
ABSTRACT
Tumor microenvironments polarize neutrophils to protumoral phenotypes. Here, we demonstrate that the angiotensin converting enzyme inhibitors (ACEis) and angiotensin II type 1 receptor (AGTR1) antagonist attenuate tumor growth via polarization of neutrophils toward an antitumoral phenotype. The ACEis or AGTR1 antagonist enhanced hypersegmentation of human neutrophils and increased neutrophil cytotoxicity against tumor cells. This neutrophil hypersegmentation was dependent on the mTOR pathway. In a murine tumor model, ACEis and AGTR1 antagonist attenuated tumor growth and enhanced neutrophil hypersegmentation. ACEis inhibited tumor-induced polarization of neutrophils to a protumoral phenotype. Neutrophil depletion reduced the antitumor effect of ACEi. Together, these data suggest that the modulation of Ang II pathway attenuates tumor growth via polarization of neutrophils to an antitumoral phenotype.
Introduction
Although increased neutrophil proliferation and subsequent recruitment have been observed in several types of human and animal tumors, the role of neutrophils in tumor immunology is still under debate. Due to their highly toxic arsenals, activated neutrophils can kill tumor cells, thereby playing an antitumor role in the host (reviewed in Piccard et alCitation1 and Brandau et alCitation2). Neutrophil infiltration into tumors is associated with a favorable prognosis in some human tumors.Citation3,4 On the other hand, studies have revealed protumoral functions of neutrophils during tumor progression (reviewed in Gabrilovich et alCitation5 and Brandau et alCitation2). An increased neutrophil-to-lymphocyte ratio and the presence of intratumoral neutrophils have been associated with a poor prognosis in several human studies.Citation6-9 Since cancer-related inflammation is recognized as a central mechanism of tumor progression,Citation10 neutrophils are considered a major player during carcinogenesis and tumor progression.Citation2 This discrepancy regarding the role of neutrophils in tumor immunology reflects the plasticity of neutrophils within the tumor microenvironment.
The protumoral function of neutrophils is reflected by the immunosuppressive functions of neutrophils from tumor-bearing hosts. Neutrophils are considered to play an important role in cancer-related inflammation during carcinogenesis and tumor progression.Citation1,2,11 In addition, recent studies indicate that myeloid-derived suppressor cells (MDSCs) is responsible for immunosuppression in tumor-bearing hosts.Citation1,2,12 Among immunosuppressive immune cells, granulocytic-myeloid derived suppressor cells (G-MDSCs) share distinct features of neutrophils, including surface markers (Ly6G and CD11b) and morphology (ring-shaped nucleus). G-MDSCs suppress the immune response in tumor-bearing hosts by expressing high levels of arginase 1 and facilitate tumor progression, invasion and metastasis.Citation2,13-15
Recently, regulation of neutrophil plasticity in the tumor microenvironment has been reported.Citation16,17 Fridlender et alCitation16 demonstrated the existence of dual neutrophil phenotypes within tumors: N1 (antitumoral) and N2 (protumoral) tumor-associated neutrophils (TANs). They showed that the tumor-driven cytokine tumor growth factor (TGF)-β prevents the generation of antitumoral N1 neutrophils and induces polarization of neutrophils to the protumoral N2 phenotype. The inhibition of TGF-β reverses this protumoral phenotype polarization. They suggest that hypersegmentation of nuclei is a distinct characteristic of antitumoral neutrophils; antitumoral neutrophils have more lobulated, hypersegmented nuclei, while protumoral neutrophils have round, circular nuclei.Citation16
Although these studies strongly suggest the plasticity of neutrophils in tumor-bearing hosts, the mechanisms or agonists that drive the phenotype polarization of neutrophils remain largely unknown. Recently, Cortez Retamozo et al suggested an interesting mechanism for protumoral phenotype polarization of monocytes.Citation18 They showed that increased levels of angiotensin II (Ang II), a main peptide hormone of renin-angiotensin system (RAS), in tumor-bearing mice were responsible for the amplification of macrophage progenitor cells in the spleen and for the polarization of monocytes toward a tumor-promoting phenotype. However, the role of Ang II in the polarization of neutrophils remains unknown. Interestingly, an increased number of hypersegmented neutrophils has been reported in the peripheral blood from children receiving angiotensin-converting-enzyme inhibitors (ACEis).Citation19 Since hypersegmentation is considered a distinct characteristic of antitumoral neutrophils, we hypothesized that ACEi may attenuate tumor growth by inducing neutrophil polarization to an antitumoral phenotype. To evaluate this hypothesis, we examined the effects of ACEi on neutrophil hypersegmentation and investigated the effects of hypersegmented neutrophils on tumor growth in a mouse tumor model.
Results
ACEi and AGTR1 antagonist induce human neutrophil hypersegmentation and enhance neutrophil cytotoxicity against tumor cells
We examined whether captopril, an ACEi, induces hypersegmentation in human neutrophils. Neutrophils were exposed to various concentrations of captopril, and mean lobe counts were calculated. Captopril increased mean lobe counts in a concentration- and time-dependent fashion (). Neutrophils with more than five distinct lobes were classified as hypersegmented, and the percentage of hypersegmented neutrophils was significantly increased following captopril treatment (). depicts representative images of control and captopril-treated neutrophils. Neutrophils have been reported to gain more segments during differentiation; thus, hypersegmented neutrophils could represent “old” neutrophils.Citation15 Thus, we examined the effect of captopril on neutrophil survival. Neutrophils were exposed to 500 μM captopril for 12 h, and survival rates were determined using annexin V/propidium iodide (PI) staining. Captopril treatment affected neither apoptosis (annexin V-positive only) nor necrosis (double positive for annexin-V and PtdIns) rates in neutrophils (Fig. S1).
Figure 1. ACEis and AGTR1 antagonist induce hypersegmentation of human neutrophils and enhance neutrophil cytotoxicity against tumor cells. (A)–(C) Neutrophils were stimulated with different concentrations of captopril. (A) Mean lobe counts of neutrophils stimulated with captopril. n = 10 donors; ** p < 0.01 compared to zero values in each time point; *** p < 0.001 compared to zero values in each time point. (B) Percentage of hypersegmented neutrophils after stimulation with captopril. n = 10 donors; ** p < 0.01 compared to zero values in each time point; ***p < 0.001 compared to zero values in each time point. (C) Representative images of Wright-Geimsa staining of neutrophils stimulated with captopril for 4 h. Hypersegmented neutrophils are denoted by arrows. (D) Top, RT-PCR analysis of ACE expression in human neutrophils: representative pictures from three healthy volunteers. Bottom, Ang II concentration in conditioned medium from neutrophils treated with either vehicle or 500 μM captopril. n = 5 donors; ***p < 0.001. (E) Mean lobe counts of neutrophils exposed to 500 μM captopril in the presence or absence of 100 nM AngII. n = 3 donors; ***p < 0.001. (F) Mean lobe counts of neutrophils subjected to ACE silencing. Neutrophils were treated with control siRNA (siCtrl) or siRNA specific for ACE (siACE) for 24 h and were further stimulated with 500 μM captopril for 4 h. Vehicle (Veh) denotes neutrophils without any stimulation. n = 3 donors; ***p < 0.001. (G) Percentage of tumor cells that survived after exposure to neutrophils. Calcein-AM-stained tumor cells were exposed to neutrophils at a ratio of 1:10 for 8 h. n = 3 donors; *p < 0.05; ***p < 0.001. (H), (I) The effect of Ang II inhibition on neutrophil hypersegmentation. Neutrophils were stimulated with either losartan, enalapril or fosinopril for 4 h. n = 3 donors; ***p < 00.001 versus vehicle. All results are shown as means ± SEMs.
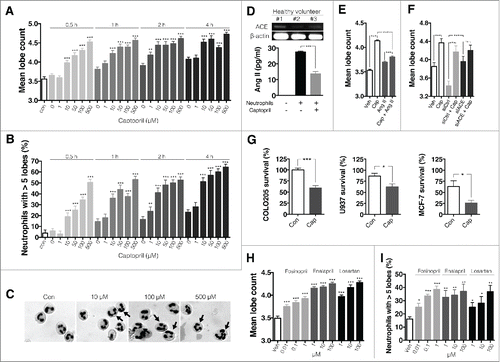
Previous studies suggest that a mobile RAS present on leukocytes contributes to circulating Ang II levels.Citation20-22 We hypothesized this mobile RAS might be responsible for the effect of captopril on neutrophil hypersegmentation. To identify RAS components in human neutrophils, the presence of ACE in human neutrophils was examined by reverse transcription polymerase chain reaction (RT-PCR). We found that neutrophils constitutively express ACE (, upper panel). We further investigated whether neutrophils locally produce Ang II. We detected Ang II in neutrophil-conditioned medium (25.4 ± 0.6 pg/mL mean ± SEM), but no detectable Ang II was present in control medium (, lower panel). Captopril treatment (500 μM) significantly decreased Ang II concentrations in conditioned medium (13.6 ± 1.5 pg/mL, mean ± SEM).
We next examined whether Ang II-treatment inhibits ACEi-induced hypersegmentation. Neutrophils were treated with vehicle or 500 μM captopril in the presence or absence of 100 nM Ang II for 4 h, and then mean lobe counts were examined. The addition of Ang II attenuated captopril-induced hypersegmentation (). We further silenced ACE using small interfering RNAs (siRNAs). Silencing procedure did not have significant effects on phenotype, function, and survival of neutrophils (Fig. S7). The transfection efficiency using FITC-conjugated control siRNA was 39.9 ± 6.8% (mean ± SEM) (Fig. S8). Interestingly, ACE silencing increased basal neutrophil lobe counts and attenuated captopril-induced hypersegmentation (). These findings suggest that constitutively generated Ang II from neutrophils plays a suppressive role in hypersegmentation.
To evaluate the function of hypersegmented neutrophils, cytotoxicity against various tumor cells was examined. COLO-205 (human colon adenocarcinoma), U937 (human histiocytic lymphoma), and MCF-7 (human breast cancer) cells were exposed to neutrophils at a ratio of 1:10 tumor cells to neutrophils. After 8 h, cells were washed and fluorescence levels in the surviving tumor cells were measured. Neutrophils were treated with either vehicle or 500 μM captopril for 4 h before exposure to tumor cells. Captopril-treated neutrophils showed increased cytotoxicity against COLO205, U937 and MCF-7 cells (). These results suggest that ACEi induces neutrophil hypersegmentation and enhances cytotoxicity of neutrophils against various tumor cells.
Since ACEis are categorized into three groups according to their structure, we examined the effects of representative agents from each category of ACEi and also examined the effect of losartan, an angiotensin II type 1 receptor (AGTR1) antagonist. Fosinopril (a phosphonate-containing ACEi), enalapril (a dicarboxylate-containing ACEi), and losartan treatment resulted in increased mean neutrophil lobe counts of neutrophils () and enhanced the percentage of hypersegmented neutrophils ().
Neutrophils are primary effectors in the inhibitory effect of ACEi and AGTR1 antagonist on tumor growth
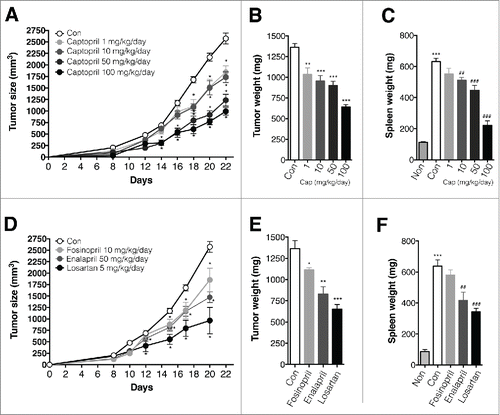
We next examined the effects of ACEi and AGTR1 antagonist in a murine tumor model. BALB/c mice were injected in the right leg with 1 × 105 4T1 cells. Tumor-bearing mice were administered captopril in their drinking water at 1, 10, 50, or 100 mg/kg/d; these are typical, murine therapeutic dosages.Citation23,24 Captopril attenuated tumor growth in a dose-dependent fashion (). On day 21, tumors and spleens from tumor-bearing mice were harvested, and their weights were determined. Both tumor and spleen weight were significantly reduced in captopril-treated mice compared to control mice (). Fosinopril (10 mg/kg/d, dissolved in drinking water), enalapril (50 mg/kg/d, dissolved in drinking water), and losartan (10 mg/kg/d, intraperitoneally, i.p.) also attenuated tumor growth () and reduced both tumor () and spleen () weight.
Figure 2. ACE is and AGTR1 antagonist attenuate tumor growth in a murine tumor model. (A)–(C) Effect of captopril on tumor growth. n = 10–15 mice for each group. (A) Growth curve for tumors in control and captopril-treated mice. *p < 0.05 compared to controls. (B) Tumor weight in control and captopril-treated mice. **p < 0.01; ***p < 00.001 compared to controls. (C) Spleen weight in control and captopril-treated mice. ***p < 0.001 compared to non-tumor-bearing mice; ##p < 0.01; ###p < 0.001 compared to controls. (D)–(F) Effect of AngII inhibition on tumor growth. n = 5–10 mice for each group. (D) Growth curve for tumors in control and AngII inhibitor-treated mice. *p < 0.05 compared to controls. (E) Tumor weight in controls and AngII inhibitor-treated mice. *p < 0.05; **p < 0.01; ***p < 0.001 compared to controls. (F) Spleen weight in control and Ang II inhibitor-treated mice. ***p < 0.001 compared to non-tumor-bearing mice; ##p < 0.01; ###p < 0.001 compared to controls. All results are shown as means ± SEMs.
Mouse neutrophils express specific surface marker Ly6G and CD11b. To evaluate the effect of captopril on the population of neutrophils in tumor-bearing mice, cells from bone marrow, blood, spleen and tumor were harvested and stained with Ly6G and CD11b. The percentage of Ly6G+ CD11b+ cells in blood, bone marrow and spleen were dramatically increased in tumor-bearing mice (Fig. S2). Captopril-treatment reduced the percentage Ly6G+ CD11b+ cells in spleen and tumors in a dose-dependent fashion while it did not reduce the percentage of neutrophils in blood and bone marrow (Fig. S2). Since ACEi did not enhance apoptosis of neutrophils (Fig. S1), the reduction of Ly6G+ CD11b+ cells in spleen and tumors could reflect the consequences of a smaller tumor burden.
We next examined the effects of neutrophil depletion on captopril-induced tumor growth inhibition. Starting 10 d after injection of mice with 4T1 cells, groups of tumor-bearing mice were treated with 50 mg/kg/d captopril (PostCap 50). To deplete neutrophils in tumor-bearing mice, mice were injected with anti-Ly6G monoclonal antibody 1A8 i.p. every 3 d, as previously described.Citation16 For comparison, a group of mice was treated with isotype-matched control IgG antibody every 3 d. Neutrophil-depletion blocked captopril-induced inhibition of tumor growth (), suggesting a crucial role for neutrophils in this process. On day 21 after 4T1 injection, tumors and spleens from tumor-bearing mice were harvested, and their weights were measured. Neutrophil depletion blocked captopril-induced reduction of tumor () and spleen () weights.
Figure 3. Neutrophil depletion reverses the captopril-induced tumor growth inhibition. (A)–(C) Effect of neutrophil depletion on tumor growth in captopril-treated mice. Starting 10 d after injection of 4T1 cells, tumor-bearing mice were administrated with 50 mg/kg/d captopril (PostCap50). Simultaneously, mice were injected with either 100 μg of anti-Ly6G antibody 1A8 (anti-Ly6G) or control IgG antibody (ctrl IgG) i.p. every 3 d. n = 10–15 for each group (A) Growth curve for tumors. *p < 0.05 for PostCap50 + ctrl IgG compared to controls; #p < 0.05 for PostCap50 + anti-Ly6G compared to PostCap50 + ctrl IgG. (B), (C) Weight of tumor and spleen. ***p < 0.001. (D) Effect of captopril on serum cytokine concentrations. Sea from naive, control, and captopril-treated mice were collected and TGF-β1, IL-2, IL-4, IFNγ and TNF-α concentrations were measured using ELISA. n = 10; n.s., not significant; *p < 0.05; **p < 0.01. All results are shown as means ± SEMs.
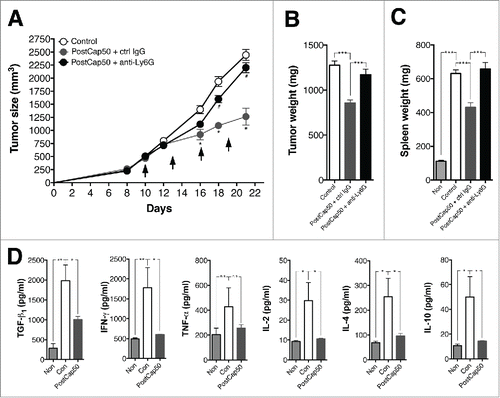
To evaluate whether captopril treatment alters the tumor microenvironment, we examined its effect on serum cytokine concentration. Sera from naive, control, and captopril-treated mice were collected, and the concentrations of TGF-β1, interleukin (IL)-2, IL-4, interferon (IFN)γ, and tumor necrosis factor (TNF)-α were measured. Serum TGF-β1, IL-2, IL-4, and IFNγ levels in tumor-bearing mice were significantly higher than in naive mice (). Captopril treatment reversed the significant increase in concentrations of these cytokines ().
ACEi polarizes splenic neutrophils toward an antitumor phenotype
To investigate whether ACEi also induced hypersegmentation of neutrophils in a mouse tumor model, the morphology of neutrophils was examined. Ly6G+ cells were isolated from the blood, bone marrow, spleen, and tumors of control tumor-bearing mice and captopril-treated tumor-bearing mice. All Ly6G+ cells from naive mice showed a clear neutrophil-like morphology with characteristic circular nuclei and light pink/purple cytoplasm (, left panel). However, all Ly6G+ cells from tumor-bearing control mice showed an increased mean lobe count compared to those from naive mice (, right panel). Interestingly, a higher percentage of splenic and intratumoral Ly6G+ cells from captopril-treated mice showed increased hypersegmented nuclei with oval-shaped nuclei and decreased cell diameter than was observed in cells from tumor-bearing control mice (, left panel). In addition, their mean lobe counts were significantly increased compared to those from control mice (, right panel).
Figure 4. Captopril induces neutrophil hypersegmentation in a murine tumor model. (A) Left panel, representative images of Wright-Geimsa staining of Ly6G+ cells. Right panel, mean lobe counts of Ly6G+ cells. Non, Ly6G+ cells from naive mice; Con, Ly6G+ cells from control tumor-bearing mice; Cap, Ly6G+ cells from captopril-treated tumor-bearing mice. (B)–(C) Function of hypersegmented Ly6G+ cells. Ly6G+ cells from tumor-bearing control mice or captopril-treated tumor-bearing mice were isolated and exposed to PMA for 4 h. (B) PMA-induced ROS generation from Ly6G+ cells. (C) PMA-induced NETs formation. (D) Cytotoxicity of Ly6G+ cells against 4T1 cells. Ly6G+ cells from control tumor-bearing mice or captopril-treated tumor-bearing mice were isolated and exposed to 4T1 cells at a ratio of 10:1 for 18 h. (E) Effects of captopril on Ly6G+ cells from naive mice. Blood, bone marrow, and splenic Ly6G+ cells were isolated from naive mice and exposed to 500 μM captopril for 4 h, and then mean lobe counts were measured. Veh, vehicle-treatment. (F) Cytotoxicity of captopril-treated neutrophils against 4T1 cells. Blood, bone marrow, and splenic Ly6G+ cells were isolated from naive mice and exposed to 4T1 cells at a ratio of 10:1 for 18 h in the presence or absence of 500 μM captopril. n = 5–10 mice for each group. n.s. not significant; *p < 0.05; **p < 0.01; ***p < 0.001. All results are shown as means ± SEMs.
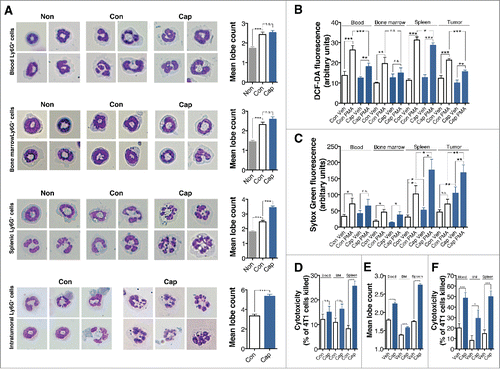
We further evaluate the functions of hypersegmented neutrophils. Neutrophil generation of reactive oxygen species (ROS) and formation of neutrophil extracellular traps (NETs) were examined. Isolated neutrophils were exposed to 1 μg/mL phorbol 12-myristate 13-acetate (PMA) for 4 h. Blood, splenic, and intratumoral Ly6G+ cells from captopril-treated mice showed decreased amounts of ROS generation in response to PMA (). However, splenic and intratumoral Ly6G+ cells from captopril-treated mice showed increased levels of basal NETs formation and PMA-induced NETs formation (). We also investigated the cytotoxicity of hypersegmented neutrophils against 4T1 cells. 4T1 cells were exposed to Ly6G+ cells at a ratio of 1:10 4T1 cells to neutrophils. After 16 h, cells were washed, and the percentage of 4T1 cells killed was calculated. Captopril treatment enhanced the cytotoxicity of splenic Ly6G+ cells against 4T1 cells ().
Next, we investigated the effect of captopril on neutrophils from naive mice. Ly6G+ cells were isolated from the blood, bone marrow and spleens of naive mice and exposed to 500 μM captopril for 4 h. Then, mean lobe counts were measured. Captopril treatment enhanced hypersegmentation of Ly6G+ cells and enhanced their mean lobe counts, regardless of their origins (). We also examined the cytotoxicity of hypersegmented Ly6G+ cells in vitro. Again, regardless of their origins, all captopril-treated Ly6G+ cells showed increased cytotoxicity against 4T1 cells ().
Our next goal was to phenotypically define ACEi-induced polarized neutrophils. Thus, we examined the expression of a number of phenotypic markers in captopril-treated neutrophils. Phenotypic markers were categorized into the following subgroups: (i) differentiation markers: Ly6C, CD14, CD15, CD16, CD62L, CD83, and CD45R; (ii) Toll-like receptors: CD282 and CD284; (iii) complement-associated molecules: CD55, CD21/35, and CD88; (iv) TNF signaling-associated molecules: CD40, CD120, CD256, 4-1BBL, and CD137; (v) adhesion and metabolism-associated molecules: CD43, CD98, CD101, CD44, and CD38; (vi) Fas and Fc receptor-associated molecules: CD178, CD95, and CD64a; and (vii) other unspecified molecules: CD1d, CD80, and TIM-3. Total cells from blood, bone marrow, spleen, and tumor were harvested and red blood cells (RBCs) were removed. Then, cells were fixed and labeled with Ly6G- and CD11b-specific antibodies and subjected to flow cytometric analysis. Granulocytes were gated based on forward and side scatter profiles. Neutrophil subsets were further gated based on Ly6G and CD11b expression. Our gating strategy is described in Fig. S3.
All neutrophils from non-tumor-bearing mice were Ly6Cneg CD14neg CD15+ CD62Lneg CD83neg CD45R+ CD282+ CD284neg CD21/35neg 4-1BBLneg CD44+ CD38neg CD178neg CD95neg CD64neg CD1dneg TIM3neg ( and S4). These phenotypic markers did not change in neutrophils from tumor-bearing mice. Additionally, intratumoral neutrophils from control tumor-bearing mice showed similar phenotypic characteristics as neutrophils from non-tumor-bearing mice. These markers thus represent basic phenotypic characteristics of mouse neutrophils.
Figure 5. Phenotype characterization of captopril-induced polarized neutrophils. (A) Basic phenotypic characteristics of mouse neutrophils. + indicates expression and - indicates lack of expression. (B) Phenotypic characterization of neutrophils. Left, the representative flow cytometry histogram for each phenotypic marker. Right, quantification of each phenotypic marker's expression. Gray, isotype-matched control IgG staining; Black, Ly6Ghigh CD11b+ cells from non-tumor-bearing mice; Blue, Ly6Ghigh CD11b+ cells from tumor-bearing mice; Red, Ly6Ghigh CD11b+ cells from captopril-treated tumor-bearing mice. n = 5–10 mice per each group; *p < 0.05; **p < 0.01; ***p < 0.001. All results are shown as means ± SEMs.
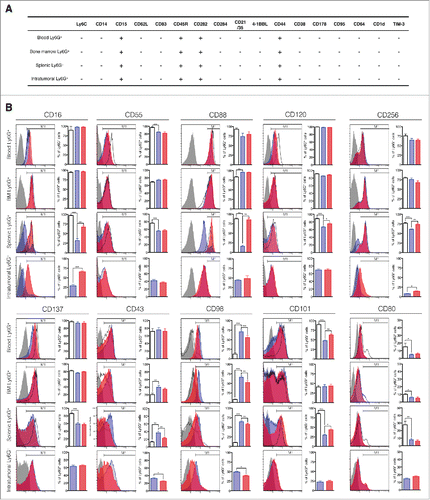
Blood and bone marrow neutrophils from tumor-bearing mice and non-tumor-bearing mice showed few differences in the surface expression of phenotypic markers (). However, blood neutrophils from tumor-bearing mice did show decreased expression of CD101 and CD80 and increased expression of CD98. In addition, bone marrow neutrophils from tumor-bearing mice showed decreased expression of CD80 and increased expression of CD43 and CD98.
By contrast, splenic and intratumoral neutrophils from tumor-bearing mice and non-tumor-bearing mice showed significant differences with regard to surface markers (). Compared to splenic neutrophils from non-tumor-bearing mice, splenic neutrophils from tumor-bearing mice showed decreased expression of CD16, CD55, CD88, CD120, CD256, CD137, CD101, and CD80. They also showed increased expression of CD43 and CD98. Intratumoral neutrophils showed similar phenotypic marker expression patterns as splenic neutrophils from tumor-bearing mice, with the exception of CD256, which was drastically reduced in intratumoral neutrophils. Interestingly, captopril treatment reversed the decreased expression of CD16, CD88, CD120, CD256, and CD101 in splenic neutrophils. The increased expression of CD43 in splenic neutrophils was also reversed by captopril treatment. Intratumoral neutrophils from captopril-treated mice showed increased expression of CD16 and CD256 with little decrease in CD43 and CD98 levels. These findings suggest that captopril treatment in tumor-bearing mice prevents the tumor-induced phenotype polarization of splenic neutrophils.
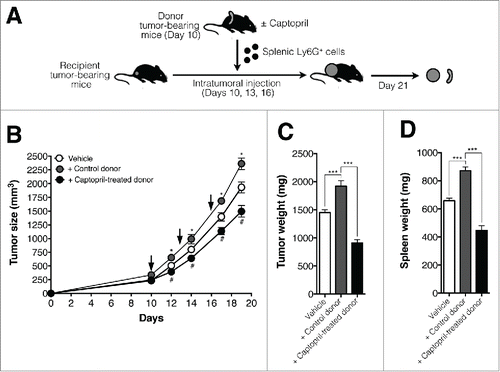
To confirm the antitumoral effects of captopril-induced polarized splenic neutrophils, we examined the effects of intratumoral (i.t.) injection of captopril-induced polarized neutrophils or control neutrophils on tumor growth in recipient mice. Recipient mice harboring 4T1 tumors were inoculated i.t. with 5 × 106 splenic Ly6G+ cells from donor mice on days 10, 13, and 16 post-tumor inoculations. Donor mice had previously been injected with 1 × 105 4T1 cells and treated with either captopril or vehicle for 10 d (). As shown in , the transfer of splenic neutrophils from vehicle-treated donor (+control donor) enhanced the growth of tumor in recipient tumor-bearing mice. Interestingly, the transfer of splenic neutrophils from captopril-treated donor (+captopril-treated donor) significantly attenuated the tumor growth in recipient mice (). Tumor and spleen weight were also reduced in recipient mice treated with neutrophils from captopril-treated donors ().
Figure 6. The transfer of splenic neutrophils from captopril-treated donor mice attenuates the tumor growth in recipient mice. (A) Procedure used to inject splenic neutrophils from donor tumor-bearing mice to recipient tumor-bearing mice. Donor mice harboring tumors were treated with either captopril or vehicle for 10 d. Donor splenic neutrophils were delivered to recipient tumor-bearing mice i.t. 10, 13, and 16 d after tumors were inoculated in recipient mice. (B) Growth curve for tumors in recipient mice. Recipient tumor-bearing mice were injected with Ly6G+ cells from control donor or captopril-treated donors 10, 13, and 16 d after tumor inoculation (Arrow). Control denotes tumor growth in mice injected with vehicle (RPMI). n = 5–10 mice for each group; *p < 0.05 for +Control donor versus. Vehicle; #p < 00.05 for +Captopril-treated donor versus +Control donor. (C)–(D) Tumor and spleen weights in recipient mice. n = 5–10 mice for each group; ***p < 0.001. All results are shown as means ± SEMs.
Previously, G-MDSCs from tumor-bearing mice were reported to impair TGF-β1-induced differentiation of CD4+ CD25+ Foxp3+ inducible Treg (iTreg) from CD4+ CD25− Foxp3− T cells.Citation25 The serum concentration of TGF-β1 and the percentage of intratumoral CD4+ CD25+ Foxp3+ regulatory T (Treg) cells were decreased in captopril-treated tumor-bearing mice (Fig. S5). Therefore, we examined the effect of captopril-treated polarized neutrophils in the generation of iTregs. Splenic neutrophils were isolated from either control tumor-bearing mice or captopril (50 mg/kg/d)-treated tumor-bearing mice, and exposed to CD4+ CD25− lymphocytes with anti-CD3, anti-CD28, IL-2, and TGF-β1 as previously described.Citation25 Since neutrophils are dying cells and the half-life of neutrophils with ex vivo manipulation is thought to be less than 24 h,Citation26 we examined the effect of Ly6G+ cells on iTreg induction within 24 h. Day 1 denotes the experimental procedure that CD4+ CD25− lymphocytes were exposed to Ly6G+ cells for 24 h. Day 4 denotes that CD4+ CD25− lymphocytes were allowed for induction of iTreg for 3 d and exposed to Ly6G+ cells for further 24 h. At day 1, splenic Ly6G+ cells from control mice increased the induction of iTregs, while splenic Ly6G+ cells from captopril-treated mice inhibited the induction of iTregs (Fig. S5). However, at day 4, neither splenic Ly6G+ cells from control mice nor captopril-treated mice did affect the induction of iTregs. Therefore, the amount of inhibitory effect of captopril-induced polarized neutrophils on the generation of iTreg seems to be negligible.
Involvement of the mTOR pathway in neutrophil hypersegmentation
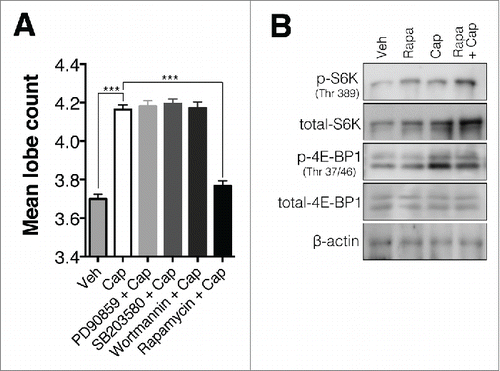
To explore the mechanism underlying captopril-induced neutrophil hypersegmentation, we examined the effects of the ERK inhibitor PD90859, the p38 inhibitor SB203580, the phosphatidylinositol 3-kinase inhibitor wortmannin, and the mTOR inhibitor rapamycin. PD98059 (10 μM), SB203580 (10 μM) and wortmannin (1 μM) treatment did not inhibit captopril-induced hypersegmentation. However, rapamycin (1 μM) completely inhibited captopril-induced hypersegmentation (). We also examined the effect of ACE on the expression of 4E-BP1 and S6K, downstream signaling molecules in the mTOR pathway. Captopril treatment enhanced phosphorylation of 4E-BP1, but not S6K. Rapamycin treatment inhibited captopril-induced 4E-BP1 phosphorylation ().
Figure 7. The mTOR pathway is involved in neutrophil hypersegmentation. (A) Inhibitory effect of rapamycin on captopril-induced hypersegmentation. Neutrophils were exposed to 500 μM captopril in the presence of PD90859 (10 μM), SB203580 (10 μM), wortmannin (1 μM), and rapamcyin (1 μM). n = 4 for each group; ***p < 0.001 compared to vehicle-treated cells; ###p < 0.001 compared to captopril-treated cells. Results are shown as means ± SEMs. (B) Western blot analysis of phosphorylation of 4E-BP1 and S6K in captopril-treated neutrophils. Neutrophils were treated with 500 μM captopril for 4 h in the presence or absence of 1 μg/mL rapamycin.
Discussion
Ang II, the main peptide hormone of RAS, is involved in several events during the inflammatory process (reviewed in Suzuki et alCitation27). Recently, increased Ang II concentration has been reported in animal tumor modelsCitation18 and some tumors are known to contain components of RAS, such as renin, ACE, or receptors for Ang II.Citation28 The inhibition of Ang II generation by ACEi decreases the growth of various tumors.Citation29 In addition, a retrospective study of hypertensive patients showed a decreased relative risk of incident, fatal cancer among ACEi-receiving groups.Citation28 Since Ang II is known to stimulate neovascularization in the tumor microenvironment, inhibition of tumor-induced neovascularization was considered a possible mechanism for these effects.Citation28 In the current study, we found an additional role of Ang II: phenotype polarization of neutrophils. We demonstrated the presence of a functional component of RAS and the constitutive generation of Ang II in human neutrophils (). Moreover, the treatment with either an ACEi or an AGTR1 antagonist induced the hypersegmentation of both human () and murine () neutrophils, and these hypersegmented neutrophils showed increased cytotoxicity against tumor cells () with increased NETs formation with PMA stimulation (). Additionally, ACEi-treatment reversed tumor-induced phenotypic marker changes in splenic and intratumoral neutrophils (). These results demonstrate the role of Ang II in phenotype polarization of neutrophils and also suggest possible therapeutic benefits of modulating this pathway on the phenotype polarization of neutrophils.
The RAS is known to mediate systemic Ang II production. Recent studies indicate that immune cells locally produce Ang II. Human T cell and natural killer (NK) cells display functional RAS and deliver Ang II to sites of inflammation.Citation20 Alveolar macrophages express ACE and produce Ang II in human atherosclerotic plaques.Citation21 Circulating rat leukocytes are also reported to secrete angiotensinogen,Citation22 and human mononuclear leukocytes possess functional components of RAS that can generate Ang II locally.Citation30,31 These studies suggest that a mobile RAS present on leukocytes contributes to circulating Ang II levels. In the current study, we have found that neutrophils also have a functional component of RAS and constitutively generate Ang II (). The inhibition of Ang II generation induced the neutrophil hypersegmentation () and the addition of Ang II completely reversed neutrophil hypersegmentation (). Furthermore, ACEi-induced neutrophil hypersegmentation was completely blocked by ACE silencing (). These findings demonstrate the presence of a mobile RAS on neutrophils and its role in phenotype polarization.
Neutrophils are generally considered to be fully differentiated cells. During later stages of differentiation, neutrophils show increasing segmentation of nucleus. Variations in neutrophil nuclear morphology serves as useful diagnostic indicators for several pathological conditions.Citation32 Therefore, the existence of a single hypersegmented neutrophil in peripheral blood smear is considered as diagnostic criteria for megaloblastic anemia. Recently, a line of studies suggested the hypersegmentation of neutrophils as a distinct hallmark of antitumoral phenotype of neutrophils.Citation16,17 Hypersegmented neutrophils showed increased cytotoxicity against tumor cells,Citation16 and attenuated tumor growth.Citation16,33 However, little is known regarding the induction mechanism of this neutrophil phenotype. In the current study, we found the role of Ang II on the polarization of neutrophils into antitumoral phenotype. The treatment with ACEis and AGTR1 antagonist induced hypersegmentation of both human () and murine neutrophils (). Consistent with previous studies, these hypersegmented neutrophils showed increased cytotoxicity against tumor cells (). Additionally, the inhibitory effect of ACEi on tumor growth was reversed by neutrophil depletion (). Taken together, these data demonstrate therapeutic potential of treating tumor by modulating Ang II-mediated neutrophil phenotype polarization.
Although hypersegmentation is considered to be a distinct characteristic of the antitumoral phenotype, several articles have indicated that nuclear morphology alone is not a sufficient indicator of neutrophil phenotypes.Citation15,34 Therefore, we examined the different phenotypes of neutrophils by evaluating the surface expression of a number of markers (). Neutrophils are known to polarize into protumoral phenotype in tumor-bearing hostsCitation16,17 and most of these immunosuppressive neutrophils are found in the spleen and tumors.Citation1,15 Consistent with these findings, we found that splenic and intratumoral neutrophils from control tumor-bearing mice showed significant differences in surface marker expression compared with those from naive mice. Splenic neutrophils from control tumor-bearing mice showed the decreased expression of CD16 (Fcγ receptor III), CD55 (Decay-accelerating factor), CD88 (C5a receptor), CD120a/b (TNF receptor), CD256 (TNF ligand superfamily member 13), CD137 (CD137, a TNF receptor family member), CD101 (immunoglobulin superfamily member 2), CD80 (co-stimulating molecule), and the increased expression of CD43 (leukosialin) and CD98 (large neutral amino acid transporter). Intratumoral neutrophils showed a similar pattern of phenotypic marker expression as splenic neutrophils. In contrast to these changes observed in splenic and intratumoral neutrophils, phenotypic changes in blood and bone marrow neutrophils were negligible. Captopril-treatment reversed most of phenotypic marker changes in splenic and intratumoral neutrophils.
The hypersegmented neutrophils showed increased cytotoxicity against tumor cells (). The transfer of splenic neutrophils from captopril-treated mice into recipient tumor-bearing mice attenuated tumor growth (). Although these results suggest the antitumor activity of hypersegmented neutrophils, it is still unclear how hypersegmented neutrophils killed tumor cells. Due to the highly-toxic arsenals of neutrophils, activated neutrophils are considered to kill tumor cells.Citation1,12 The proposed mechanisms regarding antitumor activity of neutrophils are oxidative damage caused by ROS generation, antibody-dependent tumor cell lysis, and NETs formation.Citation1,12,35 The tumor destruction caused by ROS generation is unlikely, because hypersegmented neutrophils showed decreased ROS generation in response to PMA stimulation (). However, hypersegmented neutrophil showed increased surface expression of Fcγ receptor III (CD16) (). Previous studies indicated that neutrophils mediate antibody-dependent cellular cytolysis of tumor cells via Fc receptor activation.Citation1,2,36 We believe that the reversed expression of CD16 in captopril-treated neutrophils might represent a plausible mechanism of antitumoral activity of hypersegmented neutrophils. Finally, hypersegmented neutrophils have shown increased NETs formation in response to PMA stimulation (). Recently, an interesting role of NETs in tumor immunology has been proposed.Citation35 The existence of NETs-releasing neutrophils was found in Ewing Sarcoma and authors suggested the hypothesis regarding the role of NETs in tumor. Since NETs contains several components which are highly cytotoxic to tumor cells,Citation36,37 we believe that increased NETs formation from hypersegmented neutrophils could be another possible mechanism for antitumor activity of neutrophils.
Our data also show that mTOR pathway is responsible for the hypersegmentation (). The hypersegmentation induced by ACEi was inhibited by rapamycin (), and captopril enhanced phosphorylation of 4E-BP1, a signaling molecule downstream of mTOR (). The mTOR pathway mediates several neutrophil functions, such as chemotaxis,Citation38-40 NETs formation,Citation41 and inflammatory cytokine release.Citation42 Notably, the relationship between ACE and mTOR activity has been reported.Citation43 The overexpression of ACE in a mouse skeletal muscle-derived C2C12 cells suppressed phosphorylation of mTOR, and captopril treatment enhanced phosphorylation of mTOR.Citation43 Consistent with this study, our results also illustrate the relationship between ACE and mTOR signaling in neutrophils.
In summary, our results give important new insights into phenotype polarization of neutrophils. We found the involvement of Ang II in the polarization of neutrophils in tumor microenvironment. The inhibition by ACEi or AGTR1 antagonist polarizes neutrophils toward an antitumoral phenotype. Therefore, these results a novel approach for tumor therapy by inducing phenotype polarization of neutrophils into antitumor phenotype.
Materials and methods
Animals and cells
BALB/c (female, 4–6 weeks old) mice were purchased from SAMTAKO (Osan, Republic of Korea). Procedures of animal experiments were approved by the Institutional Animal Care and Use Committee of Hallym University. 4T1, AB12, COLO-205, MCF-7, and U937 were purchased from the American Type Culture and European Collection of Cell Culture (Manassas, VA). Cells were cultured in DMEM (Gibco, Carlsbad, CA) supplemented with 10% FBS (Gibco) and 10 mg/L penicillin/streptomycin (Sigma-Aldrich, St.Louis., MO).
Isolation of human neutrophils
Venous blood was taken from healthy volunteers in accordance with a protocol approved by Ethnic committee of Hallym University. Informed consent was obtained from all participating persons, in compliance with the Declaration of Helsinki. Human neutrophils were purified using histopaque centrifugation followed by Dextran sedimentation.Citation44 Briefly, human venous blood was drawn from healthy male volunteers into vials containing anti-coagulant and layered over an equal volume of histopaque 1,077 (Sigma-Aldrich), followed by centrifugation at 2,500 rpm for 30 mins at RT. The lower layer containing neutrophils was collected and sedimented with 5% (w/v) dextran (Pharmacosmos, Holbaek, Denmark) for 30 min. The upper neutrophil-rich layer was collected and remaining RBCs were removed using hypotonic lysis. The cells were finally resuspended in RPMI 1640 (Gibco) supplemented with 5% Fetal bovine serum (Gibco) at 1 × 107/mL. The purity of neutrophils was determined by Wright–Geimsa staining. The purity was consistently greater than 95%.
Induction of hypersegmentation in human neutrophils
Neutrophils (2 × 106 cells/mL) were seeded on 24-well plates and subsequently stimulated with captopril (Sigma-Aldrich), enalapril (Sigma-Aldrich), fosinopril (Sigma-Aldrich), and losartan (Sigma-Aldrich) at different concentration. To inhibit the mTOR pathway, neutrophils were stimulated with captopril in the presence of rapamycin (Tokyo chemical industry, Tokyo, Japan). Neutrophils were collected and stained with either Hemacolor® stain (Merck Millipore, Germany) or Diff-quik® stain (Sysmex Inc., Kobe, Japan) to determine the polymorphic forms of the nuclei. Mean neutrophil lobe counts were measured by more than two researchers in a blinded manner.
Detection of ACE on human neutrophils
Total RNA was isolated from human neutrophils using TRIzol reagent (Life technologies, Carlsbad, CA) according to manufacturer's protocol. The first strand cDNA was synthesized with SuperScript II (Life technologies), and one-tenth of the cDNA was used for each PCR. The sequence of the PCR primers were as follows : ACE, 5′-GGTGGTGTGGAACGAGTATG-3′ (forward) and 5′-TCGGGTAAAACTGGAGGATG-3′ (reverse); β-actin, 5′-TGGAGTCCTGTGGCATCCACGAAAC-3′ (forward) and 5′-AAGCATTTGCGGTGGACGATGGAG-3′ (reverse). The cycling conditions was 94°C for 30 s, electrophoresed on 1.8% agarose gel stained with ethidium bromide.
Detection of Angiotensin II in neutrophil-conditioned medium
Human neutrophils (2 × 106 cells/mL) were seeded on 24-well plates in RPMI medium for 30 min and the conditioned medium was collected. The presence of Ang II was assessed by enzyme-linked immunosorbent assay (ELISA, Abcam Inc., Cambridge, MA) according to manufacturer's protocol.
Silencing of ACE in human neutrophils
Neutrophils were transfected with ACE siRNA using Lonza 4D Nucleofector using human monocyte transfector kit (Lonza, Walkersville, MD) as previously described.Citation44,45 5 × 106 neutrophils were suspended in nucleofector solution, followed by 3 μg siRNA against ACE (ACE siRNA, sc-270350, Santa Cruz Biotechnology, CA), control siRNA (control siRNA-A, sc-37007, Santa Cruz Biotechnology, CA) or FITC-conjugated control siRNA (sc-36869, Santa Cruz Biotechnology, CA). Transfection was performed in Lonza 4D Nucleofector using program Y-100. Immediately afterward, neutrophils were diluted in 2.5 mL RPMI supplemented with 5% FBS and incubated for 24 h. Neutrophils were harvested and 2 × 106 neutrophils were further stimulated with 500 μM captopril for 4 h.
Neutrophil cytotoxicity against tumors
Neutrophil cytotoxicity was measured as reported previously with minor modification.Citation46-48 In brief, COLO-205, MCF-7 and U937 were stained with 3 μg/mL Calcein-AM (Abcam Inc.) for 30 min and exposed to human neutrophils at a ratio of 1:10. Neutrophils were exposed to 500 μM captopril for 4 h before the exposure. After 8 h, cells were washed with 1 × PBS and remaining fluorescence was measured with fluorescence microplate reader (Spectramax M2/e, Molecular Devices, Sunnyvale, CA). The percentages of survived tumor cells were calculated as 100 × (1 – fluorescence in tumor cells after exposure to neutrophils / fluorescence of tumors cells without exposure to neutrophils).
Murine tumor model
BALB/c mice were injected on the right leg with 1 × 105 4T1 cells. Tumor growth was measured every 3 d. Tumors were allowed to growth for 21 d. Tumor-bearing mice were administrated with captopril (1, 10, 50, 100 mg/kg/d), enalapril (50 mg/kg/d) and fosinopril (10 mg/kg/d) in drinking water. Losartan were administrated intraperitoneally daily at a dose of 5 mg/kg/d.
Neutrophil depletion
Neutrophil depletion was performed as previously describedCitation16 with slight modification. Starting day 10, tumor-bearing mice were administrated intraperitoneally either 100 μg of purified monoclonal anti-Ly6G antibody 1A8 (BD Biosciences) or 100 μg of control IgG antibody every 3 d. Retro-orbital blood was drawn once a week and neutrophil depletion was examined with flow cytometry.
ELISA
Serum concentrations of cytokines were examined using ELISA kit. TGF-β1, IL-2, IL-4, IFNγ and TNF-α ELISA kits were purchased from Abcam. Procedures were conducted according to manufacturer's protocol.
Isolation of mouse neutrophils
Neutrophils were isolated by positive selection using a Midi MACS separators and Ly6G+ microbead kit (Miltenyi Biotec). For evaluation of neutrophil function, neutrophils were isolated by negative selection using either neutrophil isolation kit (Miltenyi Biotec) or EasySep™ mouse neutrophil enrichment kit (StemCell technologies, Vancouver, Canada).
Quantification of intracellular ROG generation and extracellular NETs formation
Isolated mouse neutrophils (2 × 106 cells/mL) were seeded in 96-well plates and further stimulated with PMA (1 μg/mL) for 4 h. Intracellular ROS generation was determined using dichloro-dihydro-fluorescein diacetate (DCFH-DA, Invitrogen) assay as previously described.Citation49,50 Neutrophils were washed and incubated at 37 °C in serum-free RPMI media in the presence of 3 μM DCFH-DA). After 30 min, the cells were washed and DCFH-DA fluorescence was analyzed using fluorescence microplate reader (Molecular Devices) at an excitation wavelength of 490 nm and an emission wavelength of 520 nm.
Extracellular NETs formation was measured using Sytox Green, a cell-impermeable DNA binding dye as previously described.Citation51-53 After stimulation with PMA (1 μg/mL) for 4 h, neutrophils were stained with Sytox Green (3 μM) for 30 min. Sytox Green fluorescence was measured using fluorescence microplate reader (Spectramax M2/e, Molecular Devices) at an excitation wavelength of 492 nm, and an emission wavelength of 530 nm.
Flow cytometry analysis
Tumors were harvested, minced with MACS dissociator (Miltenyi Biotec, Bergisch-Gladbach, Germany). Spleens were removed and grounded in RPMI with slide glass. Bone marrow cells were obtained from right tibia and femur. Blood was collected from cardiac puncture using heparinized syringe and serum was collected. Cells were further digested with 2 mg/mL DNase I (Sigma-Aldrich) and 4 mg/mL collagenase type IV (Sigma-Aldrich) for 1 h. Then, RBCs were removed with Pharm Lyse (BD Biosciences, San Jose, CA). Cells were further fixed with Cytofix (BD Biosciences) and resuspended in staining buffer (BD Biosciences) at 5 × 106/mL. Cells were stained with Ly6G (FITC; 551460, PE; 551461, BD BiosciencesBiosciences), CD11b (PerCP, 550993, BD Biosciences) and each phenotypic markers. Phenotypic markers were categorized into the following subgroups: (i) differentiation markers: Ly6C, CD14, CD15, CD16, CD62L, CD83, and CD45R; (ii) Toll-like receptors: CD282 and CD284; (iii) complement-associated molecules: CD55, CD21/35, and CD88; (iv) TNF signaling-associated molecules: CD40, CD120, CD256, 4-1BBL, and CD137; (v) adhesion and metabolism-associated molecules: CD43, CD98, CD101, CD44, and CD38; (vi) Fas and Fc receptor-associated molecules: CD178, CD95, and CD64a; and (vii) other unspecified molecules: CD1d, CD80, and TIM-3. Neutrophils are gated based on forward/side scatter profiles and further gated based on Ly6G and CD11b expression. Surface expression levels of each phenotypic marker were expressed as positive cells compared to an isotype-matched control. Phenotypic markers used are Ly6C (PE, 553128, BD Biosciences), CD14 (PE, 561711, BD Biosciences), CD15 (Alexa Fluor® 647, 562277, BD Biosciences), CD16 (PE, 12-0161-81, eBiosciences, San Diego, CA), CD62L (PE-cy7, 560516, BD Biosciences), CD83 (PE, 12-0831-80, eBiosciences), CD45R (FITC, 560976, BD Biosciences), CD282 (Alexa Fluor® 647, 562625, BD Biosciences), CD284 (PE, 558294, BD Biosciences), CD55 (PE, 558037. BD Biosciences), CD21/35 (PE-cy7, 25-0211-80, eBiosciences), CD88 (APC, 135807, BD Biosciences), CD40 (APC, 558695, BD Biosciences), CD120 (PE, 550088, BD Biosciences), CD256 (PE, 136703, BD Biosciences), 4-1BBL (PE, 12-5901-81, eBiosciences), CD137 (PE, 558976, BD Biosciences), CD43 (PE, 12-0431-81, eBiosciences), CD98 (PE, 12-0981-81, eBiosciences), CD101 (PE, 12-1011-80, eBiosciences), CD44 (PE, 553134, BD Biosciences), CD38 (APC, 17-0381-81, eBiosciences), CD178 (APC, 17-5911-80, eBiosciences), CD95 (APC, 17-0959-41, eBiosciences), CD64a (Alexa Fluor® 647, 558539, BD Biosciences), CD1d (FITC, 11-0011, eBiosciences), CD80 (PE, 12-0801-81, eBiosciences), and TIM-3 (PE, 12-5870-81, eBiosciences). Data acquisition was performed with BD FACSCalibur and data were analyzed with FlowJo software (TreeStar Inc., Ashland, OR).
Deliver of splenic neutrophils from donor tumor-bearing mice into recipient tumor-bearing mice
Donor BALB/c mice were injected on the right leg with 1 × 105 4T1 cells, and tumors were allowed to establish for 10 d. Donor mice were treated with either captopril 50 mg/kg/d or vehicle (distilled water). Spleens from donor mice were harvested and Ly6G+ cells were isolated by positive selection using a Ly6G+ microbead kit (Miltenyi Biotec). Recipient BALB/c mice were injected on the right leg with 1 × 105 4T1 cells on day 0. Recipient mice were treated by i.t. injection of splenic Ly6G+ cells from donor mice (5 × 106 cells/mice) in a volume of 100 μL (RPMI) on days 10, 13 and 16 post-tumor inoculation. Tumor size was measured every 2–4 d.
Western blot analysis
Proteins were obtained by lysing the cell in RIPA lysis buffer (50–60 μL per 1 × 107 neutrophils) that constitutes 1 × RIPA lysis buffer conjugated with protease inhibitor and phosphatase inhibitor. Lysate were incubated in cold (4°C) for 1 h followed by bath type sonication for 1 min and centrifugation at 12000 rpm for 20 mins at 4°C. Protein concentration of the supernatant was determined by using Biorad DC-Protein assay kit (Biorad, Hemel Hempstead, UK). 15 μg of each protein samples were separated on SDS PAGE, blotted into PVDF membranes. Membranes were blocked with 5% skim milk in TBST for 1 h at RT and incubated with 1:1000 dilutions of primary antibodies in blocking buffer overnight at 4°C with gentle agitation. After washing, membranes were incubated with 1:2000 dilution of horseradish peroxidase conjugated secondary antibodies in TBST for 1 h at RT. After few washes in TBST, the immunocomplexes were detected using Fusion Fx Chamber.
Statistical analysis
All statistical data were analyzed by Graphpad prism 5.0 (Graphpad software, San Diego, CA). Data were analyzed either by two-tailed Student's t test or ANOVA. Either bonferroni test or Tukey's test were used for post hoc comparison. Values of p < 0.05 were considered to indicate statistical significance.
Author contributions
Contribution: S.S. and J.N. designed the research, performed data collection, analysis, and wrote the paper; S.K., H.H., Y.K., Y.Y., and M.Kim. performed experiments; M.Kwon and D.S. contributed reagents and provided key advice in research design; and C.H. conceived and designed the research, analyzed the data, wrote the paper, provided financial support, and approved the final paper.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1067744_supplemental_figures.doc
Download MS Word (3.7 MB)Funding
This research is supported by grant NRF-2012R1A6A3A04040639 and 2012-0000312 from National Research Foundation of Korea (NRF) and HURF-2014-58 by Hallym University.
References
- Piccard H, Muschel RJ, Opdenakker G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit Rev Oncol Hematol 2012; 82:296-309; PMID:21798756; http://dx.doi.org/10.1016/j.critrevonc.2011.06.004
- Brandau S, Dumitru CA, Lang S. Protumor and antitumor functions of neutrophil granulocytes. Semin Immunopathol 2013; 35:163-76; PMID:23007469; http://dx.doi.org/10.1007/s00281-012-0344-6
- Bässler R, Dittmann AM, Dittrich M. Mononuclear stromal reactions in mammary carcinoma, with special reference to medullary carcinomas with a lymphoid infiltrate. Analysis of 108 cases. Virchows Arch A Pathol Anat Histol 1981; 393:75-91; PMID:7347444; http://dx.doi.org/10.1007/BF00430872
- Caruso RA, Bellocco R, Pagano M, Bertoli G, Rigoli L, Inferrera C. Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in northern Italy. Mod Pathol 2002; 15:831-7; PMID:12181268; http://dx.doi.org/10.1097/01.MP.0000020391.98998.6B
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Donskov F, Maase von der H. Impact of immune parameters on long-term survival in metastatic renal cell carcinoma. J Clin Oncol 2006; 24:1997-2005; PMID:16648500; http://dx.doi.org/10.1200/JCO.2005.03.9594
- Kuang D-M, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, Yin X-Y, Zheng L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol 2011; 54:948-55; PMID:21145847; http://dx.doi.org/10.1016/j.jhep.2010.08.041
- Li Y-W, Qiu S-J, Fan J, Zhou J, Gao Q, Xiao Y-S, Xu Y-F. Intratumoral neutrophils: a poor prognostic factor for hepatocellular carcinoma following resection. J Hepatol 2011; 54:497-505; PMID:21112656; http://dx.doi.org/10.1016/j.jhep.2010.07.044
- Jensen TO, Schmidt H, Müller HJ, Donskov F, Hüyer M, Sjoegren P, Christensen IJ, Steiniche T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer 2012; 118:2476-85; PMID:21953023; http://dx.doi.org/10.1002/cncr.26511
- Hanahan D, Weinberg RA. Hallmarks of Cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Souto JC, Vila L, Brú A. Polymorphonuclear neutrophils and cancer: Intense and sustained neutrophilia as a treatment against solid tumors. Med Res Rev 2011; 31:311-63; PMID:19967776; http://dx.doi.org/10.1002/med.20185
- Brandau S. Seminars in Cancer Biology. Semin Cancer Biol 2013; 23:139-40; PMID:23454237; http://dx.doi.org/10.1016/j.semcancer.2013.02.008
- Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, Horng W, Fridlender G, Bayuh R, Worthen GS et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One 2012; 7:e31524; PMID:22348096; http://dx.doi.org/10.1371/journal.pone.0031524
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer 2013; 13:739-52; PMID:24060865; http://dx.doi.org/10.1038/nrc3581
- Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell Mol Life Sci 2013; 70:3813-27; PMID:23423530; http://dx.doi.org/10.1007/s00018-013-1286-4
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009; 16:183-94; PMID:19732719; http://dx.doi.org/10.1016/j.ccr.2009.06.017
- Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest 2010; 120:1151-64; PMID:20237412; http://dx.doi.org/10.1172/JCI37223
- Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW, Kuswanto W, Rauch PJ, Chudnovskiy A, Iwamoto Y et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity 2013; 38:296-308; PMID:23333075; http://dx.doi.org/10.1016/j.immuni.2012.10.015
- Okutan V, Kürekçi AE, Sarici SU, Atay AA, Yozgat Y, Azik F, Lenk MK, Ozcan O. Neutrophil hypersegmentation in children receiving angiotensin converting enzyme inhibitors. Turk J Pediatr 2008; 50:438-42; PMID:19102047
- Jurewicz M, McDermott DH, Sechler JM, Tinckam K, Takakura A, Carpenter CB, Milford E, Abdi R. Human T and natural killer cells possess a functional renin-angiotensin system: further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol 2007; 18:1093-102; PMID:17329576; http://dx.doi.org/10.1681/ASN.2006070707
- Diet F, Pratt RE, Berry GJ, Momose N, Gibbons GH, Dzau VJ. Increased accumulation of tissue ACE in human atherosclerotic coronary artery disease. Circulation 1996; 94:2756-67; PMID:8941100; http://dx.doi.org/10.1161/01.CIR.94.11.2756
- Gomez RA, Norling LL, Wilfong N, Isakson P, Lynch KR, Hock R, Quesenberry P. Leukocytes synthesize angiotensinogen. Hypertension 1993; 21:470-5; PMID:7681424; http://dx.doi.org/10.1161/01.HYP.21.4.470
- Albuquerque D, Nihei J, Cardillo F, Singh R. The ACE inhibitors enalapril and captopril modulate cytokine responses in Balb/c and C57Bl/6 normal mice and increase CD4(+)CD103(+)CD25(negative) splenic T cell numbers. Cell Immunol 2010; 260:92-7; PMID:19854435; http://dx.doi.org/10.1016/j.cellimm.2009.09.006
- Hii SI, Nicol DL, Gotley DC, Thompson LC, Green MK, Jonsson JR. Captopril inhibits tumour growth in a xenograft model of human renal cell carcinoma. Br J Cancer 1998; 77:880-3; PMID:9528828; http://dx.doi.org/10.1038/bjc.1998.145
- Centuori SM, Trad M, LaCasse CJ, Alizadeh D, Larmonier CB, Hanke NT, Kartchner J, Janikashvili N, Bonnotte B, Larmonier N et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF- -induced differentiation of CD4+CD25+FoxP3+ Tregs from CD4+CD25-FoxP3- T cells. J Leukoc Biol 2012; 92:987-97; PMID:22891289; http://dx.doi.org/10.1189/jlb.0911465
- Pillay J, Braber den I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, Tesselaar K, Koenderman L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010; 116:625-7; PMID:20410504; http://dx.doi.org/10.1182/blood-2010-01-259028
- Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol 2003; 35:881-900; PMID:12676174; http://dx.doi.org/10.1016/S1357-2725(02)00271-6
- Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson JW. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 1998; 352:179-84; PMID:9683206; http://dx.doi.org/10.1016/S0140-6736(98)03228-0
- Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis 2008; 29:1675-84; PMID:18632755; http://dx.doi.org/10.1093/carcin/bgn171
- Jankowski V, Vanholder R, van der Giet M, Henning L, Tolle M, Schonfelder G, Krakow A, Karadogan S, Gustavsson N, Gobom J et al. Detection of angiotensin II in supernatants of stimulated mononuclear leukocytes by matrix-assisted laser desorption ionization time-of-flight/time-of-flight mass analysis. Hypertension 2005; 46:591-7; PMID:16087781; http://dx.doi.org/10.1161/01.HYP.0000177436.09733.d4
- Kitazono T, Padgett RC, Armstrong ML, Tompkins PK, Heistad DD. Evidence that angiotensin II is present in human monocytes. Circulation 1995; 91:1129-34; PMID:7850951; http://dx.doi.org/10.1161/01.CIR.91.4.1129
- Sanchez JA, Wangh LJ. New insights into the mechanisms of nuclear segmentation in human neutrophils. J Cell Biochem 1999; 73:1-10; PMID:10088718; http://dx.doi.org/10.1002/(SICI)1097-4644(19990401)73:1%3c1::AID-JCB1%3e3.0.CO;2-S
- Gregory AD, McGarry Houghton A. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res 2011; 71:2411-6; PMID:21427354; http://dx.doi.org/10.1158/0008-5472.CAN-10-2583
- Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother 2012; 61:1155-67; PMID:22692756; http://dx.doi.org/10.1007/s00262-012-1294-5
- Berger-Achituv S, Brinkmann V, Abed UA, Kühn LI, Ben-Ezra J, Elhasid R, Zychlinsky A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol 2013; 4:48; PMID:23508552; http://dx.doi.org/10.3389/fimmu.2013.00048
- Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood 2001; 97:339-45; PMID:11154206; http://dx.doi.org/10.1182/blood.V97.2.339
- Odajima T, Onishi M, Hayama E, Motoji N, Momose Y, Shigematsu A. Cytolysis of B-16 melanoma tumor cells mediated by the myeloperoxidase and lactoperoxidase systems. Biol Chem 1996; 377:689-93; PMID:8960369; http://dx.doi.org/10.1515/bchm3.1996.377.11.689
- Foldenauer MEB, McClellan SA, Berger EA, Hazlett LD. Mammalian target of rapamycin regulates IL-10 and resistance to pseudomonas aeruginosa corneal infection. The J Immunology 2013; 190:5649-58; PMID:23626014; http://dx.doi.org/10.4049/jimmunol.1203094
- Gomez-Cambronero J. Rapamycin inhibits GM-CSF-induced neutrophil migration. FEBS Lett 2003; 550:94-100; PMID:12935893; http://dx.doi.org/10.1016/S0014-5793(03)00828-7
- Lehman JA. Mechanism of ribosomal p70s6 kinase activation by granulocyte macrophage colony-stimulating factor in neutrophils: cooperation of a mek-related, thr421/ser424 kinase and a rapamycin-sensitive, mtor-related thr389 kinase. J Biol Chem 2003; 278:28130-8; PMID:12740386; http://dx.doi.org/10.1074/jbc.M300376200
- McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, Yost CC. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 2012; 120:3118-25; PMID:22919032; http://dx.doi.org/10.1182/blood-2012-01-405993
- Lindemann SW, Yost CC, Denis MM, McIntyre TM, Weyrich AS, Zimmerman GA. Neutrophils alter the inflammatory milieu by signal-dependent translation of constitutive messenger RNAs. Proc Natl Acad Sci USA 2004; 101:7076-81; PMID:15118085; http://dx.doi.org/10.1073/pnas.0401901101
- Mori S, Tokuyama K. ACE activity affects myogenic differentiation via mTOR signaling. Biochem Biophys Res Commun 2007; 363:597-602; PMID:17892857; http://dx.doi.org/10.1016/j.bbrc.2007.09.006
- Hong C-W, Kim T-K, Ham H-Y, Nam J-S, Kim YH, Zheng H, Pang B, Min T-K, Jung J-S, Lee S-N et al. Lysophosphatidylcholine increases neutrophil bactericidal activity by enhancement of azurophil granule-phagosome fusion via glycine.GlyR alpha 2/TRPM2/p38 MAPK signaling. J Immunol 2010; 184:4401-13; PMID:20237295; http://dx.doi.org/10.4049/jimmunol.0902814
- Ham H-Y, Lee S-N, Kwon M-S, Kim Y-J, Song D-K. Sulfur mustard primes human neutrophils for increased degranulation and stimulates cytokine release via TRPM2/p38 MAPK signaling. Toxicol Appl Pharmacol 2012; 258:82-8; PMID:22036725; http://dx.doi.org/10.1016/j.taap.2011.10.010
- Clark AJ, Diamond M, Elfline M, Petty HR. Calicum microdomains form within neutrophils at the neutrophil–tumor cell synapse: role in antibody-dependent target cell apoptosis. Cancer Immunol Immunother 2009; 59:149-59; PMID:19593564; http://dx.doi.org/10.1007/s00262-009-0735-2
- Metelitsa LS, Naidenko OV, Kant A, Wu HW, Loza MJ, Perussia B, Kronenberg M, Seeger RC. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J Immunol 2001; 167:3114-22; PMID:11544296; http://dx.doi.org/10.4049/jimmunol.167.6.3114
- Chen RL, Reynolds CP, Seeger RC. Neutrophils are cytotoxic and growth-inhibiting for neuroblastoma cells with an anti-GD2 antibody but, without cytotoxicity, can be growth-stimulating. Cancer Immunol Immunother 2000; 48:603-12; PMID:10663607; http://dx.doi.org/10.1007/s002620050008
- Dyugovskaya L, Berger S, Polyakov A, Lavie L. The development of giant phagocytes in long-term neutrophil cultures. J Leukoc Biol 2014; 96:511-21; PMID:24577569; http://dx.doi.org/10.1189/jlb.0813437
- Youn J-I, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol 2013; 14:211-20; PMID:23354483; http://dx.doi.org/10.1038/ni.2526
- Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FHY, Surette MG, Sugai M et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to staphylococcus aureus. J Immunol 2010; 185:7413-25; PMID:21098229; http://dx.doi.org/10.4049/jimmunol.1000675
- Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, Pittet JF, Tracey K, Thannickal VJ, Abraham E et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. AJP: Lung Cellular and Molecular Physiology 2013; 304:L342-9; PMID:23316068; http://dx.doi.org/10.1152/ajplung.00151.2012
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 2007; 176:231-41; PMID:17210947; http://dx.doi.org/10.1083/jcb.200606027