
abstract
G protein-coupled chemoattractant receptors (GPCRs) have been implicated in cancer progression. Formylpeptide receptor 1 (FPR1) was originally identified as a GPCR mediating anti-microbial host defense. However, the role of FPR1 in tumorigenesis remains poorly understood. The current study aims to investigate the potential of FPR1 to regulate human hepatoma growth and invasion. We found the FPR1 gene and protein expression in human intratumoral and peritumoral tissues of hepatocellular carcinoma (HCC) specimens and in human hepatoma cell lines. FPR1 activation mediated the migration, calcium mobilization and ERK-dependent IL-8 production by hepatic cancer cells. FPR1 knockdown substantially reduced the tumorigenicity of hepatoma cells in nude mice. Necrotic hepatic tumor cells released factor(s) that activated FPR1 in live tumor cells. Our results indicate a critical role of FPR1 in the progression of malignant human hepatic cancer. FPR1 thus may represent a molecular target for the development of novel anti-hepatoma therapeutics.
Abbreviations
| CT | = | cholera toxin |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| DMEM | = | Dulbecco's modified Eagle medium |
| ELISA | = | enzyme-linked immunosorbent assays |
| ERK | = | extracellular signal-regulated kinases |
| EGFR | = | epidermal growth factor receptor |
| ETFR | = | epitope-tagged FPR |
| FACS | = | fluorescence-activated cell sorting |
| FCS | = | fetal calf serum |
| FITC | = | fluorescein isothiocyanate |
| fMLF | = | N-formylmethionyl-leucyl-phenylalanine |
| FPR | = | formylpeptide receptor |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| GPCR | = | G protein-coupled receptor |
| HBSS | = | Hank's Balanced Salt Solution |
| HBV | = | hepatitis B virus |
| HCV | = | hepatitis C virus |
| HE | = | the hematoxylin and eosin stain |
| HUVECs | = | human umbilical cord vein endothelial cells |
| IL | = | interleukin |
| MAPK | = | Mitogen-Activated Protein Kinase |
| MMPs | = | matrix metallo proteinases |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBMC | = | peripheral blood mononuclear cells |
| PBS | = | phosphate buffered saline |
| PT | = | pertussis toxin |
| RFUs | = | fluorescence units |
| RT-PCR | = | reverse transcription-polymerase chain reaction |
| shRNA | = | small hairpin RNA |
| tBoc-MLF | = | t-Butyloxycarbonyl-methionylleucyl-phenylalanine |
| TNF-α | = | tumor necrosis factor-α |
| VEGF | = | vascular endothelial growth factor |
| WT | = | wild type. |
Introduction
HCC is the fifth most common cancer with increasing incidence worldwide.Citation1 It is also one of the most lethal human malignancies due to the difficulty of early detection, rapid progression and chemoresistance. Hepatoma is characterized by vigorous angiogenesis and metastasis which account for high rate of postsurgical recurrence and extremely poor prognosis.Citation1 HCC generally develops from chronic liver injury, leading to inflammation, matrix remodeling, fibrosis and cirrhosis.Citation2 Chronic infections by hepatitis viruses such as hepatitis B virus (HBV) and hepatitis C virus (HCV) are major risk factors for HCC development.Citation3
FPR1 is a G protein-coupled 7 transmembrane cell surface receptor (GPCR), originally identified in phagocytic leukocytes, that mediates cell chemotaxis and activation in response to the bacterial formylated chemotactic peptides.Citation4 FPR1 is involved in a broad spectrum of pathophysiologic processes including inflammation,Citation5 wound healing,Citation6 glioblastoma progressionCitation7,8 and the host defense against HCV infection.Citation9
As one of the most vascularized solid tumors, the HCC progression and prognosis correlate with the status of angiogenesis,Citation10 due largely to elevated production of angiogenic factors, such as interleukin-8 (IL-8, CXCL8) by tumor cells.Citation11 Previous reports have implicated IL-8 in the growth and angiogenesis of malignant tumors.Citation12 In cancer models of pancreas, colorectum, melanoma and liver, IL-8 functions as an autocrine growth factor.Citation11 In human hepatoma, clinical investigation has reported that high level of IL-8 is associated with higher frequency invasion of portal vein venous vessels and bile duct by tumor.Citation13 IL-8 production was also associated with severe hepatitis and the development of hepatocellular carcinoma.Citation14 Upregulation of the IL-8 receptor CXC chemokine receptor 2 (CXCR2) was found in HCC and was correlated with intrahepatic metastasis.Citation15 In vitro experiments showed IL-8 production by HCC cell lines in response to tumor necrosis factor-α (TNF-α).Citation16
Previous studies reported that N-formylmethionyl-leucyl-phenylalanine (fMLF), a bacterial chemotactic peptide activating FPR1, increased chemotaxis and production of angiogenic factor IL-8 by human gliobstoma.Citation7,17 FPR1 in glioblastoma cells also interacts with agonists released by necrotic tumor cells,Citation7 suggesting that tumor cells may utilize FPR1 to recognize agonists produced in the tumor microenviroment for their advantage. Since hepatocarcinogenesis involves a highly orchestrated interplay of injury, chronic inflammation and neovascularization,Citation2 the multitude of FPR1 suggests that it may also play a role in the development of hepatic cancer.Citation5-7, Citation9,17 In the present study, we report that FPR1 was expressed by HCC tissues from patients and the human hepatoma cell lines. Hepatoma cells responded to the FPR1 agonist fMLF by increased motility, proliferation and enhanced IL-8 production. FPR1 small hairpin RNA (shRNA) substantially reduced the tumorigenicity of hepatoma cells in nude mice. Our study thus demonstrates a significant role of FPR1 in the carcinogenesis of human hepatoma.
Results
The expression of FPR1 on human hepatocellular carcinoma tissues
We performed histologic and immune fluorescence staining of FPR1 in tumor tissues from HCC patients. In surgical specimens, hematoxylin-eosin (H&E)-staining revealed poorly () and moderately () differentiated HCC with a trabecular pattern. In the high-grade intratumor specimens, multiple tumors of intrahepatic metastases and portal vein invasion were observed. The cellular and nuclear pleomorphism, intracellular vacuoles, mitotic patterns, vessel formation and the necrosis in central tumor tissues were also demonstrated (, upper right panels). The peritumor (, lower right panels) liver tissue showed a chronic inflammatory infiltration in the fibrous stroma, diagnosed as hepatic cirrhosis. Strong FPR1 signal was detected in grade III HCC specimens (, left panels), and positive staining was enriched in intratumor area , upper left panels). In contrast, the lesser aggressive grade II hepatoma specimens showed intermediate staining intensity of FPR1 in intratumoral tissues (, upper left panels) and very low FPR1 expression in peritumoral tissues (, lower left panels). We then examined whether FPR1 expression is selectively enhanced in hepatocellular carcinoma. shows that protein was detectable in human normal liver tissues adjacent to HCC. However, the levels were far lower than that in HCC tissues. Very few FPR1-positive cells were found in the adjacent normal liver tissues, demonstrating that FPR1 expression is selective in HCC and in particular in intratumor tissues.
Figure 1. FPR1 expression in human hepatocellular carcinoma tissues. Sections of 20 samples from grade III (A) and II (B) hepatocellular carcinoma and 10 samples of adjacent normal liver tissues (C) were stained with an antibody against FPR1 (green) and counterstained with DAPI (blue). Representative intratumor (upper panels) and peritumor (lower panels) immunofluorescence staining (left panels) and corresponding H&E staining (right panels) are shown. Bar = 100 μm. (D), quantitative PCR analysis showing the level of FPR1 gene in intratumor or peritumor tissues of grade III and II hepatocellular carcinoma and adjacent normal liver tissues. The data was shown as the mean-fold changes of FPR1 expression levels (± SEM) after intra-sample normalization to the levels of GAPDH. * p <0.05, statistically significant difference vs. adjacent normal liver tissues in values.
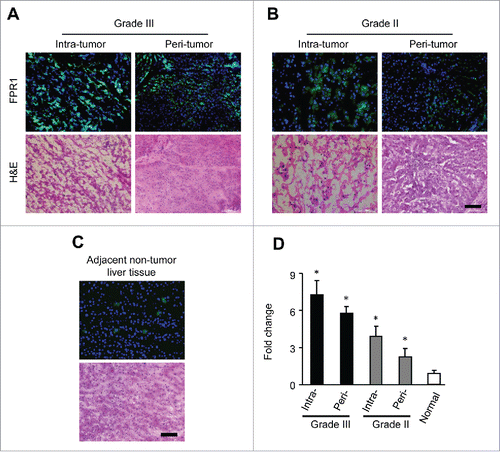
We next measured FPR1 RNA in HCC tissues and found FPR1 mRNA was higher in grade III than in grade II carcinoma specimens. The highest mRNA expression was found in the poorly-differentiated intratumor samples ( and Fig. S1A and B), consistent with the results obtained with histology analysis. These results demonstrate the expression of FPR1 by human hepatocellular carcinoma.
The expression of FPR1 by human hepatoma cell lines
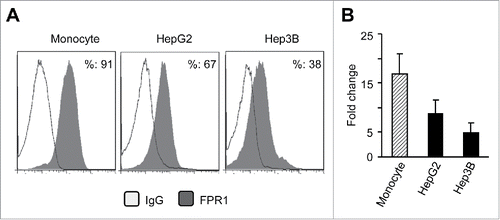
To further determine the biological function of FPR1 in hepatic cancer cells, we used established human hepatic cancer cell lines HepG2 and Hep3B. Fluorescence- activated cell sorting (FACS) analysis shows that both HepG2 and Hep3B cells expressed FPR1 (). HepG2 cells expressed higher levels of FPR1 on cell surface than Hep3B cells (). HepG2 and Hep3B cells also expressed FPR1 gene with higher levels in HepG2 cells ( and Fig. S1C and D).
Figure 2. The expression of FPR1 protein and mRNA in human hepatoma cell lines. (A) FACS analysis of FPR1 expression on hepatic cancer cells lines HepG2 and Hep3B. Human monocytes were used as controls. The results are from representative of three independent experiments. (B) the expression of FPR1 mRNA in hepatoma cell lines. Representative quantitative PCR results from three independent experiments are shown as the mean-fold changes of FPR1 expression levels (± SEM) after intra-sample normalization to the levels of GAPDH.
FPR1 promotes human hepatoma cell chemotaxis and invasion
We next tested the capacity of the FPR1 ligand fMLF to induce directional migration of human hepatoma cell lines. Both HerG2 and Hep3B cells migrated in response to fMLF with a bell-shaped dose-response curve ( and Fig. S2A), typical of the cell response to chemoattractants.Citation18 We also tested the capacity of Ac2–26, a specific cognate ligand for FPR1,Citation19 to induce the migration of human hepatoma cell lines. Both HepG2 and Hep3B cells migrated in response to this FPR1 agonist (Fig. S2B-D). However, addition fo fMLF and Ac2–26 simultaneously to the cells in the upper wells of the chemotaxis chamber abrogated cell migration induced by equal concentrations of fMLF and Ac2–26 in the lower wells (data not shown). Thus, FPR1-induced migration of human hepatoma cell lines was based on chemotaxis rather than chemokinesis. Furthermore, the migration of HepG2 and Hep3B in response to fMLF was completely inhibited by pretreatment of the cells with the Gi protein inhibitor pertussis toxin (PT) and a FPR1-specific antagonist tBoc-MLF,Citation20 but not cholera toxin (CT) or herbimycin A, a protein tyrosine kinase inhibitor ( and Fig. S2A-E), suggesting the involvement of a G-protein of the Gi-type coupled receptor.Citation21
Figure 3. Responsiveness of human hepatoma cell lines to fMLF. (A) chemotaxis of HepG2 (upper panels) and Hep3B (lower panels) cells in response to 100 nM fMLF. (B) inhibition of HepG2 cell migration in response to 100 nM fMLF by pretreatment of the cells with PT (100 ng/mL) or the FPR1-specific antagonist tBoc-MLF (1 µM) for 30 min at 37°C. Representative results from three independent experiments are shown. (C) percentage of HepG2 cell migration in response to different concentrations of fMLF (in the presence or absence of PT or tBoc-MLF) over total loading cells. (D) and (E), motility in wound-healing model. HepG2 cells grown to confluence on plastic were scratched to create a wound. p < 0.05, vehicle-treated vs. fMLF-treated cells. (D) cells in 10 % FCS/DMEM were photographed at 0 and 8 h. The results are representative of three independent experiments. (E) the mean distance (mm) of leading cells moving toward the ‘wound’ area was assessed. *Indicates significantly slower locomotion of HepG2 cells treated with vehicle (n = 3) * p < 0.05, vehicle-treated vs. fMLF-treated cells.
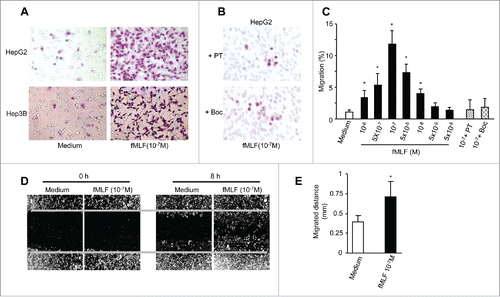
To more precisely examine the contribution of FPR1 to liver tumor cell motility and invasiveness, we used a wound-healing model by creating a gap in a confluent HepG2 cell monolayer. HepG2 cells treated with fMLF showed more rapid locomotion than vehicle-treated cells toward the center of the gap on cell monolayer (). Addition of cyclosporin H (a FPR1 specific antagonist) and PT to the cells blocked the capacity of fMLF to induce human hepatoma cell migration in scratch wound-healing assays, confirming FPR1-mediated cell motility (Fig. S3A and B). Thus FPR1 activation enables HepG2 cells to exhibit higher motility.
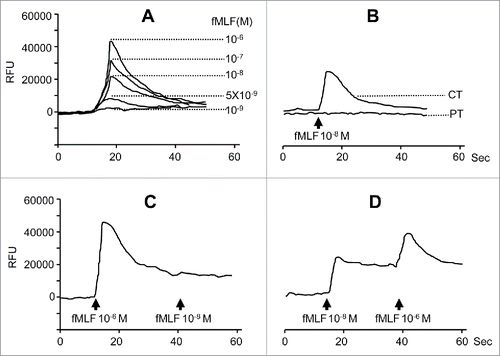
In addition to promoting cell motility, fMLF elicited a potent dose-dependent and PT-sensitive Ca2+ mobilization in HepG2 and Hep3B cells ( and Fig. S4A and B). Sequential stimulation of HepG2 cells with fMLF at high and low concentrations or vice versa resulted in bidirectional desensitization (). However, pretreatment with FPR1 antagonists cyclosporin H or tBoc-MLF reduced the response of HepG2 and Hep3B cells induced by fMLF (Fig. S4D-I). These results further support the specificity of FPR1 expressed by HCC cell lines in response to fMLF
Figure 4. Calcium (Ca2+) mobilization in human hepatoma cells induced by fMLF. Ca2+ mobilization in HepG2 cells was measured with a FlexStation II384 system using Fura-3 AM. Changes in intracellular calcium concentration in response to agonists were recorded as relative fluorescence units (RFUs). (A) dose response of fMLF-induced Ca2+ flux in HepG2 cells. (B) inhibition of HepG2 cell response to 10 nM fMLF by pretreatment of the cells with PT (100 ng/mL) but not CT (100 ng/mL). (C) and (D), cross-desensitization of Ca2+ flux in human hepatoma cells. (C), desensitization of 10 nM fMLF induced Ca2+ flux by 1 μM fMLF in HepG2 cells. (D), 1 μM fMLF-treated HepG2 cell response to second challenge by 10 nM fMLF. Representative results from three independent experiments are shown.
FPR1 mediates MAPK activation in hepatocellular carcinoma cells
Since FPR1 has been reported to mediate phosphorylation of MAP kinases in various cell types,Citation22-24 we stimulated serum-starved HepG2 and Hep3B cells with 100 nM fMLF and determined MAPK phosphorylation at different time points. fMLF stimulated ERK1/2 phosphorylation with a maximum effect at 5 min. ( and Fig. S5A and B). Similar to fMLF, Ac2–26 induced the phosphorylation of ERK1/2 and MAPK in HepG2 and Hep3B cells (Fig. S5A-D). However, pre-treatment of the cells with FPR1 antagonists cyclosporin H or Boc markedly inhibited of fMLF- or Ac2–26-stimulated ERK1/2 phosphorylation (Fig. S5A-D). Therefore, ERK1/2 MAPK is coupled to FPR1 in HCC cells.
Figure 5. FPR1-mediated ERK1/2 activation and IL-8 production by HepG2 hepatoma cells. (A), lysates of HepG2 cells stimulated with 100 nM fMLF for 15 min were examined for phosphorylated ERK1/2 by Western blotting. Total ERK1/2 expression was used as a control. (B) production of IL-8 by fMLF-stimulated hepatoma cells. Levels of IL-8 protein in the supernatants of HepG2 cells treated with different concentrations of fMLF were measured by ELISA. HepG2 cells were also pre-incubated with the MEK1 inhibitor PD98059 (PD) at 50 μ M for 60 min then treated with 100 nM fMLF at 37°C to collect culture medium at 24 and 48 h. The experiments were repeated three times and each time point consisted of three replicate samples. The results are presented as the mean ± SEM. p < 0.05, vehicle-treated vs. fMLF-treated cells. (C), formation of capillary-like structures on Matrigel by HepG2 cells. Culture supernatants from HepG2 cells treated with 100 nM fMLF for 72 h were incubated with an anti-IL-8 antibody (+) or IgG (-) (each at 1 μg/mL) for 45 min. HUVECs were then mixed with the supernatants and examined for tubule formation on Matrigel after 20 h. Photomicrographs were taken with a phase-contrast microscope. Representative results from 3 independent experiments are shown. (D) branching points and total length of tubule structure were quantified. Untreated or fMLF-treated HepG2 cells were pre-incubated in the presence or absence of a neutralizing anti-IL-8 antibody. The differences were examined for statistical significance with Student's t test (n = 5). Values represent the mean ± SEM. p < 0.05, vehicle-treated vs. fMLF-treated cells.
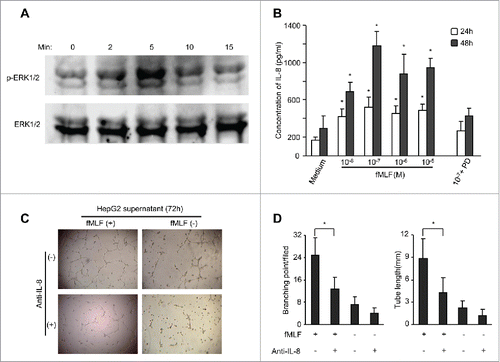
Activation of FPR1 promotes IL-8 production by human hepatoma HepG2 cells
Since activation of FPR1 induces the production of IL-8,Citation11,25 we tested whether FPR1 also promotes IL-8 production by hepatoma cells. Untreated HepG2 and Hep3B cells produced low levels of IL-8 protein and treatment of fMLF and Ac2–26 increased IL-8 production (), reaching a maximum at 48 h after stimulation with 100 nM fMLF (). Addition of the ERK inhibitors PD98059, U0126 and CsH, but not p38 MAPK inhibitor SB203580, inhibited fMLF- and Ac2–26-stimulated IL-8 production by HCC cells (). Thus, only the ERK1/2 MAPK pathway appears to be crucial for FPR agonist-induced IL-8 expression by hepatoma cells.
IL-8 contained in the culture medium of fMLF-stimulated HepG2 cells was biologically active. When cultured with conditioned medium from fMLF-treated HepG2 cells and fMLF+SB-treated HepG2 cell, but not fMLF+CsH- nor fMLF+U0126-treated HepG2 cells, HUVECs formed capillary-like structures on a Matrigel surface (), which was inhibited by the addition of a monoclonal antibody against human IL-8 (). Thus, IL-8 in the conditioned medium from FPR1-activated HepG2 cells induces endothelial cells to form capillary-like structures, a key event associated with neovascularization.
FPR1 knockdown by shRNA reduces the tumorigenicity of hepatic cancer cells
To evaluate the role of FPR1 in hepatoma tumorigenicity, we used shRNA to delete FPR1 in HepG2 cells. After stable transfection of FPR1 shRNA of HepG2 cells, the expression of FPR1 mRNA () and fMLF-induced chemotaxis () were abolished. In addition, the ability of fMLF to induce IL-8 production by HepG2 cells () was abrogated. Further, the cells failed to respond to the proliferation stimulating activity of fMLF ().
Figure 6. The effect of FPR1 shRNA on tumorigenicity of HepG2 cells in athymic mice. (A) the expression of FPR1 mRNA. The levels of FPR1 mRNA were examined by RT-PCR in HepG2 cells stably transfected with FPR1 shRNA. Wild-type (WT, nontransfected) and mock-transfected HepG2 cells were used as control. GAPDH PCR product was used as a loading control. Representative results from three independent experiments are shown. (B) migration of FPR1 shRNA-transfected HepG2 cells in response to fMLF. Percentage of FPR1-mediated cell migration in total loaded cells was calculated. Values represent the mean ± SEM (n = 3). * p < 0.05, compared with mock-transfected cells. (C) the effect of FPR1 shRNA on IL-8 production by HepG2 cells. IL-8 in the supernatants of HepG2 cells cultured in the presence of fMLF was measured by ELISA after 24 and 48 h. The experiments were repeated three times, and each time point consisted of three replicate samples. Values represent the mean ± SEM. * p < 0.05, mock-transfected vs. shRNA-transfected cells. (D) growth curves of mock- and shRNA-transfected HepG2 cells in response to 100 nM fMLF. Cell growth was measured by MTT assays and the results were expressed as the mean ± SEM OD values. Asterisk indicates significantly increased proliferation of HepG2 cells stimulated by fMLF, as compared to non-stimulated cells (p < 0.05). (E) tumor formation. HepG2 cells (at 1 × 106 cells in 100 μL of PBS per mouse) were injected subcutaneously into the flanks of athymic mice (10 mice per group). Mice were examined for tumor formation at indicated times. (F) representative images of xenograft tumors of each group at 45 d after HepG2 cell injection. * p < 0.05, tumors formed by mock- vs. FPR shRNA-transfected cells. (G) tumor growth. Tumor size 35 and 45 d after implantation of HepG2 cells is presented as the mean volume (mm3) of tumors from 10 mice per group. Values represent the mean ± SEM. * p < 0.05, compared with the tumors formed by mock-transfected cells. (H) survival rate of tumor-bearing mice was shown by Kaplan–Meier survival curves. * p < 0.05, mock- vs. FPR shRNA-transfected cells.
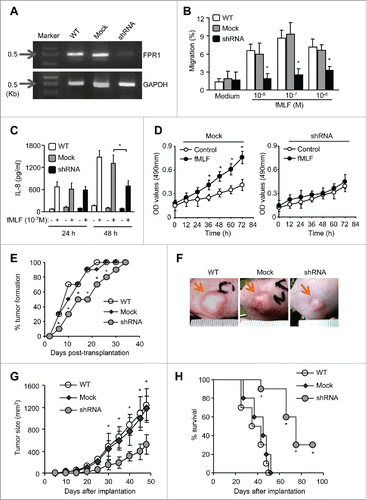
We then injected HepG2 cells transfected with FPR1 shRNA into the flanks of athymic mice. Tumor nodules formed by HEPG2 cells transfected with FPR1 shRNA appeared later () and grew more slowly than those formed by wild-type HepG2 cells or by mock-transfected cells (). By day 52, all mice implanted with wild-type or mock-transfected HepG2 cells were dead. In contrast, 90% of the mice bearing tumors formed by FPR1 shRNA-transfected HepG2 cells survived to the day 66 after implantation (). To conform the role of FPR1 in HCC progression by in vivo experiments using murine cancer cell lines in immunocompetent mice, we examined the effect of FPR1 tumorigenicity in H22 tumor model of BALB/c mice following the method described by another group.Citation26 We show that FPR1-shRNA had similar antitumor effects in immunocompetent mice shown in immunocompromised mice with human cell line (Fig. S6A, B and C). These results indicate that depletion of FPR1 markedly reduced the ability of HepG2 cells to form tumors, confirming the contribution of FPR1 to the tumorigenicity of human HCC cells.
The production of FPR1 agonist activity by necrotic hepatoma cells
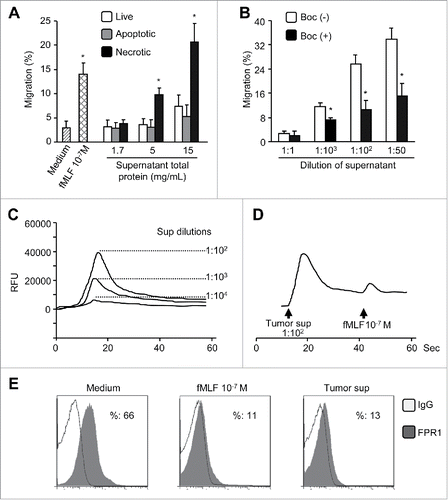
Necrotic cell death has been reported to promote hepatocarcinogenesis,Citation27,28 and mitochondria of ruptured cells contain chemotactic formylpeptides that activate FPR1 in myeloid cells.Citation29 We thus investigated whether necrotic hepatoma cells and tissues might produce agonist(s) recognized by FPR1 on live hepatic cancer cells. HepG2 cells and HepG2 tumors formed in athymic mice released potent chemotactic activity for live HepG2 cells () and ETFR cells overexpressing FPR1 (data not shown). The FPR1 agonist activity released by necrotic HepG2 cells and tumor tissues was blocked by an anti-FPR1 antibody or by the FPR1-specific antagonist tBoc-MLF (, and data not shown). Necrotic hepatoma cell supernatant also induced a robust intracellular Ca2+ mobilization in live HepG2 cells () which attenuated HepG2 cell to response to subsequently administered fMLF (). These results suggest that the agonist(s) contained in the supernatants of necrotic HepG2 cells shares FPR1 with fMLF on HepG2.Citation30 We additionally observed that necrotic hepatoma supernatants inhibited the expression of FPR1 on the surface of ETFR cells with an efficacy comparable to that of 103 nM fMLF (). Thus, necrotic hepatic cancer cells produce FPR1 agonist(s) that interacts with FPR1 on live tumor cells.
Figure 7. FPR1 agonist activity in necrotic tumor supernatants. (A) chemotactic activity. Supernatants of HepG2 cells after three cycles of freezing and thawing (necrotic) were assayed for chemotactic activity on live HepG2 cells. Supernatants from apoptotic or live HepG2cells were used as controls. Percentage of cell migration in response to supernatants in total loaded cells was calculated. Data are expressed as the mean ± SEM (n = 3). * p < 0.05, compared with control medium. (B) chemotactic activity of supernatants of tumor tissue extracts. The supernatants from necrotic HepG2 tumor tissues were assessed for chemotactic activity on live HepG2 cells, which were pre-incubated in the presence or absence of the FPR1-specific antagonist tBoc-MLF at 37 °C for 30 min. Values represent the mean ± SEM (n = 3). * p < 0.05, compared with untreated (-) cells. (C), Ca2+ flux induced in HepG2 cells by necrotic tumor cell supernatants or fMLF. (D) desensitization of fMLF-induced Ca2+ flux by necrotic tumor cell supernatants in HepG2 cells. Representative results from three independent experiments are shown. (E) inhibition of FPR1 expression on ETFR cells by necrotic tumor cell supernatants. The level of FPR1 on the surface of ETFR cells, treated as indicated, was measured by flow cytometry with an anti-FPR1 antibody or normal IgG. Representative results from three independent experiments are shown as a percentage of FPR1-positive cells.
Discussion
In this article, we showed that the classic leukocyte chemoattractant receptor FPR1 is expressed by human HCC tissues and hepatoma cell lines. To our knowledge, this is the first demonstration that FPR1 may contribute to the progression of HCC by mediating tumor cell chemotaxis, proliferation, and production of IL-8 in response to endogenous agonist(s). A growing body of evidence has suggested important roles of chemoattractant GPCRs in cancer initiation and progression by influencing aberrant cell growth and survival.Citation31 GPCRs are also implicated in the invasion and metastasis of cancer cells and contribute to the establishment and maintenance of a permissive tumor microenvironment.Citation31 The FPR1 involvement in cancer malignancy has been shown by its roles in glioblastoma tumorigenicity.Citation7 FPR1 expressed by highly malignant human glioma cells promoted the motility, growth, and the production of angiogenic factors.Citation7,32 The glioblastoma-promoting activity of FPR1 is mediated in part by transactivation of the epidermal growth factor receptor (EGFR),Citation8 associated with increased methylation of p53 gene.Citation33 FPR1 in glioblastoma cells is stimulated by Annexin 1 released by necrotic tumor cells.Citation19 Our present study extended the functional scope of FPR1 to its potential role in promoting the growth of malignant human hepatoma.
As a primary malignancy that emerges on a background of chronic liver diseases,Citation34 HCC is the fifth most common cancer worldwide with an extremely poor prognosis.Citation35 Chronic inflammation and improper wound healing following hepatic injury are crucial causative factors for cancer in the liver.Citation2 The link between an inflammatory state and cancer can be viewed from an extrinsic perspective for which infection and subsequent chronic inflammation drive oncogenesis.Citation36 In that case, cancers associated with inflammation are generally aggressive.Citation11 Functional FPR1 has reportedly been detected in hepatocytes, and it promoted the production of acute phase proteins in response to fMLF.Citation37 Thus, FPR1 expressed on hepatocytes might participate in the inflammatory courses in liver. It has been shown that the expression levels of chemokines in the liver are positively correlated with the severity of hepatic inflammation in chronic HCV infection.Citation38,39 The elevated expression of chemokines in a severely inflamed liver could efficiently attract leukocytes to the liver.Citation39,40 Our previous studies also revealed that HCV peptide (C5A) is able to activate FPR1 on phagocytic leukocytes.Citation9 Given that HCC is an example of inflammation-related cancer, and the chronic infections with HBV and HCV are major risk factors for HCC development,Citation41 our findings unveiling the hepatoma-promoting properties of FPR1 may represent a novel mechanism by which the pro-inflammatory response is linked to tumor initiation.
A hallmark in the progression of malignant tumors is increased angiogenesis. The hyper-vasculature in chronic liver diseases facilitates the progression from small dysplastic nodules, through neoplastic lesions to large hepatocellular carcinoma.Citation34 It has been reported that FPR2 (FPRL1) in human corneal epithelial cells was activated by an anti-microbial peptide LL-37 to stimulated IL-8 production by the cells resulting in enhanced vascularization.Citation42 As a proinflammatory chemokine, IL-8 mediates the recruitment of leukocytes, which in turn produce angiogenic factors recruiting vascular endothelial cells and promoting their proliferation.Citation11 IL-8 has been shown to increase the motility of a variety of tumor cells from glioblastoma, melanoma, pancreatic cancer to colon cancer.Citation11,17 IL-8 production was correlated with the severity of hepatitis and the development of hepatocellular carcinoma. IL-8 was elevated in patients with advanced HCC with distant metastasis.Citation14,43 In addition, upregulated IL-8 in the drainage vein of colorectal cancer is linked to the occurrence of hepatic metastasis.Citation44 Our present study showing the induction of IL-8 by FPR1 activation in human hepatoma cells supports the adverse effect of this chemokine on hepatoma growth and angiogenesis.
The expression of IL-8 in normal cells is low, but it was highly inducible by a variety of pro-inflammatory stimulants.Citation45 In primary human hepatocytes, IL-8 is induced by adipokine,Citation46 whereas HCC produced IL-8 after stimulation by TNF-α.Citation16 In rat cornea, IL-8 stimulates angiogenesis by promoting the chemotaxis and growth of vascular endothelial cells.Citation47 IL-8 also inhibits the apoptosis of endothelial cells, upregulates endothelial cell production of matrix metalloproteinase-2 and -9, mimics the function of vascular endothelial growth factor (VEGF), and trans-activates VEGF-R2.Citation11 In vitro mitochondrial proteins from necrotic HepG2 cells stimulate FPR1 in monocyte to release IL-8.Citation25 Thus, the current study provides evidence for IL-8 as a molecule down-stream of FPR1 signaling pathway to amplify the effect of FPR1 on hepatic tumor growth.
Malignant tumors exploit their microenvironment to favor their survival, growth, invasion, and metastasis.Citation48 For instance, tumor cells produce aberrant levels of regulatory molecules to increase their proliferation. Tumor cells also produce high levels of IL-8 to enable cancer cells to survive and to recruit endothelial cells for vascularization.Citation11 In addition, malignant tumor cells express receptors that interact with agonists that are present in the vicinity of the tumor or produced by distant organs to increase tumor cell motility to favor tumor cell invasion and metastasis. In the present study, FPR1 was expressed at a higher level by advanced hepatocellular carcinoma. A positive correlation between FPR1 expression and the grade of HCC suggests FPR1 as a biomarker for more aggressive tumor cells. Identification of FPR1 agonist(s) in the supernatants of necrotic tumor cells provides evidence that this receptor may interact with endogenous agonists produced in tumor lesions, presumably in the necrotic area frequently associated with highly malignant hepatoma or in surrounding tissues that are compressed by growing tumor in a limited anatomical compartment. Previous studies have demonstrated FPR1-dependent necrotaxis as the molecular mechanisms underlying the neutrophil recruitment to sites of sterile focal hepatic necrosis.Citation49 It is thus plausible that FPR1 in live tumor cells may serve as a sensor for the agonists produced in a “paracrine” manner in the tumor microenvironment to promote cell migration, survival, proliferation and the production of IL-8.
Further study is required to more precisely define the relationship between the FPR1 expression and the progression of human primary HCC and to identify the mechanistic basis for the control of FPR1 expression in highly malignant human hepatic cancer cells. In addition, the pathogenesis of human hepatoma is likely to be complex, and FPR1 may not be the sole factor that regulates the progression of malignant liver tumor. Additional experiments are needed to address the possible cross-talk between FPR1 and other growth receptors such as EGFRCitation8 in exacerbating the malignant phenotype of cancer cells. Also, the relationship between FPR1 expression and the survival of hepatoma patients after treatment remains to be established. Nevertheless, the present study implicates the role of FPR1 in the rapid progression of highly malignant human hepatocellular carcinoma, thus raises the possibility that FPR1 may be a candidate molecular target for developing novel therapeutics.
Materials and methods
Patients and specimens
Tumor samples were obtained from patients with pathologically confirmed HCC at the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China. All samples were coded anonymously in accordance with local ethical guidelines (as stipulated by the Declaration of Helsinki), and written informed consent was obtained. The protocol was approved by the Review Board of the First Affiliated Hospital, Sun Yat-sen University. Detailed information is described in Supplementary Materials and Methods.
Cells
Detailed information about cell lines including human hepatic cancer cells HepG2 and Hep3B, rat basophilic leukemia cells stably transfected with epitope-tagged FPR (ETFR), human umbilical cord vein endothelial cells (HUVECs), human peripheral blood mononuclear cells (PBMC) and monocytes is described in Supplementary Materials and Methods.
Chemotaxis and Ca2+ flux
Assays for tumor cell chemotaxis and Ca2+ mobilization were performed according to the described procedures.Citation18,50 Details can be obtained from Supplementary Materials and Methods.
Tumorigenesis
To generate xenografts with hepatoma cells transfected with FPR1 shRNA, 1 × 106 cells were injected into the flank of each 4-week-old female athymic Ncr-nu/nu mouse. Animal care was provided in accordance with the Guide for the Care and Use of Laboratory Animals. Detailed information about FPR1 knockdown in HepG2 cells and tumor implantation is described in Supplementary Materials and Methods.
Additional Materials and Methods
A full description of the methods including histology and immunofluorescence staining, RT-PCR, flow cytometry analysis, wound-healing assays, Western blotting, ELISA, formation of capillary-like structures, MTT assay, generation of tumor cell supernatant and tissue extracts, and statistical analyses can be found in Supplementary Materials and Methods.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1078055_Supplementary_Material.docx
Download MS Word (1.3 MB)Acknowledgment
The authors thank Dr. Xiuwu Bian (Institute of Pathology and Southwest Cancer Center, Southwest Hospital, Third Military Medical University, Chongqing, China) for his helpful critique of the manuscript.
Funding
This work is supported by the grants from National Nature Science Foundation of China 81200674 (Q.L) and 31170861 (S.B.S). Chen K and JM Wang were funded in part by Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and were supported in part by the Intramural Research Program of the NCI, NIH, United States of America.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; http://dx.doi.org/3322/caac.20107
- Bishayee A. The inflammation and liver cancer. Adv Exp Med Biol 2014; 816:401-35; PMID:24818732; http://dx.doi.org/10.1007/978-3-0348-0837-8_16
- Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012; 379:1245-55; PMID:22353262; http://dx.doi.org/10.1016/S0140-6736(11)61347-0
- Le YY, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol 2002; 23:541-8; PMID:12401407; http://dx.doi.org/10.1016/S1471-4906(02)02316-5
- Le YY, Oppenheim JJ, Wang JM. Pleiotropic roles of formyl peptide receptors. Cytokine Growth F R 2001; 12:91-105; PMID:11312121; http://dx.doi.org/10.1016/S1359-6101(01)00003-X.
- Liu MY, Chen KQ, Yoshimura T, Liu Y, Gong WH, Le YY, Gao JL, Zhao JH, Wang JM, Wang AM. Formylpeptide Receptors Mediate Rapid Neutrophil Mobilization to Accelerate Wound Healing. PloS one 2014; 9.; PMID:24603667; http://dx.doi.org/10.1371/journal.pone.0090613
- Zhou Y, Bian XW, Le YY, Gong WH, Hu JY, Zhang X, Wang LH, Iribarren P, Salcedo R, Howard OMZ et al. Formylpeptide receptor FPR and the rapid growth of malignant human gliomas. J Natl Cancer I 2005; 97:823-35; PMID:15928303; http://dx.doi.org/10.1093/jnci/dji142
- Huang J, Hu J, Bian X, Chen K, Gong W, Dunlop NM, Howard OM, Wang JM. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant behavior of human glioblastoma cells. Cancer Res 2007; 67:5906-13; PMID:17575160; http://dx.doi.org/10.1158/0008-5472.CAN-07-0691
- Lin Q, Fang D, Hou XW, Le YY, Fang JH, Wen F, Gong WH, Chen KQ, Wang JM, Su SB. HCV Peptide (C5A), an Amphipathic α-Helical Peptide of Hepatitis Virus C, Is an Activator of N-Formyl Peptide Receptor in Human Phagocytes. J Immunol 2011; 186:2087-94; PMID:21228351; http://dx.doi.org/10.4049/jimmunol.1002340
- Yang ZF, Poon RTP. Vascular changes in hepatocellular carcinoma. Anat Rec 2008; 291:721-34; PMID:18484619; http://dx.doi.org/10.1002/ar.20668
- Gales D, Clark C, Manne U, Samuel T. The Chemokine CXCL8 in Carcinogenesis and Drug Response. ISRN Oncol 2013; 2013:859154; PMID:24224100; http://dx.doi.org/10.1155/2013/859154
- Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncol 2005; 7:122-33; PMID:15831231; http://dx.doi.org/10.1215/S1152851704001061
- Akiba J, Yano H, Ogasawara S, Higaki K, Kojiro M. Expression and function of interleukin-8 in human hepatocellular carcinoma. Int J Oncol 2001; 18:257-64; PMID:11172590
- Tachibana Y, Nakamoto Y, Mukaida N, Kaneko S. Intrahepatic interleukin-8 production during disease progression of chronic hepatitis C. Cancer Lett 2007; 251:36-42; PMID:17240051; http://dx.doi.org/10.1016/j.canlet.2006.10.028
- Liu Z, Yang L, Xu J, Zhang X, Wang B. Enhanced expression and clinical significance of chemokine receptor CXCR2 in hepatocellular carcinoma. J Surg Res 2011; 166:241-6; PMID:20018298; http://dx.doi.org/10.1016/j.jss.2009.07.014
- Wang Y, Wang W, Wang L, Wang X, Xia J. Regulatory mechanisms of interleukin-8 production induced by tumour necrosis factor-α in human hepatocellular carcinoma cells. J Cell Mol Med 2012; 16:496-506; PMID:21545687; http://dx.doi.org/10.1111/j.1582-4934.2011.01337.x
- Yao XH, Ping YF, Chen JH, Chen DL, Xu CP, Zheng J, Wang JM, Bian XW. Production of angiogenic factors by human glioblastoma cells following activation of the G-protein coupled formylpeptide receptor FPR. J Neurooncol 2008; 86:47-53; PMID:17611713; http://dx.doi.org/10.1007/s11060-007-9443-y
- Le Y, Hu J, Gong W, Shen W, Li B, Dunlop NM, Halverson DO, Blair DG, Wang JM. Expression of functional formyl peptide receptors by human astrocytoma cell lines. J Neuroimmunol 2000; 111:102-8; PMID:11063827; http://dx.doi.org/10.1016/S0165-5728(00)00373-8
- Yang Y, Liu Y, Yao X, Ping Y, Jiang T, Liu Q, Xu S, Huang J, Mou H, Gong W et al. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am J Pathol 2011; 179:1504-12; PMID:21782780; http://dx.doi.org/10.1016/j.ajpath.2011.05.059
- Fu H, Karlsson J, Bylund J, Movitz C, Karlsson A, Dahlgren C. Ligand recognition and activation of formyl peptide receptors in neutrophils. J Leukoc Biol 2006; 79:247-56; PMID:16365159; http://dx.doi.org/10.1189/jlb.0905498
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 2009; 61:119-61; PMID:19498085; http://dx.doi.org/10.1124/pr.109.001578
- Kim SD, Kim JM, Jo SH, Lee HY, Lee SY, Shim JW, Seo SK, Yun J, Bae YS. Functional expression of formyl peptide receptor family in human NK cells. J Immunol 2009; 183:5511-7; PMID:19843937; http://dx.doi.org/10.4049/jimmunol.0802986
- Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y et al. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nat Immunol 2012; 13:457-64; PMID:22447027; http://dx.doi.org/10.1038/ni.2258
- Rane MJ, Carrithers SL, Arthur JM, Klein JB, McLeish KR. Formyl peptide receptors are coupled to multiple mitogen-activated protein kinase cascades by distinct signal transduction pathways: role in activation of reduced nicotinamide adenine dinucleotide oxidase. J Immunol 1997; 159:5070-8; PMID:9366435
- Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S, Huang Q, Wewers MD. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit Care Med 2009; 37:2000-9; PMID:19384205; http://dx.doi.org/10.1097/CCM.0b013e3181a001ae
- Wang R, Zhang XW, Wang GQ, Chen XC, Tian L, Yang HS, Hu M, Peng F, Yang JL, He QM et al. Systemic inhibition of tumor growth by soluble Flk-1 gene therapy combined with cisplatin. Cancer Gene Ther 2006; 13:940-7; PMID:16799469; http://dx.doi.org/10.1038/sj.cgt.7700958
- Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol 2004; 4:641-8; PMID:15286730; http://dx.doi.org/10.1038/nri1415
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005; 121:977-90; PMID:15989949; http://dx.doi.org/10.1016/j.cell.2005.04.014
- Carp H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med 1982; 155:264-75; PMID:6274994; http://dx.doi.org/10.1084/jem.155.1.264
- Le Y, Gong W, Li B, Dunlop NM, Shen W, Su SB, Ye RD, Wang JM. Utilization of two seven-transmembrane, G protein-coupled receptors, formyl peptide receptor-like 1 and formyl peptide receptor, by the synthetic hexapeptide WKYMVm for human phagocyte activation. J Immunol 1999; 163:6777-84; PMID:10586077
- Huang J, Chen K, Gong W, Dunlop NM, Wang JM. G-protein coupled chemoattractant receptors and cancer. Front Biosci 2008; 13:3352-63; PMID:18508437; http://dx.doi.org/10.2741/2930
- Huang J, Chen K, Chen J, Gong W, Dunlop NM, Howard OM, Gao Y, Bian XW, Wang JM. The G-protein-coupled formylpeptide receptor FPR confers a more invasive phenotype on human glioblastoma cells. Br J Cancer 2010; 102:1052-60; PMID:20197768; http://dx.doi.org/10.1038/sj.bjc.6605591
- Huang J, Chen K, Huang J, Gong W, Dunlop NM, Howard OM, Bian X, Gao Y, Wang JM. Regulation of the leucocyte chemoattractant receptor FPR in glioblastoma cells by cell differentiation. Carcinogenesis 2009; 30:348-55; PMID:19037090; http://dx.doi.org/10.1093/carcin/bgn266
- Coulon S, Heindryckx F, Geerts A, Van Steenkiste C, Colle I, Van Vlierberghe H. Angiogenesis in chronic liver disease and its complications. Liver Int 2011; 31:146-62; PMID:21073649; http://dx.doi.org/10.1111/j.1478-3231.2010.02369.x
- Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003; 362:1907-17; PMID:14667750; http://dx.doi.org/10.1016/S0140-6736(03)14964-1
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008; 454:436-44; PMID:18650914; http://dx.doi.org/10.1038/nature07205
- McCoy R, Haviland DL, Molmenti EP, Ziambaras T, Wetsel RA, Perlmutter DH. N-formylpeptide and complement C5a receptors are expressed in liver cells and mediate hepatic acute phase gene regulation. J Exp Med 1995; 182:207-17; PMID:7540650; http://dx.doi.org/10.1084/jem.182.1.207
- Shields PL, Morland CM, Salmon M, Qin S, Hubscher SG, Adams DH. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J Immunol 1999; 163:6236-43; PMID:10570316
- Apolinario A, Majano PL, Alvarez-Perez E, Saez A, Lozano C, Vargas J, Garcia-Monzon C. Increased expression of T cell chemokines and their receptors in chronic hepatitis C: relationship with the histological activity of liver disease. Am J Gastroenterol 2002; 97:2861-70; PMID:12425561; http://dx.doi.org/10.1111/j.1572-0241.2002.07054.x
- Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol 2001; 2:123-8; PMID:11175804; http://dx.doi.org/10.1038/84219
- Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, Zazzeroni F, Alesse E. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int 2013; 2013:187204; PMID:23533994; http://dx.doi.org/10.1155/2013/187204
- Koczulla R, von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest 2003; 111:1665-72; PMID:12782669; http://dx.doi.org/10.1172/JCI17545
- Harimoto N, Shirabe K, Abe T, Kajiyama K, Nagaie T, Gion T, Kuroda Y, Maehara Y. Interleukin-8 producing hepatocellular carcinoma with pyrexia. HPB Surgery 2009; 2009:461492; PMID:19707535; http://dx.doi.org/10.1155/2009/461492
- Haraguchi M, Komuta K, Akashi A, Matsuzaki S, Furui J, Kanematsu T. Elevated IL-8 levels in the drainage vein of resectable Dukes' C colorectal cancer indicate high risk for developing hepatic metastasis. Oncol Rep 2002; 9:159-65; PMID:11748475
- Mukaida N, Mahe Y, Matsushima K. Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J Biol Chem 1990; 265:21128-33; PMID:2250017
- Wanninger J, Neumeier M, Weigert J, Bauer S, Weiss TS, Schaffler A, Krempl C, Bleyl C, Aslanidis C, Scholmerich J et al. Adiponectin-stimulated CXCL8 release in primary human hepatocytes is regulated by ERK1/ERK2, p38 MAPK, NF-kappaB, and STAT3 signaling pathways. Am J Physiol Gastrointest Liver Physiol 2009; 297:G611-8; PMID:19608729; http://dx.doi.org/10.1152/ajpgi.90644.2008
- Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H, Kuwano M. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor α-dependent angiogenesis. Mol Cell Biol 1997; 17:4015-23; PMID:9199336
- Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature 2001; 411:375-9; PMID:11357145; http://dx.doi.org/10.1038/35077241
- McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010; 330:362-6; PMID:20947763; http://dx.doi.org/10.1126/science.1195491
- Zhu J, Wang O, Ruan L, Hou X, Cui Y, Wang JM, Le Y. The green tea polyphenol (-)-epigallocatechin-3-gallate inhibits leukocyte activation by bacterial formylpeptide through the receptor FPR. Int Immunopharmacol 2009; 9:1126-30; PMID:19426837; http://dx.doi.org/10.1016/j.intimp.2009.05.002