
Abstract
The major hurdle to the creation of cancer-specific monoclonal antibodies (mAb) exhibiting limited cross-reactivity with healthy human cells is the paucity of known tumor-specific or mutated protein epitopes expressed on the cancer cell surface. Mutated and overexpressed oncoproteins are typically cytoplasmic or nuclear. Cells can present peptides from these distinguishing proteins on their cell surface in the context of human leukocyte antigen (HLA). T cell receptor mimic (TCRm) mAb can be discovered that react specifically to these complexes, allowing for selective targeting of cancer cells. The state-of-the-art for TCRm and the challenges and opportunities are discussed. Several such TCRm are moving toward clinical trials now.
Cancer Antigens and T Cell Receptor-Mimic Monoclonal Antibodies
Spontaneous antitumor T cell responses occur frequently in cancer patients and analyses of these responses have led to successful identification of tumor antigens recognized by T cells in the context of HLA.Citation1 Tumor specific antigens can originate from mutated gene products, such as Ras and BCR/ABL fusion proteins.Citation2,3 Although the neoantigens resulting from these mutations are strictly unique to tumor cells, the chances that a peptide displaying such a mutation will bind the patient's HLA and be displayed are small, and few are documented.Citation4 Tumor associated antigens, on the other hand, include proteins that are overexpressed in tumor cells, and therefore are displayed at a far higher rate on the surface of cancer cells (). CD8+ T cells of the immune system can identify antigenic peptides presented by HLA class I molecules. Peptides recognized as non-self, such as those derived from mutated, oncofetal or viral genes, can be detected by T cells, which will then kill the antigen presenting tumor cell.
Table 1. Classifications of tumor associated antigens
Strategies for enhancing T cell responses to these antigens include vaccination of cancer patients using DNA, peptides, whole proteins derived from tumor antigens, and dendritic cells loaded with peptides or incorporated with mRNAs.Citation18 Unfortunately, therapeutic results to date have not been robust.Citation19 Current approaches to primarily enhance tumor-specific T cell immunity by vaccination appear inadequate to maintain an effective antitumor immune response, likely because it is difficult to vaccinate patients against self-antigens. As our understanding of the complex interaction between tumors and the immune system has improved, alternative approaches have been exploited to enhance the therapeutic efficacy of T cells. One approach is to adoptively transfer T cells that have been engineered to express high affinity T cell receptors (TCRs) specific for tumor antigens.Citation20 Another approach is to engineer T cells with chimeric antigen receptors (CARs).Citation21 These constructs link antigen-specific mAb with one or more intracellular T cell co-stimulatory molecules. Transducing such constructs into polyclonal T cells directs T cell cytotoxicity to tumor cells. However, CAR T cells have been largely generated to recognized differentiation antigens that are already well recognized by mAbs. The CAR T cell may offer far more effective T cell therapy by bypassing immune tolerance to a predetermined antigen.
The other arm of adaptive immunity is circulating immunoglobulins, which have been effectively exploited therapeutically as mAbs. MAbs mediate their activity by direct cytotoxicity by blocking or activating signaling pathways, complement-dependent cytolysis, antibody-dependent cell cytotoxicity (ADCC), or by activating the immune response. The FDA has approved nearly 20 mAbs for the treatment of various hematological and solid tumors. Targets include primarily lineage and differentiation antigens such epidermal growth factor receptor, vascular endothelia growth factor, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), PD1, CD20, CD30, and CD52.Citation22 Immunoglobulins can also serve as carrier vehicles for targeted delivery of more potent cytotoxic agents, such as toxins, drugs, and radiation. However, all the marketed therapeutic mAbs have been limited to cell surface or extracellular proteins found on healthy cells and tissues, resulting in off-target toxicity. Additionally, as the vast majority of tumor specific and tumor associated antigens are intracellular, these important antigens cannot be targeted by conventional mAb therapy.
A TCRm mAb would be able to combine recognition of intracellular proteins, analogous to that of a TCR, with the therapeutic potency and versatility of a mAb. Biological and technical issues were major obstacles to this approach until recently. The antigenic density of a peptide within a HLA class I molecule on the cancer cell surface (perhaps 10 to a few thousand molecules) is substantially lower than for most expressed cell surface targets (ranging from tens of thousands to a million molecules). In addition, the presented peptide is buried in the groove of the HLA molecule, a protein found in large numbers on the surface of all nucleated cells. Therefore, it was extremely difficult to generate a mAb with both high specificity and high affinity by traditional hybridoma techniques. Phage display library technology to select such mAbs has now made it possible to easily identify rare mAbs against epitopes such as peptide/HLA-A complexes, from a large library.Citation23 As an example, we have successfully generated a fully human TCRm mAb, ESK1, specific for epitopes derived from WT1, an intracellular oncofetal antigen and transcription factor overexpressed in a wide range of cancer types.Citation24 The mAb showed potent therapeutic activity against WT1-expressing leukemia and solid tumors, both in vitro and in vivo, via antibody-dependent cellular cytotoxicity.Citation24-26
TCRm mAbs Under Investigation
Numerous TCRm based constructs have been created over the past 15 years, recognizing both viral and self-antigens in the context of HLA-A, most commonly in HLA-A*02:01, and HLA-A*24:02.Citation23 The majority of TCRm are used as biochemical tools to study antigen presentation, structure, and recognition. Many of these TCRm mAbs target viral antigen presentation, such as Env183/A2 (Hep B/HLA-A*02:01),Citation27 KP14/1 and KP15/11 (HIV envelope gp160/HLA-A*02:01),Citation28 and RL36A (West Nile Virus/mouse H-2Db).Citation29 TCRm derivatives have also been designed for cancer therapy. In addition, TCR-like and TCR transduced T cells aimed at cancer antigens overexpressed in tumors, such as MAGE-A1 or WT1 respectively, have been described for engineered T cell therapy.Citation5,30 TCRm Fab linked immunotoxin conjugates have been constructed as well. However, none of these TCRm have yet advanced to the clinical setting.Citation12,31
The generation of full-length functional TCRm IgGs for the purpose of cancer therapy has rarely been accomplished. Three groups have reported on such TCRm. The Weidanz lab has created three antibodies: RL6A (p68 RNA helicase/HLA-A*02:01),Citation32 RL4B (hCG-β/HLA-A*02:01),Citation33,34 and RL1B (Her2-E75/HLA-A*02:01).Citation35 The Molldrem group created the TCRm mAb 8F4, targeting PR-1 in context of HLA-A*02:01.Citation16,17 Finally, our lab has generated the antibody ESK1, and its Fc enhanced form, ESKM, directed to WT1/HLA-A*02:01 ().Citation24-26 The latter TCRm is a human IgG, allowing its use immediately in patients. While all of these TCRm antibodies have shown promising results in vitro and in vivo, clinical trials in humans are still pending.
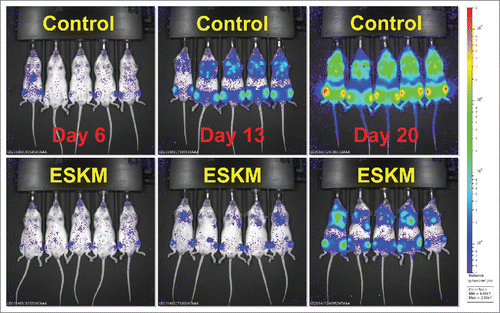
Figure 1. Therapy of disseminated SET2 (human AML cell line) in NSG mice by use of ESKM TCRm. Mice were treated as describedCitation26 starting on day 6 with biweekly ESKM 100 µg IP (top row) or no therapy (bottom row). Luciferase bioluminescent imaging was used to quantify the leukemia cells. Leukemia burden was reduced more than 30 fold in this experiment.
Regulation of TCRm Epitope Expression
A potential hurdle to effective use of TCRm mAb is the relative lack of epitope density on the cell surface, which may be extremely low (100–1,000 sites per cell), as compared to the high (20,000–500,000 sites per cell) number of sites per cell for traditional mAbs. TCRm Abs have been shown to trigger complement mediated killing or ADCC even at low epitope density. Potency appears to be correlated with numbers of target sites, stressing the importance of examining epitope regulation.Citation24-26
Level of protein expression, protein half-life, peptide processing, HLA levels, and HLA presentation of the peptide all dictate TCRm target epitope density. The protein must first be translated in sufficient quantities to facilitate peptide processing, and stability plays an important role. For instance, defective ribosomal products (DRiPs) may accumulate due to errors in transcription, translation, or protein folding. Various reports suggest DRiPs are rapidly degraded and constitute a significant percentage of peptides presented on HLA.Citation36,37 Short lived proteins also appear to be more likely to be presented than those with a longer half-life.Citation38 To initiate antigen processing, proteins are cleaved into random-sized peptides by proteasome-mediated degradation in the cytosol. These peptides then enter the endoplasmic reticulum through TAP, where they are trimmed by aminopeptidases, loaded onto HLA, and transported to the cell surface. Peptide HLA loading is governed by the binding affinity of the HLA protein to the peptide. The amino acid residues at position number 2 and 9 in the peptide determine binding affinity to most HLA alleles. Although several peptides may briefly bind a single HLA molecule, chaperones such as tapasin allow rapid peptide exchange ensuring the replacement of low-affinity peptides by those with high affinity.Citation39 The resulting product is trafficked to the cell surface. Unstable peptides dissociate from HLA on the cell surface, resulting in significantly shortened half-lives.Citation40 Moreover, specific peptides have been shown to cluster on the cell surface, which increases the avidity of the epitope and allows for increased activation of recruited effectors.Citation41 However, some tumors have been observed to significantly downregulate surface HLA expression.Citation42,43 Such tumors will be far less susceptible to TCRm based therapy due to target downregulation.
To overcome the issue of low target density, pharmacological modulation of key steps in peptide presentation should be considered. For instance, specific cytokines, such as IFNγ and TNFα, increase HLA presentation.Citation44 Specific chemotherapeutic agents and radiation can also enhance HLA expression.Citation45 Thus, modulation through exogenous treatment or cytokine release in the local tumor environment may increase epitope density and augment TCRm efficacy. Moreover, because tools for predicting affinity and immunogenicity of potential peptide epitopes are imprecise, development of robust tools will allow for greater accuracy in deciding which peptides will be presented in adequate numbers and hence lead to potential targets to pursue with TCRm Abs.
Determining Ideal TCRm Targets
The power of TCRm comes from their ability to exquisitely target cells based on expression of specific, otherwise undruggable, intracellular proteins. Ideal TCRm targets for cancer therapy are thus tumor-specific peptide/HLA complexes found in abundance on the cell surface. These antigens are most likely to be found in overexpressed proteins with short half-lives that are cleaved and processed into peptides with high affinity to the patient's HLA.Citation38,46
Targets meeting the above criteria can be grouped into three general categories. The first category is antigens presented only by the tumor itself, such as peptides derived from commonly mutated proteins (e.g. KRAS G12V/D)Citation47 or fusion proteins.Citation48 Another group is re-expressed oncofetal proteins (e.g. Carcinoembryonic antigen)Citation49 and overexpressed genes (e.g. MART-1/Melan-A,Citation10 MAGE,Citation50 PSACitation51) (). Targeting commonly mutated proteins is the most conceptually interesting as it may enable the holy grail of cancer therapy: a drug which selectively kills transformed cells across a wide array of cancer types. The disadvantage of these common mutations is that not all tumors will bear susceptible mutations—or the correct HLA type. Developing TCRm against re-expressed oncofetal proteins (as we have done with WT1) or proteins overexpressed in cancer may be a more general strategy, in which the limited expression on healthy tissue must be addressed in each case.Citation52
A third broad category of possible TCRm targets are tumor associated cells of the microenvironment, such as regulatory T cells,Citation53 tumor-associated macrophages (TAMs),Citation54 and cells involved in angiogenesis. It is too early to tell whether targeting these tumor infiltrates will be an effective antitumor strategy, but it is notable that a phase I clinical trial targeting macrophages systematically with an anti-CSF-1R antibody has shown early positive results.Citation55 As genes that distinguish tumor-infiltrating cells from their systemic counterparts are discovered—TCRm, which do not rely on surface marker expression—may allow for more specific targeting of only the tumor infiltrating cells. TCRm developed against populations of normal cells may have other applications, such as in diseases of immune dysregulation.
TCRm Versus TCR
While TCRm antibodies react with a peptide sequence similar to that which the TCR reacts with, (because both see an epitope within the peptide carried by HLA, as well as the HLA molecule itself) there are differences between the structures of the two molecules that lead to different binding affinities and practical applications. TCRm typically have far stronger binding affinities compared to their TCR counterparts ().Citation24,56
Table 2. A Comparison of TCR and TCRm
Typically this avidity difference can be 1,000 fold, between micromolar for TCR and nanomolar for TCRm. This will have a large impact on pharmacologic dosing, scheduling and off rates of the molecules. In addition, TCR are typically monovalent versus the bivalent TCRm, which may also greatly affect the pharmacology. Interestingly, the TCR and TCRm interact with similar binding surface areas of the peptide HLA (pHLA) complex (∼1000 Å2), so this is unlikely to be the source of binding affinity difference.Citation57 The difference in affinity is more likely due to the rather constrained in vivo selection process for TCR structures, versus the ex vivo unrestricted structures available for mAbs. TCR are selected against high-affinity binding to self-antigens in the thymus, compared to the in vitro selection for TCRm either by phage display or hybridomas.Citation58 This may explain the higher affinities observed for TCRm mAb. The high-affinity of TCRm may thus be useful for targeting the low-abundance epitopes presented on HLA-I molecules, since the number of epitopes presented may be very low, on the order of a few hundred sites. TCR, when presented in the context of a T cell, have the advantage of having cell surface expression along with co-stimulatory molecules, such as CD27 and CD8+ to aid in binding to HLA-I.Citation59,60 Therefore, the avidity of the effector cell to target interaction may be quite high. Structural analyses have shown that TCR bind in a diagonal manner to the peptide/HLA-I complex.Citation61-63 TCRm structures have shown a much more diverse set of orientations, either similar to canonical TCR binding, or binding in orientations and regions completely different from naturally occurring TCR.Citation57,64 These structural differences open up avenues for targeting regions of the pHLA complex not accessible to T cell based therapies.
TCRm mAb have more practical advantages. The main approaches utilizing TCR-based therapy are use of peptide vaccines to elicit CD8+ T cell responses, adoptive T cell therapy, or TCR engineered T cells.Citation65 One difficulty of vaccines is that low levels of, and low-affinity, epitope specific T cells are generally observed, and clinical benefits that have been seen in studies are not robust, with low partial and complete response rates.Citation66 Adoptive T cell and CAR therapies are far more expensive, cumbersome, logistically difficult to provide as off the shelf therapies. Furthermore, persistence of T cells is difficult to maintain in vivo.Citation20,67 TCRm mAb are simple and inexpensive to manufacture, as has been done successfully for the past two decades for many FDA approved therapeutic antibodies. Additionally, TCRm mediated ADCC, complement killing, or TCRm immunoconjugates rely on multiple effectors or no cellular effectors, respectively, and allows for the bypassing of T cell mediated immunosuppression in the cancer patients on chemotherapy.Citation68
Mechanisms of TCRm Function
TCRm mAbs have been shown to mediate Fc-independent cell death, specifically such as: RL4B, RL6A, and RL1B.Citation33,35 These TCRm antibodies activate apoptosis in target breast cancer models in which the hCG-β, p68 RNA helicase, or Her2 derived peptides are presented on HLA-A0201, respectively. This apoptosis is mediated by internalization of the antibody bound to the peptide/HLA complexes, followed by phosphorylation of the JNK protein kinase and/or p38 MAPK resulting in activation of the intrinsic caspase-dependent pathway. However, other therapeutic TCRm antibodies do not promote Fc-independent cell death.Citation17,24
Fc-dependent mechanisms of mAb action can be further broken down into two subcategories: complement mediated or effector cell mediated. Complement proteins can bind to the Fc region of therapeutic mAbs and initiate the complement cascade, resulting in CDC. RL4B, RL6A, and 8F4 TCRm antibodies have been shown to induce complement in vitro.Citation17,32,33,69 TCRm Abs also recruit immune effector cells, such as NK cells, monocytes/macrophages, and neutrophils, via Fcγ receptors, resulting in ADCC. The exact mechanism of ADCC varies depending on the type of immune effector cell activated. For example, activated NK cells secrete perforin and granzyme B, which are taken up by the target cell and result in cell lysis, similar to cytotoxic CD8+ T cells.Citation70 Meanwhile, monocytes and macrophages have been shown to phagocytose an IgG-bound target cell as the primary method of ADCC, but can also secrete cytotoxic factors, such as TNF and reactive oxygen intermediates.Citation71,72 Neutrophils have also been suggested to mediate ADCC in this manner.Citation73 ADCC has shown itself to be a widespread mechanism of action for therapeutic TCRm antibodies. RL4A, RL6A, and 8F4 have all demonstrated in vitro ADCC activity.Citation17,32,33,69 Additionally, ESK, which does not show direct apoptotic induction nor complement activation, is a potent initiator of ADCC.Citation24-26
Challenges and Opportunities with TCRm
A potential concern for the efficacy of TCRm antibodies is the generally low density of their target peptide/MHC epitopes, with only hundreds to a few thousand expressed on a target cell surface, versus the tens to hundreds of thousands of epitopes targeted by commercially available antibodies. Therefore, strategies to augment the therapeutic index should be considered. For example, Fc engineering to enhance ADCC via altered Fc glycosylation or amino acid changes in the Fc sequence have been employed.Citation25,74,75 In addition, TCRm mAbs serve as an ideal cancer targeting platform for delivery of cytotoxic payloads specifically to a tumor, including attachment of α emitting radioisotopes or potent drugs and toxins.Citation31,76,77 Lastly, the ScFv's used to reverse engineer a TCRm mAb can be formed into bispecific antibodies, bispecific T-cell engagers (BiTEs), and CARs for expression on cytotoxic T-cells.Citation78-80
The effective use of TCRm mAbs for treatment of cancer must overcome a number of hurdles, including the problems with low epitope density discussed earlier (). In addition, the determination of appropriate antigenic targets, the complete avoidance of cross-reactivity with the HLA molecule itself (found on all nucleated cells), the discovery of high specificity, high-affinity TCRm, and the resistance by tumors to ADCC are potential issues. In general, MHC molecules do not readily or rapidly internalize, reducing the efficacy of drug conjugates, so activation of immune effectors will likely be the dominant mode of cytotoxicity for TCRm. However, the low density of cell surface targets makes both ADCC and complement mediated killing far more difficult. Furthermore, some of the patients in whom the TCRm may be prescribed will not have sufficient numbers of active effector cells as a consequence of the underlying disease (such as with leukemias) or due to prior cytotoxic therapy.
Table 3. Overcoming challenges of TCRm therapy
Although the manufacture of mAb on a large scale is easily done, the selection of targets, mAb creation and verification are at this time all difficult, expensive, and time consuming. The selection of validated targets poses several challenges. HLA restriction has been posed as a major obstacle to the widespread use of TCRm. However, HLA-A*02 is found in up to 40% of Caucasians, and 10–20% of other ethnic groups around the world. The success of just one TCRm to this HLA type would treat more patients than a typical “targeted” therapy that can only reach the small fraction of patients that express a particular mutated kinase or receptor. By preparing several TCRm of different HLA restrictions, it would be possible to address a large fraction of patients of patients with many cancers.
While a number of transcription factors and proteins have been described as over-expressed in tumor cells, few are seen on the cell surface in the context of HLA in sufficient numbers to allow for TCRm guided ADCC. An approach to overcome this problem might be to utilize multiple TCRm to different epitopes presented by the same HLA type. Making multiple mAbs targeting a variety of commonly overexpressed targets in the context of a variety of the most common HLA types also would allow for increased specificity, efficacy, and population coverage. Another potential hurdle is the cross-reactivity of TCRm to epitopes that share sequence homology to the chosen 9-mer amino acid sequences. As the mAb binds to small numbers of amino acids derived from sequences from both the HLA and the peptide, other amino acid sequences (either from processed peptides that share partial homology, or from HLA that share homology) may bind the TCRm.
Conclusions
Effective, truly cancer-specific therapies do not currently exist. A major advantage of the TCRm approach is that for the first time it should allow a readily available and potent drug that binds almost exclusively to target cancer cells, a goal currently not possible with any other approved form of therapy. Currently available small molecule therapies are not specific to cancer cells and other immunological approaches, such as vaccines or cellular therapies, which might be characterized as tumor specific, are cumbersome to produce, use, or control, or they lack the potency of the mAb. Moreover, as antibody therapy does not require the host adaptive immunity system, these drugs may be used in conjunction with lymphotoxic chemotherapy. TCRm may be vialed and frozen or lyophilized, allowing widespread, immediate and inexpensive use worldwide in virtually any setting. Such accessibility is not possible with adoptive cell therapies, CAR T cell therapies, bone marrow transplants, or donor leukocyte infusions. However, as has been observed with traditional mAb therapy against widespread targets on cancer cells, mAb therapy alone is not likely to be sufficiently potent to eliminate 100% of cells when used as monotherapy, even at high effector to target ratios. Mechanisms of cancer cell resistance to antibody therapy must therefore be elucidated and remediated before the full potential of TCRm mAb therapy can be realized. Combination therapies with other anticancer agents likely will be necessary.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by the Leukemia and Lymphoma Society, NIH P30 CA 008748, NIH R01CA55349, P01CA23766, Lymphoma Foundation, Tudor and Glades funds, the MSKCC Technology Development Fund and the Experimental Therapeutics Center. L Dubrovsky was supported by NIH T32CA62948-18. E Brea and RS Gejman were supported by a Medical Scientist Training Program NIH T32GM007739. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14:135-46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24457417; PMID:24457417
- Kubuschok B, Neumann F, Breit R, Sester M, Schormann C, Wagner C, Sester U, Hartmann F, Wagner M, Remberger K et al. Naturally occurring T-cell response against mutated p21 ras oncoprotein in pancreatic cancer. Clin Cancer Res 2006; 12:1365-72. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16489095; PMID:16489095
- Kessler JH, Bres-Vloemans SA, van Veelen PA, de Ru A, Huijbers IJG, Camps M, Mulder A, Offringa R, Drijfhout JW, Leeksma OC et al. BCR-ABL fusion regions as a source of multiple leukemia-specific CD8+ T-cell epitopes. Leukemia 2006; 20:1738-50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16932347; PMID:16932347
- Pinilla-Ibarz J, Cathcart K, Korontsvit T, Soignet S, Bocchia M, Caggiano J, Lai L, Jimenez J, Kolitz J, Scheinberg DA. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood 2000; 95:1781-7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10688838; PMID:10688838
- Asai H, Fujiwara H, An J, Ochi T, Miyazaki Y, Nagai K, Okamoto S, Mineno J, Kuzushima K, Shiku H et al. Co-introduced functional CCR2 potentiates in vivo anti-lung cancer functionality mediated by T cells double gene-modified to express WT1-specific T-cell receptor. PLoS One 2013; 8:e56820. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3575507&tool=pmcentrez&rendertype=abstract; PMID:23441216; http://dx.doi.org/10.1371/journal.pone.0056820
- Imai N, Ikeda H, Shiku H. [Targeting cancer antigen (MAGE-A4, NY-ESO-1) for immunotherapy]. Nihon Rinsho 2012; 70:2125-9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23259384; PMID:23259384
- Pinilla-Ibarz J, May RJ, Korontsvit T, Gomez M, Kappel B, Zakhaleva V, Zhang RH, Scheinberg DA. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia 2006; 20:2025-33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16990779; PMID:16990779
- May RJ, Dao T, Pinilla-Ibarz J, Korontsvit T, Zakhaleva V, Zhang RH, Maslak P, Scheinberg DA. Peptide epitopes from the Wilms' tumor 1 oncoprotein stimulate CD4+ and CD8+ T cells that recognize and kill human malignant mesothelioma tumor cells. Clin Cancer Res 2007; 13:4547-55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17671141; PMID:17671141
- Dermime S, Armstrong A, Hawkins RE, Stern PL. Cancer vaccines and immunotherapy. Br Med Bull 2002; 62:149-62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12176857; PMID:12176857
- Castelli C, Storkus WJ, Maeurer MJ, Martin DM, Huang EC, Pramanik BN, Nagabhushan TL, Parmiani G, Lotze MT. Mass spectrometric identification of a naturally processed melanoma peptide recognized by CD8+ cytotoxic T lymphocytes. J Exp Med 1995; 181:363-8. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2191826&tool=pmcentrez&rendertype=abstract; PMID:7807017
- Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J, Han H, He W, Wang W, Zhao Y et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep 2014; 4:3571. Available from: http://www.nature.com/srep/2014/140106/srep03571/full/srep03571.html; PMID:24389689
- Klechevsky E, Gallegos M, Denkberg G, Palucka K, Banchereau J, Cohen C, Reiter Y. Antitumor activity of immunotoxins with T-cell receptor-like specificity against human melanoma xenografts. Cancer Res 2008; 68:6360-7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2728007&tool=pmcentrez&rendertype=abstract; PMID:18676861
- Disis ML, Cheever MA. HER-2/neu protein: a target for antigen-specific immunotherapy of human cancer. Adv Cancer Res 1997; 71:343-71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9111870; PMID:9111870
- Hörig H, Lee CSD, Kaufman HL. Prostate-specific antigen vaccines for prostate cancer. Expert Opin Biol Ther 2002; 2:395-408. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11955277; PMID:11955277
- LeMaistre CF, Meneghetti C, Howes L, Osborne CK. Targeting the EGF receptor in breast cancer treatment. Breast Cancer Res Treat 1994; 32:97-103. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7819590; PMID:7819590
- Alatrash G, Mittendorf EA, Sergeeva A, Sukhumalchandra P, Qiao N, Zhang M, St John LS, Ruisaard K, Haugen CE, Al-Atrache Z et al. Broad cross-presentation of the hematopoietically derived PR1 antigen on solid tumors leads to susceptibility to PR1-targeted immunotherapy. J Immunol 2012; 189:5476-84. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3504175&tool=pmcentrez&rendertype=abstract; PMID:23105141
- Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, McIntyre BW, Ma Q, Li D, St John L et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood 2011; 117:4262-72. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3087478&tool=pmcentrez&rendertype=abstract; PMID:21296998; http://dx.doi.org/10.1182/blood-2010-07-299248
- Disis ML. Mechanism of action of immunotherapy. Semin Oncol 2014; 41(Suppl 5):S3-13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25438997; PMID:25438997
- Baxevanis CN, Papamichail M, Perez SA. Therapeutic cancer vaccines: a long and winding road to success. Expert Rev Vaccines 2014; 13:131-44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24224539; PMID:24224539
- Schmitt TM, Aggen DH, Stromnes IM, Dossett ML, Richman SA, Kranz DM, Greenberg PD. Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood 2013; 122:348-56. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3716200&tool=pmcentrez&rendertype=abstract; PMID:23673862; http://dx.doi.org/10.1182/blood-2013-01-478164
- Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 2015; 263:68-89. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25510272; PMID:25510272
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012; 12:278-87. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22437872; PMID:22437872
- Dahan R, Reiter Y. T-cell-receptor-like antibodies - generation, function and applications. Expert Rev Mol Med 2012; 14:e6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22361332; PMID:22361332
- Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, Scott A, Whitten J, Maslak P, Casey E et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med 2013; 5:176ra33. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3963696&tool=pmcentrez&rendertype=abstract; PMID:23486779
- Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L, Whitten JA, Park S-M, Korontsvit T, Zakhaleva V et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin Cancer Res 2014; 20:4036-46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24850840; PMID:24850840
- Dubrovsky L, Pankov D, Brea EJ, Dao T, Scott A, Yan S, O'Reilly RJ, Liu C, Scheinberg DA. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood 2014; 123:3296-304. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24723681; PMID:24723681; http://dx.doi.org/10.1182/blood-2014-01-549022
- Sastry KSR, Too CT, Kaur K, Gehring AJ, Low L, Javiad A, Pollicino T, Li L, Kennedy PTF, Lopatin U et al. Targeting hepatitis B virus-infected cells with a T-cell receptor-like antibody. J Virol 2011; 85:1935-42. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3067764&tool=pmcentrez&rendertype=abstract; PMID:21159876
- Polakova K, Plaksin D, Chung DH, Belyakov IM, Berzofsky JA, Margulies DH. Antibodies Directed Against the MHC-I Molecule H-2Dd Complexed with an Antigenic Peptide: Similarities to a T Cell Receptor with the Same Specificity. J Immunol 2000; 165:5703-12. Available from: http://www.jimmunol.org/content/165/10/5703.full; PMID:11067928
- Kim S, Pinto AK, Myers NB, Hawkins O, Doll K, Kaabinejadian S, Netland J, Bevan MJ, Weidanz JA, Hildebrand WH et al. A novel T-cell receptor mimic defines dendritic cells that present an immunodominant West Nile virus epitope in mice. Eur J Immunol 2014; 44:1936-46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24723377; PMID:24723377
- Willemsen RA, Ronteltap C, Chames P, Debets R, Bolhuis RLH. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J Immunol 2005; 174:7853-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15944290; PMID:15944290
- Epel M, Carmi I, Soueid-Baumgarten S, Oh SK, Bera T, Pastan I, Berzofsky J, Reiter Y. Targeting TARP, a novel breast and prostate tumor-associated antigen, with T cell receptor-like human recombinant antibodies. Eur J Immunol 2008; 38:1706-20. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2682370&tool=pmcentrez&rendertype=abstract; PMID:18446790
- Verma B, Hawkins OE, Neethling FA, Caseltine SL, Largo SR, Hildebrand WH, Weidanz JA. Direct discovery and validation of a peptide/MHC epitope expressed in primary human breast cancer cells using a TCRm monoclonal antibody with profound antitumor properties. Cancer Immunol Immunother 2010; 59:563-73. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19779714; PMID:19779714; http://dx.doi.org/10.1007/s00262-009-0774-8
- Verma B, Jain R, Caseltine S, Rennels A, Bhattacharya R, Markiewski MM, Rawat A, Neethling F, Bickel U, Weidanz JA. TCR mimic monoclonal antibodies induce apoptosis of tumor cells via immune effector-independent mechanisms. J Immunol 2011; 186:3265-76. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21282517; PMID:21282517; http://dx.doi.org/10.4049/jimmunol.1002376
- Verma B, Neethling FA, Caseltine S, Fabrizio G, Largo S, Duty JA, Tabaczewski P, Weidanz JA. TCR mimic monoclonal antibody targets a specific peptide/HLA class I complex and significantly impedes tumor growth in vivo using breast cancer models. J Immunol 2010; 184:2156-65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20065111; PMID:20065111; http://dx.doi.org/10.4049/jimmunol.0902414
- Jain R, Rawat A, Verma B, Markiewski MM, Weidanz JA. Antitumor activity of a monoclonal antibody targeting major histocompatibility complex class I-Her2 peptide complexes. J Natl Cancer Inst 2013; 105:202-18. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3611852&tool=pmcentrez&rendertype=abstract; PMID:23300219; http://dx.doi.org/10.1093/jnci/djs521
- Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol 2003; 3:952-61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14647477; PMID:14647477; http://dx.doi.org/10.1038/nri1250
- Bourdetsky D, Schmelzer CEH, Admon A. The nature and extent of contributions by defective ribosome products to the HLA peptidome. Proc Natl Acad Sci U S A 2014; 111:E1591-9. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4000780&tool=pmcentrez&rendertype=abstract; PMID:24715725; http://dx.doi.org/10.1073/pnas.1321902111
- Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of HLA-I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics 2015; Available from: http://www.ncbi.nlm.nih.gov/pubmed/25576301; PMID:25576301
- Praveen PVK, Yaneva R, Kalbacher H, Springer S. Tapasin edits peptides on MHC class I molecules by accelerating peptide exchange. Eur J Immunol 2010; 40:214-24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20017190; PMID:20017190; http://dx.doi.org/10.1002/eji.200939342
- Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 2011; 11:823-36. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22076556; PMID:22076556
- Lu X, Gibbs JS, Hickman HD, David A, Dolan BP, Jin Y, Kranz DM, Bennink JR, Yewdell JW, Varma R. Endogenous viral antigen processing generates peptide-specific MHC class I cell-surface clusters. Proc Natl Acad Sci U S A 2012; 109:15407-12. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3458372&tool=pmcentrez&rendertype=abstract; PMID:22949678; http://dx.doi.org/10.1073/pnas.1208696109
- Khanna R. Tumour surveillance: missing peptides and MHC molecules. Immunol Cell Biol 1998; 76:20-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9553772; PMID:9553772; http://dx.doi.org/10.1046/j.1440-1711.1998.00717.x
- Angell TE, Lechner MG, Jang JK, LoPresti JS, Epstein AL. MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in vitro. Clin Cancer Res 2014; 20:6034-44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25294906; PMID:25294906; http://dx.doi.org/10.1158/1078-0432.CCR-14-0879
- Hallermalm K, Seki K, Wei C, Castelli C, Rivoltini L, Kiessling R, Levitskaya J. Tumor necrosis factor-α induces coordinated changes in major histocompatibility class I presentation pathway, resulting in increased stability of class I complexes at the cell surface. Blood 2001; 98:1108-15; PMID:11493458; http://dx.doi.org/10.1182/blood.V98.4.1108
- Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class i expression through elevated interferon-beta signaling in breast cancer cells. PLoS One 2012; 7:e32542; PMID:22396773; http://dx.doi.org/10.1371/annotation/3155a3e9-5fbe-435c-a07a-e9a4846ec0b6
- Fortier M-H, Caron E, Hardy M-P, Voisin G, Lemieux S, Perreault C, Thibault P. The MHC class I peptide repertoire is molded by the transcriptome. J Exp Med 2008; 205:595-610. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2275383&tool=pmcentrez&rendertype=abstract; PMID:18299400; http://dx.doi.org/10.1084/jem.20071985
- Warren RL, Holt RA. A census of predicted mutational epitopes suitable for immunologic cancer control. Hum Immunol 2010; 71:245-54. Available from: http://www.sciencedirect.com/science/article/pii/S0198885909006612; PMID:20035814; http://dx.doi.org/10.1016/j.humimm.2009.12.007
- Worley BS, van den Broeke LT, Goletz TJ, Pendleton CD, Daschbach EM, Thomas EK, Marincola FM, Helman LJ, Berzofsky JA. Antigenicity of fusion proteins from sarcoma-associated chromosomal translocations. Cancer Res 2001; 61:6868-75. Available from: http://cancerres.aacrjournals.org/content/61/18/6868.full; PMID:11559563
- Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst 1995; 87:982-90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7629885; PMID:7629885; http://dx.doi.org/10.1093/jnci/87.13.982
- Van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254:1643-7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1840703; PMID:1840703; http://dx.doi.org/10.1126/science.1840703
- Correale P, Walmsley K, Nieroda C, Zaremba S, Zhu M, Schlom J, Tsang KY. In vitro generation of human cytotoxic T lymphocytes specific for peptides derived from prostate-specific antigen. J Natl Cancer Inst 1997; 89:293-300. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9048833; PMID:9048833; http://dx.doi.org/10.1093/jnci/89.4.293
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013; 36:133-51. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3581823&tool=pmcentrez&rendertype=abstract; PMID:23377668; http://dx.doi.org/10.1097/CJI.0b013e3182829903
- Oleinika K, Nibbs RJ, Graham GJ, Fraser A. R. Suppression, subversion and escape: The role of regulatory T cells in cancer progression. Clin Exp Immunol 2013; 171:36-5; PMID:23199321; http://dx.doi.org/10.1111/j.1365-2249.2012.04657.x
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity [Internet] 2014; 41:49-61; PMID:25035953; http://dx.doi.org/10.1016/j.immuni.2014.06.010
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014; 25:846-59; PMID:24898549; http://dx.doi.org/10.1016/j.ccr.2014.05.016
- Matsui K, Boniface J, Reay P, Schild H, Fazekas de St Groth B, Davis M. Low affinity interaction of peptide-MHC complexes with T cell receptors. Science 1991; 254:1788-91. Available from: http://www.sciencemag.org/content/254/5039/1788.short; PMID:1763329; http://dx.doi.org/10.1126/science.1763329
- Mareeva T, Martinez-Hackert E, Sykulev Y. How a T cell receptor-like antibody recognizes major histocompatibility complex-bound peptide. J Biol Chem 2008; 283:29053-9. Available from: http://www.jbc.org/content/283/43/29053.long; PMID:18703505; http://dx.doi.org/10.1074/jbc.M804996200
- Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NR. T-cell-receptor affinity and thymocyte positive selection. Nature 1996; 381:616-20; PMID:8637599; http://dx.doi.org/10.1038/381616a0
- Devine L, Sun J, Barr MR, Kavathas PB. Orientation of the Ig domains of CD8 alpha beta relative to MHC class I. J Immunol 1999; 162:846-51. Available from: http://www.jimmunol.org/content/162/2/846.full; PMID:9916707
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996; 14:233-58; PMID:8717514; http://dx.doi.org/10.1146/annurev.immunol.14.1.233
- Ding Y-H, Smith KJ, Garboczi DN, Utz U, Biddison WE, Wiley DC. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity 1998; 8:403-11. Available from: http://www.sciencedirect.com/science/article/pii/S1074761300805464; PMID:9586631; http://dx.doi.org/10.1016/S1074-7613(00)80546-4
- Garcia KC. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science 1998; 279:1166-72. Available from: http://www.sciencemag.org/content/279/5354/1166.long; PMID:9469799
- Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 1996; 384:134-41; PMID:8906788; http://dx.doi.org/10.1038/384134a0
- Stewart-jones G, Hombach A, Shenderov E, Held G, Fischer E, Kleber S, Stenner-liewen F, Mcmichael A, Knuth A, Abken H et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci 2009; 106:10872-2; PMID:19307587; http://dx.doi.org/10.1073/pnas.0905399106
- Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med 2013; 5:174ra27. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3678970&tool=pmcentrez&rendertype=abstract; PMID:23447018
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10:909-15; PMID:15340416; http://dx.doi.org/10.1038/nm1100
- June CH. Principles of adoptive T cell cancer therapy. J Clin Invest 2007; 17:1204-12. Available from: http://www.jci.org/articles/view/31446#SEC6; PMID:17476350
- Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013; 138:105-15. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3575763&tool=pmcentrez&rendertype=abstract; PMID:23216602; http://dx.doi.org/10.1111/imm.12036
- Wittman VP, Woodburn D, Nguyen T, Neethling FA, Wright S, Weidanz JA. Antibody targeting to a class I MHC-Peptide epitope promotes tumor cell death. J Immunol 2006; 177:4187-95. Available from: http://www.jimmunol.org/content/177/6/4187.full; PMID:16951384
- Kerndrup G, Meyer K, Ellegaard J, Hokland P. Natural killer (NK)-cell activity and antibody-dependent cellular cytotoxicity (ADCC) in primary preleukemic syndrome. Leuk Res 1984; 8:239-47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6717064; PMID:6717064
- Munn DH, Cheung NK. Phagocytosis of tumor cells by human monocytes cultured in recombinant macrophage colony-stimulating factor. J Exp Med 1990; 172:231-7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2188164&tool=pmcentrez&rendertype=abstract; PMID:2193096
- Van der Bij GJ, Bögels M, Otten MA, Oosterling SJ, Kuppen PJ, Meijer S, Beelen RHJ, van Egmond M. Experimentally induced liver metastases from colorectal cancer can be prevented by mononuclear phagocyte-mediated monoclonal antibody therapy. J Hepatol 2010; 53:677-85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20619916; PMID:20619916
- Dallegri F, Patrone F, Holm G, Gahrton G, Sacchetti C. Neutrophil-mediated antibody-dependent cellular cytotoxicity against erythrocytes. Mechanisms of target cell destruction. Clin Exp Immunol 1983; 52:613-9. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1536050&tool=pmcentrez&rendertype=abstract; PMID:6307559
- Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 2005; 21:11-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15903235; PMID:15903235
- Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc Natl Acad Sci U S A 2012; 109:6181-6. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3341029&tool=pmcentrez&rendertype=abstract; PMID:22474370
- McDevitt MR, Ma D, Lai LT, Simon J, Borchardt P, Frank RK, Wu K, Pellegrini V, Curcio MJ, Miederer M et al. Tumor therapy with targeted atomic nanogenerators. Science 2001; 294:1537-40. Available from: http://www.sciencemag.org/content/294/5546/1537.long; PMID:11711678; http://dx.doi.org/10.1126/science.1064126
- Maguire WF, McDevitt MR, Smith-Jones PM, Scheinberg DA. Efficient 1-step radiolabeling of monoclonal antibodies to high specific activity with 225Ac for α-particle radioimmunotherapy of cancer. J Nucl Med 2014; 55:1492-8. Available from: http://jnm.snmjournals.org/content/55/9/1492.long; PMID:24982438; http://dx.doi.org/10.2967/jnumed.114.138347
- Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? MAbs 2009; 1:539-47. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2791310&tool=pmcentrez&rendertype=abstract; PMID:20073127; http://dx.doi.org/10.4161/mabs.1.6.10015
- Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol 2014; PMID:25367186; http://dx.doi.org/10.1038/icb.2014.93
- Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med 2012; 14:405-15. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22262649; PMID:22262649; http://dx.doi.org/10.1002/jgm.2604