained with biotin-conjugated anti-Gr1, followed by staining with PE-conjugated anti-CD11b, Streptavidin-APC and analyzed by FLUOVIEW FV10 i (OLYMPUS, Tokyo, Japan). The experiments were performed independently at least three times with similar results.
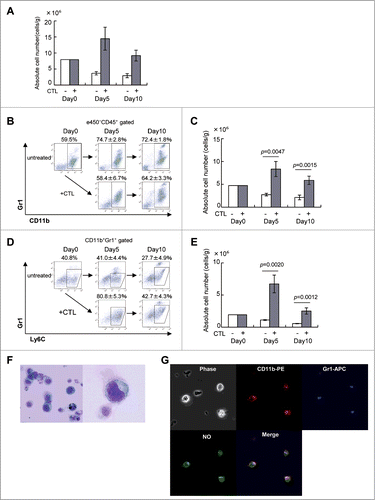
NO produced by MDSCs suppresses CTL proliferation in vitro
To test their capacity to inhibit antigen-specific CTL proliferation, MDSCs were harvested from B16 tumors 3 d after CTL transfer and positive selection by anti-CD11b magnetic beads. Their purity was 86.8% (data not shown). Pmel-1 TCR-transgenic spleen cells were labeled with CFSE and their proliferation evaluated by the shift of fluorescence as the dye is diluted 50% at each cell division. While no proliferation of pmel-1 cells was observed without stimulation by specific peptide, 94.6 ± 0.4% underwent cell division on peptide stimulation (). Proliferation of pmel-1 cells was suppressed to 38.7 ± 7.5%, when they were stimulated with peptide in the presence of MDSCs at a ratio of 1:0.3. These results indicate that MDSCs in the tumors of CTL-treated mice inhibit the proliferation of antigen-specific CTLs. Because NO is one of the immunosuppressive mediators produced by MDSCs, C-PTIO (an NO quenching reagent) and L-NMMA (an iNOS inhibitor) were tested for their ability to abrogate this suppression. In the presence of C-PTIO or L-NMMA, inhibitory activity of MDSCs was completely prevented, and 97.6 ± 0.3% or 99.4 ± 0.1% of the pmel-1 cells could now proliferate even in the presence of MDSCs (, lower lane). These results indicate that NO mediates the immunosuppressive activity of MDSCs on the proliferation of antigen-specific CTLs in B16 tumor-bearing mice treated with adoptively-transferred CTLs. Blockade of NO activity essentially abrogated the inhibitory activity of MDSCs and restored the proliferation potential of the CTLs.
Figure 3. MDSCs inhibit the proliferation of antigen-specific CTLs via NO production. C57BL/6 mice were treated as described in . Tumor-infiltrating cells were prepared from pooled B16 tumors (n = 12) 3 d after CTL transfer and CD11b+Gr1+ cells were positively selected using anti-CD11b magnetic beads. CFSE-labeled pmel-1 CTL were stimulated with hgp100 peptide in the presence or absence of CD11b+Gr1+ cells at the indicated ratio. The proliferation of pmel-1 cells was evaluated by flow cytometry. This was studied in the presence of carboxy-PTIO or L-NMMA. Numbers on the images show the percentage of gated cells (mean ±SD). All experiments shown were performed independently at least three times with similar results.
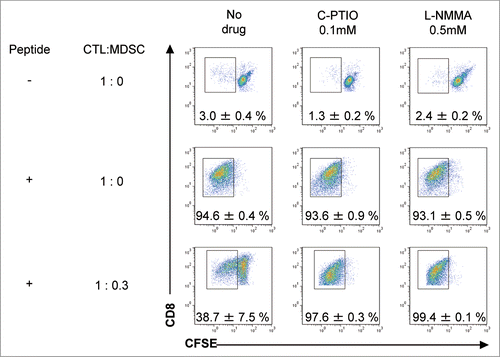
NO quenching by C-PTIO potentiates the antitumor activity of adoptive CTL therapy
To test whether modulation of NO could enhance the antitumor activity of CTL therapy, B16 tumor-bearing mice were divided into 4 groups: untreated or given−intraperitoneal injections of C-PTIO (2 mg, twice daily) alone, given 1 × 107 CTLs alone or a combination of both CTLs and C-PTIO. On day 1 through day 10 after CTL transfer, 2 mg of C-PTIO in 200 μL PBS was injected i.p. twice daily. As shown in , C-PTIO treatment alone did not suppress tumor growth; tumor volume exceeded 500 mm3 by day 4.4 ± 1.5 in untreated mice, and by 4.2 ± 1.0 days in C-PTIO-treated mice. As expected, CTL treatment suppressed tumor growth to some extent, but the combination of CTL+C-PTIO therapy further delayed it. It took 10.1 ± 1.0 days before tumor volume exceeded 500 mm3 on CTL treatment, but 13.6 ± 1.0 days in mice given both CTL and C-PTIO (). On day 11, tumor volume in CTL-treated mice was 1199 ± 480 mm3 as opposed to only 321 ± 90 mm3 in mice treated with a combination of CTL+C-PTIO (p = 0.0011). These results indicate that C-PTIO treatment augmented the antitumor activity of CTLs. To investigate the mechanism of enhancement and confirm that it was mediated via the effect of C-PTIO on NO production by MDSCs, tumors were harvested and tumor-infiltrating cells were analyzed 3 d after CTL transfer. NO production by CD11b+Gr-1+ MDSCs was evaluated as mean fluorescence intensity (MFI) of DAF-FM staining (). Compared to 68.9 ± 11.1 fluorescence units in MDSCs from CTL-treated mice, the MFI was decreased to 43.6 ± 7.8 in MDSCs from mice treated with the CTL+C-PTIO combination. These results indicate that C-PTIO indeed decreased NO production by MDSCs in CTL-treated mice.
Figure 4. Elimination of NO by C-PTIO potentiates the antitumor activity of CTLs. (A) B16 melanoma cells (1 × 106) were implanted intradermally in C57BL/6 mice. Mice were divided into four groups: untreated, Carboxy-PTIO-treated, CTL-treated or CTL and C-PTIO-treated (5–7 mice per group). Nine days later (designated as day 0), tumor-bearing mice received 1 × 107 CTLs. From day 1 to day 10, C-PTIO (2 mg/200 μl PBS) was injected twice daily i.p. Tumor growth was measured every other day. (B) Mice (n = 3 per group) were killed on day 3 and NO expression in CD11b+Gr1+ MDSCs was compared between CTL-treated mice and CTL+C-PTIO-treated mice.
The NO Scavenger C-PTIO restores the function of CTLs in vivo
Tumor-infiltrating cells were harvested on day 3 or 7 after CTL transfer (). The percentage of CTLs was similar on both days regardless of whether or not the mice had received C-PTIO (). However, while the number of CTLs in the tumor was the same on day 3, there were more of them in the CTL+C-PTIO-treated mice on day 7. To identify proliferating CTLs, 2 mg of BrdU was injected i.p. into CTL-treated mice 16 h before sacrifice on day 3 or 7 (). BrdU was incorporated into 3.1 ± 1.3% of CTLs infiltrating the tumors on day 3, while 12.2 ± 6.5% of CTLs from CTL+C-PTIO-treated mice were BrdU-positive. Although not reaching statistical significance, this difference strongly suggests more proliferation in the latter (p = 0.07). In contrast, at day 7, this difference between mice treated with CTL alone and CTL+C-PTIO-treated was no longer seen. Consistent with this, more BrdU-positive CTLs (27.4 ± 5.0%) were observed in the draining lymph node from CTL+C-PTIO-treated mice than in those treated with CTL only (13.7 ± 6.1%) on day 3 (p = 0.04), while there was no difference on day 7. These results suggest that C-PTIO caused an initial reduction in immunosuppression by scavenging NO production from MDSCs and restoring the proliferation of CTLs in vivo.
Figure 5. NO Scavenger C-PTIO restores CTL function. (A) Mice were treated as described in the legend to . TILs were harvested from tumors on days 3 or 7. CTLs in the tumor were analyzed by flow cytometry (left). The absolute number of CTLs was calculated (right). (B) BrdU incorporation by CTLs in the tumor and draining lymph node (DLN) on days 3 and 7 was analyzed by flow cytometry as described in the Materials and Methods section. (C) IFNγ production by CTLs on days 3 and 7 with or without 1 μg/mL hgp100 peptide. (D) The absolute number of IFNγ+ CTLs is shown. (E) CD107a expression on CTLs on days 3 and 7 with or without 1 μg/mL hgp100 peptide. (F) The absolute number of CD107a+ CTLs is shown.
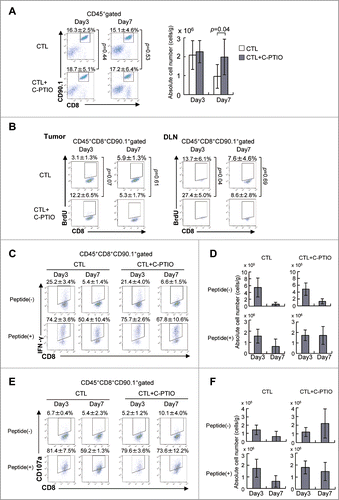
We next determined whether cytokine production was also improved by C-PTIO in vivo. To this end, IFNγ-producing CTLs in the tumor with or without gp100 peptide stimulation were monitored on day 3 and 7 after CTL transfer (). CTLs that were positive for IFNγ ex vivo without peptide stimulation indicated that they had recognized and responded to the tumor in vivo, whereas IFNγ production in response to peptide stimulation indicated the capacity of CTLs to be activated. On day 3, the percentage of CTLs in the tumor that produced IFNγ without peptide stimulation ex vivo was similar in the two treatment arms (25.2 ± 3.4% vs. 21.4 ± 4.0%). The percentage reacting to peptide stimulation was also similar (74.2 ± 3.6% vs. 75.7 ± 2.6%). In CTL-treated mice on day 7, the percentage of IFNγ-producing CTLs decreased relative to day 3, both without or with peptide stimulation (5.4 ± 1.4% or 50.4 ± 10.4%, respectively) Similarly, the percentage of IFNγ producing cells in CTL+C-PTIO-treated mice was also decreased on day 7 (6.6 ± 1.5% without peptide or 67.8 ± 10.6% with peptide). Thus, on day 7, more CTLs (67.8 ± 10.6%) produced IFNγ in response to peptide in CTL+C-PTIO-treated mice than those treated only with CTLs (50.4 ± 10.4%). Similarly, the absolute number of IFNγ-positive CTLs with or without peptide stimulation on day 3 was similar in these two groups (), but on day 7, their numbers were greater in the CTL+C-PTIO group both without (p = 0.08) and significantly with peptide stimulation (p = 0.03). These results indicate that the IFNγ producing capacity of CTLs was hampered in CTL-treated mice, but was better maintained in CTL+C-PTIO-treated mice, due to the elimination of NO.
The cytotoxic activity of CTLs in the tumor after adoptive transfer was evaluated using the CD107a translocation assay (). Consistent with the results of the IFNγ assay, similar percentages of CD107a-positive CTLs in the tumor were seen in CTL-treated and CTL+C-PTIO-treated mice on day 3, both with or without peptide stimulation. On day 7, again, the percentage of CD107a-positive CTLs was higher in the C-PTIO-treated mice, regardless of peptide stimulation. Consistently, the numbers of CD107a-positive CTLs were also higher in the CTL+C-PTIO-treated mice (). Together, these results indicate that the NO Scavenger C-PTIO restored the function of CTLs, which was otherwise impaired by the immunosuppressive activity of MDSCs in the tumor microenvironment.
Augmentation of antitumor activity of CTLs by C-PTIO
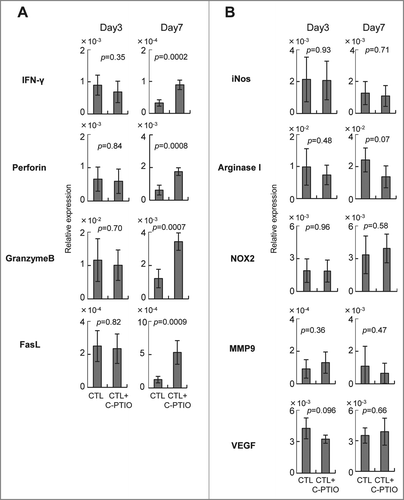
Tumor-infiltrating CTLs mediated both antitumor activity as well as enhancing the immune suppressive microenvironment in the tumor. To investigate the responsible mechanisms, quantitative RT-PCR was performed on mRNA extracted from the tumors of mice treated either with CTLs alone or CTLs+C-PTIO (). The levels of mRNAs for the effector molecules IFNγ, perforin, granzyme B, FasL and for the immunoregulatory molecules, iNOS, arginase I, NOX2, MMP9 and VEGF were compared on day 3 and day 7. On day 3, the expression of all these mRNAs in the tumor was similar in both groups of mice. However, while in mice treated only with CTLs, the expression of IFNγ, perforin, granzyme B and FasL decreased from day 3 to day 7, it was maintained or even increased in those also receiving C-PTIO. These results indicate that CTL function was hampered in the tumor due to the expression of immunosuppressive molecules, and that the NO scavenger C-PTIO maintained antitumor activity. In contrast, expression of iNOS, arginase I, NOX2, MMP9 and VEGF mRNAs in the tumor was similar in both groups of mice, both on day 3 and day 7, suggesting that C-PTIO did not affect the immunosuppressive microenvironment, but protected CTLs from their functional impairment.
Figure 6. Expression of genes related to effector (A) and immunoregulatory (B) functions. Mice were treated as described in the legend to . Tumor tissues from CTL-treated mice and CTL+C-PTIO-treated mice were harvested on days 3 and 7. Total RNA was extracted from each tumor tissue and the expression of the indicated genes determined by qRT-PCR.
Discussion
Adoptive CTL therapy can achieve robust and durable antitumor responses. However, although tumor-infiltrating CTLs produce IFNγ and mediate antitumor activity, they also induce counter-regulatory immunosuppressive mechanisms via recruitment of M-MDSCs.Citation13 Here, in a B16 melanoma/pmel-1 CTL adoptive transfer model, we show that the number of CTLs in the tumor was gradually reduced over time, and their function adversely affected, paralleled by the increased recruitment of M-MDSCs (). Consistent with a previous report that it is involved in M-MDSC-mediated immune suppression,Citation15 we found that NO inhibited the proliferation and function of CD8+ T cells and contributed to tumor growth (). To prevent the decrease in numbers and loss of function of CTLs in the tumor, we tested the NO scavenger C-PTIO and the iNOS inhibitor L-NMMA in combination with CTL transfer therapy. NO scavenging by C-PTIO restored the impaired proliferative potential and functions of CTLs, resulting in more marked the suppression of tumor growth ().
MDSCs are detected in most mouse tumor modelsCitation16 and in the PBMC of human cancer patients.Citation17 Furthermore, the presence of MDSCs correlates with stage of disease in cancer patients.Citation18 There are two major types of MDSC, granulocytic CD11b+Gr1+Ly6G+Ly6Clo (G-MDSC) and monocytic CD11b+Gr1intLy6G−Ly6C+ (M-MDSC).Citation19 In general, G-MDSC represent the major subset of circulating MDSCs expanded in cancer.Citation19 In tumors, 75% of MDSCs are G-MDSCs and the remaining 25% are M-MDSCs.Citation20 However, it has also been found that after CTL transfer, it is the M-MDSCs which are recruited into the tumor, such that they outnumber the G-MDSCs.Citation13 We have reported that the elimination of M-MDSCs or blockade of CTL-induced intratumoral M-MDSC recruitment augmented the antitumor activity of CTLs in vivo in CCR2−/− mice.Citation13 Various different strategies to inhibit MDSCs in cancer treatment have been explored including (1) inhibiting their function (e.g. PDE-5 inhibitors, NO-aspirin, COX2 inhibitors); (2) causing them to differentiate into mature myeloid cells (e.g. with ATRA, vitamin A, vitamin D3, IL-12); (3) blocking their development (e.g. with zoledronate, JAK2/STAT3 inhibitors, VEGF inhibitor); and (4) depleting them (e.g. with gemcitabine, cisplatin, paclitaxel, 5-FU, IL-6R blocker).Citation21 These strategies are now translated into clinics. Targeting immune suppression with PDE-5 inhibitor successfully augmented antitumor immunity in patients with head and neck squamous cell carcinoma and multiple myeloma.Citation22-24 Adoptive transfer of tumor infiltrating lymphocyte therapy was greatly improved by nonmyeloablative lymphodepleting chemotherapy that eliminated immunosuppressive cells, including regulatory T cells and MDSCs.Citation25 In case of cancer vaccine, we reported sunitinib that reduced the frequencies of circulating MDSCs enhanced the efficacy of dendritic-cell-based immunotherapy in metastatic renal cell carcinoma patients.Citation26 In patients with metastatic melanoma, CTLA-4 blockade with ipilimumab reduced the frequency of MDSCs and MDSCs are possible predictive biomarkers for CTLA-4 blockade therapy.Citation27,28 In the current study, we reduced the immunosuppressive activity by pharmacologically eliminating NO, the effector molecule produced by MDSC.
M-MDSCs and G-MDSCs inhibit both antigen-specific and antigen non-specific antitumor immune responses through different mechanisms, G-MDSCs mainly by producing ROS, and M-MDSCs by reactive nitrogen species (RNS).Citation19,20,29-31 M-MDSCs expressing iNOS and arginase 1 (ARG1) generate ROS and NO, both of which generate RNS such as peroxynitrite.Citation19,30,32 Peroxynitrite induces the nitration of the T-cell receptor (TCR)–CD8+ complexCitation33 and/or the binding of processed peptides to MHC molecules on tumor cells.Citation34 Either action leads to impeded antigen recognition by CTLs. Furthermore, in one study, nitrosylated chemokines failed to attract T cells to the tumor, but they could still recruit MDSCs.Citation35 Therefore, we targeted NO to inhibit the generation of RNS by M-MDSCs in the tumor.
Both the NOS inhibitor L-NMMA and the NO scavenger C-PTIO were able to restore CTL proliferation in vitro (), suggesting that NO was involved in the inhibition of CTL activity by MDSCs. We demonstrated that C-PTIO restored the function and proliferative capacity of CTLs from the tumor () and that tumor growth inhibition by CTLs was enhanced by combining CTL transfer with C-PTIO therapy (). In contrast, iNOS failed to augment antitumor activity of CTLs in vivo (data not shown). The reasons why iNOS failed in this respect remain to be elucidated, but could be related to the fact that C-PTIO directly extinguishes NO generated by NOS without affecting NOS activity itself. This contrasts with the action of the NOS inhibitor L-NMMA, which blocks the generation of NO and NO-related metabolites that mediate important physiological functions.Citation36,37 The more selective NO targeting by C-PTIO might be better for in vivo use.
Although NO scavenging by C-PTIO did restore CTL function, the effect was transient, the numbers of CTLs in the tumor eventually declined as did their function, and tumor growth resumed around day 10 after CTL transfer (). Tumors employ numerous different strategies to escape immunosurveillance; complex interactions among multiple cell types via negative costimulatory receptors on T cells, and other immunosuppressive factors and pro-apoptotic factors can all contribute to the immunosuppressive tumor microenvironment.Citation3 In addition to MDSCs, other factors might be operative in CTL-induced immunosuppression. In our model, CTLs which had infiltrated the tumor expressed PD-1 on day 5 and sustained this thereafter (data not shown). The expression of PD-L1 on tumor cells was also up-regulated by CTLs (data not shown); M-MDSCs also expressed PD-L1. Therefore, it is possible that the PD-1/PD-L1 axis also contributed to suppressing CTL activity. Thus, use of PD-1/ PD-L1 blockade together with CTL+C-PTIO treatment might further improve the antitumor activity of adoptive immunotherapy with CTLs.
In conclusion, we found that CTL therapy triggered immunosuppressive mechanisms in the tumor by facilitating the recruitment of M-MDSCs. In the presence of M-MDSCs, the numbers and functions of CTLs gradually decreased in the tumor. Our results show that the regulation of NO, which mediates immunosuppressive activity of M-MDSCs in this model, can restore CTL functions and improve the efficacy of immunotherapy.
Materials and Methods
Mice, cells and reagents
Male C57BL/6 mice at the age of 6–8 weeks were purchased from Japan SLC (Shizuoka, Japan). Pmel-1-TCR transgenic mice were obtained from The Jackson Laboratory (Bar Harbor, ME).Citation38 Their cells are Thy1.1 (CD90.1)-positive and recognize the H-2Db-restricted epitope peptide from gp100 (gp100 25–33: EGSRNQDWL). All mice were kept in a specific pathogen-free environment and all animal procedures were conducted in accordance with institutional guidelines. B16F10 is a gp100-positive spontaneous murine melanoma cell line, kindly provided by Dr N. Restifo (National Cancer Institute, Bethesda, USA) and maintained in culture medium consisting of DMEM (Wako Pure Chemical, Osaka, Japan) with 10% heat-inactivated fetal bovine serum (SIGMA-Aldrich, St Louis, MO, USA), 100 μg/mL streptomycin and 100 U/mL penicillin (Wako Pure Chemical). The H-2Db-restricted peptide human gp100 (hgp100 25–33, KVPRNQDWL) was purchased from GenScript Japan (Tokyo, Japan) at a purity of >90%, with a free amino terminal and carboxyl terminal. Carboxy-PTIO (2-[4[carboxyphenyl]-4,4,5,5,-tetramethylimidazoline-1-oxyl-3-oxide) and NG-monomethyl-L-arginine (L-NMMA) were purchased from Dojindo (Kumamoto, Japan).
CTL preparation for adoptive transfer
Bone marrow (BM)-derived dendritic cells (DCs) were prepared from tibias and femurs of C57BL/6 mice as described previously.Citation13 Briefly, BM cells were cultured in RPMI-1640 medium supplemented with 10% FCS, 12.5 mM HEPES, 5 × 10−5 M 2-mercaptoethanol, 1 × 10−5 M sodium pyruvate, 1% nonessential amino acids, 100 U/mL penicillin, 100 μg/mL streptomycin and 20 ng/mL GM-CSF (Pepro Tech, Rocky Hill, NJ, USA) for 8 d. DCs were stimulated with 1 μg/mL of lipopolysaccharide for 16 h and then pulsed with hgp100 peptide (1 μg/mL) for 3 h to obtain peptide-pulsed mature DCs. These DCs (2 × 105) were used to activate pmel-1-TCR transgenic spleen cells (1 × 107) for 3 days in medium containing 50 U/mL IL-2 (Chiron Corporation, Emeryville, CA, USA) to prepare CTLs. After 3 d in vitro stimulation, approximately 90% of the harvested cells were CD3+CD8+ CTLs. Therefore, no further purification was performed prior to their adoptive transfer.
Adoptive CTL therapy
B16F10 cells (1 × 106) were intradermally inoculated into the flanks of C57BL/6 mice on day 0. CTLs were adoptively transferred (1 × 107) on day 9, followed by C-PTIO (2 mg/200 μL PBS) injection i.p. twice daily for 10 d. Tumor growth was monitored every 2 to 3 d with calipers in a blinded fashion and was performed independently at least twice with similar results. Tumor volume was calculated by the formula π/6 × L1L2H, where L1 is the long diameter, L2 is the short diameter, and H is the height of the tumor. Tumor-bearing mice either died or had to be euthanized when the tumor volume exceeded approximately 1,500 mm3.
Flow cytometry
Tumor-infiltrating cells were prepared using a tumor dissociation kit (Miltenyi Biotec Inc., Auburn, CA, USA) according to the manufacturer's instructions. Briefly, tumors were harvested from mice at the indicated time points, cut into pieces, and transferred to gentleMACS C Tubes containing an enzyme mix (Miltenyi) and passed through a 70 μm cell strainer (BD Falcon, BD Bioscience) to obtain tumor-infiltrating cells. To eliminate dead cells, the preparations were stained with Fixable Viability Dye eFluor450 (eBioscience, San Diego, CA, USA) or Zombie Yellow (BioLegend, San Diego, CA, USA). The cells were then pretreated with Fc Block (anti-CD16/32 clone 2.4G2; BD Pharmingen), stained with antibodies and analyzed on a Gallios™ flow cytometer (Beckman Coulter, San Diego, CA, USA). The following mAbs were obtained from BioLegend (San Diego, CA) and used for flow cytometry: FITC-conjugated anti-CD45, CD90.1, PE-conjugated anti-CD11b, CD8+, PerCP-Cy5.5-conjugated anti-CD45, PE-Cy7-conjugated anti-Ly6C, APC-conjugated anti-IFNγ, CD107a, APC-Cy7-conjugated anti-CD8+, CD11b, CD45, Alexa Fluor647-conjugated anti-CD90.1, Pacific blue-conjugated anti-CD45, biotin-conjugated anti-Gr1, and Streptavidin-APC. To detect NO production, cells were incubated for 30 min at 37°C with 5 μM DAF-FM (Diaminofluorescein-FM) (SEKISUI MEDICAL). Data were processed using Kaluza software (Beckman Coulter) and analyzed with FlowJo (version 7.6.5; TreeStar, Ashland, OR). The total numbers of cells were estimated by Flow Count beads (Beckman-Coulter, Galway, Ireland).
Intracellular staining of IFNγ
Cells were stained with Fixable Viability Dye eFluor450, PerCP-Cy5.5-conjugated anti-CD45, APC-Cy7-conjugated anti-CD8+, FITC-conjugated anti-CD90.1 mAbs, followed by intracellular IFNγ staining using Intraprep permeabilization reagent (Immunotech, Marseille, France) according to the manufacturer's instructions. Cells were stained with APC-conjugated anti-IFNγ antibody or isotype control (Rat IgG2a, κ, Biolegend) at 4°C for 30 min and washed twice in FACS buffer before analysis. Cells were also stimulated for 4 h with 1 μg/mL hgp100 peptide and analyzed as described above. CD45+CD8+CD90.1+ cells were gated and IFNγ expression was determined as a measure of effector function.
The CD107a externalization assay
Cells were incubated for 4 h with 1 μg/mL hgp100 peptide in the presence of 0.4 μL of APC-conjugated anti-CD107a antibody or isotype control (Rat IgG2a, κ). Cells were stained with Fixable Viability Dye eFluor450, PerCP-Cy5.5-conjugated anti-CD45, APC-Cy7-conjugated anti-CD8+, FITC-conjugated anti-CD90.1 mAbs. CD45+ CD8+CD90.1+ cells were gated and surface expression of CD107a was evaluated as an indicator of degranulation.
In vitro cell proliferation assay
Cell proliferation was quantified in vitro as described previously.Citation13 Briefly, CD8+ cells were magnetically enriched from pmel-1 TCR-transgenic mice by negative selection using a CD8a+ T cell Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany). CD8+ pmel-1 cells were washed three times with HBSS (Life technologies) and stained with 0.6 μM carboxyfluorescein diacetate succinimidyl ester (CFSE, Dojindo) for 10 min at 37°C. MDSCs were enriched from TILs on day 3 after CTL transfer. Cells were centrifuged over the hypo-osmotic 1.077 g/mL density solution OptiPrep (AXIS-SHIELD PocAS) and CD11b+ cells were then isolated using EasySep Mouse CD11b Positive Selection Kits (STEMCELL Technologies, Vancouver, BC, Canada) according to the manufacturer's instructions. CFSE-labeled pmel-1 cells (1.5 × 105) were then stimulated for 72 h in 96-well U-bottom plates (Greiner Japan, Tokyo, Japan) with hgp100 peptide (1 μg/mL) with or without the indicated amount of MDSCs. On day 3, CD8+CD90.1+ cells were gated and fluorescence intensity of CFSE was evaluated. The assays were also performed in the presence of 0.1 mM C-PTIO or 0.5 mM L-NMMA.
In vivo cell proliferation assay
On day 3 and 7 after CTL transfer, mice were intraperitoneally injected with 2 mg BrdU (5-bromo-2′-deoxyuridine; Sigma, Saint Louis, MO) 16 h before sacrifice. TILs were isolated and stained with Fixable Viability Dye eFluor450 for dead cell elimination, APC-Cy7-conjugated anti-CD45, PE-conjugated anti-CD8+ and AlexaFluor647-conjugated anti-CD90.1 for cell surface markers. Cells were then fixed and permeabilized using Permeabilizing Solution 2 (BD Biosciences) for 10 min. at 24°C. Cells were then incubated at 37°C for 60 min in 0.15 M NaCl, 4.2 mM MgCl2, in the presence of 100 U/mL DNaseI (Roche Diagnostics, Indianapolis, IN, USA), followed by staining with anti-BrdU-FITC (eBioscience) for 20 min at 24°C 20 min and finally analyzed by FACS.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using TRIZOL according to the manufacturer's instructions (Invitrogen, Carlbad, CA, USA). The purity and RNA concentration was determined using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The RNA was converted to cDNA using the SuperScript III First-Strand Synthesis System according to the manufacturer's instructions (Invitrogen), and qRT-PCR reactions were run in a Thermal Cycler Dice Real Time System TP800 (Takara, Shiga, Japan) using the following program: 1 cycle of 95°C for 2 min, 40 cycles 95°C for 15 s, 60°C for 30 s. Results are expressed as ratios. The quantity of target mRNA was normalized to the level of GAPDH in each sample.
Cytology and Immunofluorescence
Smears were prepared from CD11b+ enriched populations, air dried, and stained with Diff-quick (Sysmex, Kobe, Japan) according to the manufacturer's instructions. Cell morphology was evaluated using bright field microscopy (OLYMPUS BX41 with Canon EOS Kiss X4 digital camera, OLYMPUS, Tokyo, Japan; magnification 400×, 1000×). CD11b+ enriched cells were also stained with 5 μM DAF-FM for 30 min at 37°C. Cells were incubated with Biotin-conjugated anti-Gr1, followed by staining with Streptavidin APC, PE-conjugated anti-CD11b for 30 min at 4°C. Cells were analyzed using a FLUOVIEW FV10i (OLYMPUS, Tokyo, Japan).
Statistical analysis
Statistical analyses were performed with JMP software, version 11.1.1 (SAS Institute Inc.., Cary, NC). Results are shown as mean ± SD. Comparison of results was carried out using the two-tailed unpaired t-test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported in part by a Grant-in-Aid for Scientific Research of the Ministry of Education, Culture, Sports, Science and Technology (Kazuhiro Kakimi). The study sponsors had no involvement in study design, collection, analysis, and interpretation of data, writing the report, or the decision to submit the report for publication. The Department of Immunotherapeutics, University of Tokyo Hospital is endowed by Medinet Co. Ltd. (Yokohama, Japan).
References
- Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013; 19:1423-37; PMID:24202395; http://dx.doi.org/10.1038/nm.3394
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5:263-74; PMID:15776005; http://dx.doi.org/10.1038/nrc1586
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011; 480:480-9; PMID:22193102; http://dx.doi.org/10.1038/nature10673
- Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013; 138:105-15; PMID:23216602; http://dx.doi.org/10.1111/imm.12036
- Chen Daniel S, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39:1-10; PMID:23890059; http://dx.doi.org/10.1016/j.immuni.2013.07.012
- Pardoll DM. Immunology beats cancer: a blueprint for successful translation. Nat Immunol 2012; 13:1129-32; PMID:23160205; http://dx.doi.org/10.1038/ni.2392
- Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy––inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res 2012; 18:6580-7; PMID:23087408; http://dx.doi.org/10.1158/1078-0432.CCR-12-1362
- Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res 2013; 19:5300-9; PMID:24089443; http://dx.doi.org/10.1158/1078-0432.CCR-13-0143
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012; 12:269-81; PMID:22437939; http://dx.doi.org/10.1038/nri3191
- Kalos M, June Carl H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013; 39:49-60; PMID:23890063; http://dx.doi.org/10.1016/j.immuni.2013.07.002
- June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotech 2012; 30:611-4; PMID:22781680; http://dx.doi.org/10.1038/nbt.2305
- Hosoi A, Matsushita H, Shimizu K, Fujii S-i, Ueha S, Abe J, Kurachi M, Maekawa R, Matsushima K, Kakimi K. Adoptive cytotoxic T lymphocyte therapy triggers a counter-regulatory immunosuppressive mechanism via recruitment of myeloid-derived suppressor cells. Int J Cancer 2014; 134:1810-22; PMID:24150772; http://dx.doi.org/10.1002/ijc.28506
- Noji S, Hosoi A, Takeda K, Matsushita H, Morishita Y, Seto Y, Kakimi K. Targeting spatiotemporal expression of CD137 on tumor-infiltrating cytotoxic T lymphocytes as a novel strategy for agonistic antibody therapy. J Immunother 2012; 35:460-72; PMID:22735804; http://dx.doi.org/10.1097/CJI.0b013e31826092db
- Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 2002; 168:689-95; PMID:11777962; http://dx.doi.org/10.4049/jimmunol.168.2.689
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499-506; PMID:19342621; http://dx.doi.org/10.4049/jimmunol.0802740
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol 2001; 166:678-89; PMID:11123353; http://dx.doi.org/10.4049/jimmunol.166.1.678
- Diaz-Montero CM, Salem M, Nishimura M, Garrett-Mayer E, Cole D, Montero A. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol Immunother 2009; 58:49-59; PMID:18446337; http://dx.doi.org/10.1007/s00262-008-0523-4
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253-68; PMID:22437938; http://dx.doi.org/10.1038/nri3175
- Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. JImmunol 2008; 181:5791-802; PMID:18832739; http://dx.doi.org/10.4049/jimmunol.181.8.5791
- Wesolowsk R, Markowitz J, Carson WE. Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. J Immunother Cancer 2013; 1:10; PMID:24829747; http://dx.doi.org/10.1186/2051-1426-1-10
- Noonan KA, Ghosh N, Rudraraju L, Bui M, Borrello I. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol Res 2014; 2:725-31; PMID:24878583; http://dx.doi.org/10.1158/2326-6066.CIR-13-0213
- Weed DT, Vella JL, Reis IM, De la Fuente AC, Gomez C, Sargi Z, Nazarian R, Califano J, Borrello I, Serafini P. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res 2015; 21:39-48; PMID:25320361; http://dx.doi.org/10.1158/1078-0432.CCR-14-1711
- Califano JA, Khan Z, Noonan KA, Rudraraju L, Zhang Z, Wang H, Goodman S, Gourin CG, Ha PK, Fakhry C et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res 2015; 21:30-8; PMID:25564570; http://dx.doi.org/10.1158/1078-0432.CCR-14-1716
- Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol 2006; 6:383-93; PMID:16622476; http://dx.doi.org/10.1038/nri1842
- Matsushita H, Enomoto Y, Kume H, Nakagawa T, Fukuhara H, Suzuki M, Fujimura T, Homma Y, Kakimi K. A pilot study of autologous tumor lysate-loaded dendritic cell vaccination combined with sunitinib for metastatic renal cell carcinoma. J Immunother Cancer 2014; 2:30; PMID:25694811; http://dx.doi.org/10.1186/s40425-014-0030-4
- Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother 2014; 63:247-57; PMID:24357148; http://dx.doi.org/10.1007/s00262-013-1508-5
- Pico de Coana Y, Poschke I, Gentilcore G, Mao Y, Nystrom M, Hansson J, Masucci GV, Kiessling R. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunol Res 2013; 1:158-62; PMID:24777678; http://dx.doi.org/10.1158/2326-6066.CIR-13-0016
- Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell–suppressive activity. Blood 2008; 111:4233-44; PMID:18272812; http://dx.doi.org/10.1182/blood-2007-07-099226
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol 2010; 40:22-35; PMID:19941314; http://dx.doi.org/10.1002/eji.200939903
- Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol 2004; 172:989-99; PMID:14707072; http://dx.doi.org/10.4049/jimmunol.172.2.989
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 2007; 13:828-35; PMID:17603493; http://dx.doi.org/10.1038/nm1609
- Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest 2011; 121:4015-29; PMID:21911941; http://dx.doi.org/10.1172/JCI45862
- Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med 2011; 208:1949-62; PMID:21930770; http://dx.doi.org/10.1084/jem.20101956
- Akaike T, Yoshida M, Miyamoto Y, Sato K, Kohno M, Sasamoto K, Miyazaki K, Ueda S, Maeda H. Antagonistic action of imidazolineoxyl N-oxides against endothelium-derived relaxing factor/.NO through a radical reaction. Biochemistry 1993; 32:827-32; PMID:8422387; http://dx.doi.org/10.1021/bi00054a013
- Maeda H, Akaike T, Yoshida M, Suga M. Multiple functions of nitric oxide in pathophysiology and microbiology: analysis by a new nitric oxide scavenger. J Leukoc Biol 1994; 56:588-92; PMID:7964166
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med 2003; 198:569-80; PMID:12925674; http://dx.doi.org/10.1084/jem.20030590