
ABSTRACT
The interaction of the MHC class I-related chain molecules A and B (MICA and MICB) and UL-16 binding protein (ULBP) family members expressed on tumor cells with the corresponding NKG2D receptor triggers cytotoxic effector functions in NK cells and γδ T cells. However, as a mechanism of tumor immune escape, NKG2D ligands (NKG2DLs) can be released from the cell surface. In this study, we investigated the NKG2DL system in different human glioblastoma (GBM) cell lines, the most lethal brain tumor in adults. Flow cytometric analysis and ELISA revealed that despite the expression of various NKG2DLs only ULBP2 is released as a soluble protein via the proteolytic activity of “a disintegrin and metalloproteases” (ADAM) 10 and 17. Moreover, we report that temozolomide (TMZ), a chemotherapeutic agent in clinical use for the treatment of GBM, increases the cell surface expression of NKG2DLs and sensitizes GBM cells to γδ T cell-mediated lysis. Both NKG2D and the T-cell receptor (TCR) are involved. The cytotoxic activity of γδ T cells toward GBM cells is strongly enhanced in a TCR-dependent manner by stimulation with pyrophosphate antigens. These data clearly demonstrate the complexity of mechanisms regulating NKG2DL expression in GBM cells and further show that treatment with TMZ can increase the immunogenicity of GBM. Thus, TMZ might enhance the potential of the adoptive transfer of ex vivo expanded γδ T cells for the treatment of malignant glioblastoma.
Abbreviations
| ADAM | = | A disintegrin and metalloprotease(s) |
| BrHPP | = | bromohydrin pyrophosphate |
| CFSE | = | carboxyfluoresceinsuccinimidyl ester |
| GBM | = | glioblastoma multiforme |
| GI | = | GI254023X |
| GW | = | GW280264X |
| MICA/B | = | MHC class I-related chain molecule A/B |
| MP | = | metalloprotease(s) |
| NKG2D | = | Natural Killer Group 2, member D |
| NKG2DL | = | NKG2D Ligand |
| sMICA/B | = | soluble MICA/B |
| sULBP1/2 | = | soluble ULBP1/2 |
| TCR | = | T-cell receptor |
| TMZ | = | temozolomide |
| ULBP 1/2 | = | UL-16 binding protein 1/2 |
Introduction
Glioblastoma multiforme, termed as glioblastoma for short (GBM) is the most lethal primary brain tumor in adults. Despite the aggressive standard therapeutic strategy including surgery and radio-chemotherapy, the median survival of GBM patients (14.6 mo) remains extremely low pointing to the urgent need for alternative treatment strategies such as immunotherapy.Citation1 Numerous studies demonstrated that ex vivo expanded immune cells could be a promising tool for the treatment of GBM.Citation2 Malignant GBM cells express several stress-inducible molecules which are sensed by the activating receptor NKG2D.Citation3 This interaction triggers cytotoxic activity in NKG2D-expressing killer cells and hence, the NKG2DL system is considered a promising target for the improvement of cell-based immunotherapies.Citation4 NKG2D is a C-type lectin-like receptor expressed on NK cells, NKT cells, γδ T cells, CD8+ T cells and a minor subset of immunoregulatory CD4+ T cells. The ligation of NKG2D triggers cytotoxicity in NK cells and co-stimulation in T-cell subsets.Citation5 Ligands for human NKG2D comprise two groups of MHC class I-related molecules, the MHC class I chain-related proteins A and B (MICA/B) and six members of the UL16-binding protein family (ULBP1-6).Citation6 Members of the ULBP family carry 2 MHC class I-like domains (α1, α2) and are either transmembrane proteins (ULBP4,5) or bound to the membrane with GPI-linkage (ULBP1,3,6). ULBP2 has the unique feature that it can be expressed at the cell surface either as a transmembrane protein or with a GPI anchor.Citation7 NKG2DLs are normally not expressed on healthy cells but expression can be induced by various types of cellular stress including viral infection, genotoxic stress or malignant transformation.Citation8 As an example, in contrast to tissues isolated from meningioma patients, 10 out of 11 GBM specimens were stained positive for NKG2DLs.Citation9 NKG2DLs are excellent targets for NKG2D-mediated cytotoxicity and higher expression levels of these ligands are associated with increased cytotoxic activity of effector cells.Citation10 However, as a mechanism of immune escape, many tumor cells release soluble NKG2DLs (sNKG2D). NKG2DLs are frequently shed by metalloproteases (MP), specifically by distinct members of the ADAM family.Citation11 ADAM10 and ADAM17 have been implicated in the shedding of MICA/B and ULBP2 in various model systems,Citation12,13 whereas GPI-anchored ULBPs (ULBP1,2,3) are known to be processed bypho sphoinositide phospholipase C or are released in association with exosomes (reviewed in Chitadze et al.Citation14). However, the exact mechanisms regulating NKG2DL in GBM cells are still elusive and require further investigation.
Current standard GBM treatment strategies include the administration of the chemotherapeutic agent TMZ in combination with radiotherapy after surgical removal of tumors. Temozolomide (TMZ) is an alkylating agent that induces apoptosis via methylation of guanine residues.Citation15 Since genotoxic stress is linked to the induction of NKG2DL expression, TMZ treatment transiently increased the expression of various NKG2DLs in TMZ-resistant GBM cell lines.Citation16
NKG2D is expressed on human γδ T cells, a minor population of peripheral blood lymphocytes (1–5%). The majority of these cells expresses a Vγ9Vδ2 TCR and recognizes microbial and eukaryotic pyrophosphate antigens (phosphoantigens) in a butyrophilin 3A1 (CD277)-dependent manner.Citation17-19 Since endogenous phosphoantigens are overproduced in transformed cells, γδ T cells can distinguish transformed cells from healthy tissues.Citation20 Furthermore, recognition of pyrophosphate antigens by γδ T cells is not restricted by MHC molecules which is advantageous in the setting of allogeneic adoptive cell transfer.Citation21 Vγ9Vδ2 T cells can be easily expanded in vitro with synthetic phosphoantigens or with nitrogen-containing bisphophonates such as zoledronate, combined with low doses of IL-2. Of note, γδ T cells elicit potent antitumor activity against a broad range of malignant cells including GBM.Citation22,23
In this study, we investigated the effects of MP inhibitors and TMZ on NKG2DL expression and shedding in several human GBM cell lines. We report that GBM cells express several NKG2DLs, but preferentially release ULBP2 into culture supernatants in an ADAM10/17-dependent manner. Moreover, we show that TMZ treatment increases the cell surface expression of NKG2DLs and also sensitizes GBM cells to γδ T cell-mediated killing involving both NKG2D- and TCR-dependent mechanisms.
Results
GBM cell lines differentially express and release NKG2D ligands
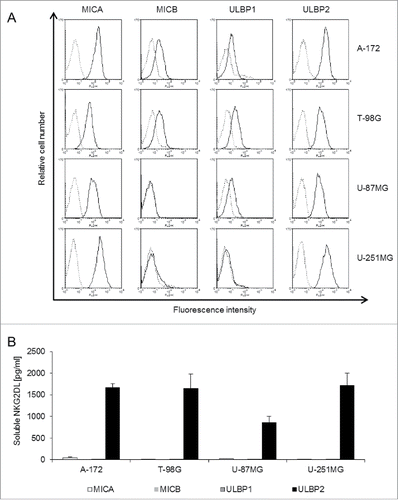
Initially we screened the GBM cell lines A-172, T-98G, U-87MG and U-251MG for cell surface expression of the endogenous NKG2DLs MICA, MICB, ULBP1 and ULBP2 by flow cytometry (). The expression levels of MICA, MICB, ULBP1 and ULBP2 varied considerably among GBM cell lines. Expression of MICA and ULBP2 was remarkably high when compared to MICB and ULBP1. Moreover, U-87MG and U-251MG cells completely lacked MICB cell surface expression (). In addition, U-251MG cells were deficient in cell surface expression of ULBP1. In parallel, levels of sNKG2DLs in cell culture supernatants were determined by ELISA to quantify the extent of shedding of MICA, MICB, ULBP1 and ULBP2 from the tested cell lines (). Interestingly, despite considerable cell surface expression of the tested NKG2DLs, only ULBP2 was detected in cell culture supernatants as a soluble molecule. Some MICA alleles (specifically MICA*008) are known to be secreted in exosomes rather than being shed by MPs.Citation24 Biochemical and sequencing analysis revealed that both A-172 and U-87MG cells indeed expressed the truncated MICA*008:01:01 allele, whereas T-98G (MICA*001) and U-251MG (MICA*002:01) expressed full length MICA (not shown). In line, MICA was detected in exosome preparations of A-172 and U-87MG but not T-89G and U-251MG cells (not shown). It has been reported that ULBP2 can be either released in association with exosomes or by the activity of MPs.Citation25 To address this issue, we immunoprecipitated ULBP2 from cellular lysates, culture supernatants and isolated exosomes of GBM cell lines. As expected from ELISA results presented in , western blot analysis revealed that ULBP2 was predominantly detected in the culture supernatants and not in the exosome preparations. However, a very weak signal at high exposure time was observed in T-98G-derived exosomal preparations. (Fig. S1).
Figure 1. GBM cell lines express various NKG2DLs and release soluble ULBP2 into the supernatant. (A) Cell surface expression of MICA, MICB, ULBP1 and ULBP2 (solid lines) was determined by flow cytometry in A-172, T-98G, U-87MG and U-251MG cells. Dotted lines represent the respective isotype controls. (B) The amount of sMICA (white), sMICB (light gray), sULBP1 (dark gray) and sULBP2 (black) in cell culture supernatants was determined by ELISA. Data are presented as mean values of three independent experiments +/− SEM.
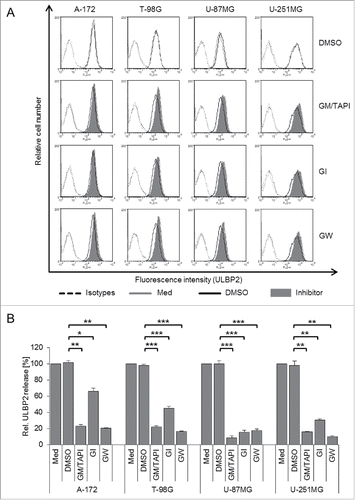
Metalloproteases, particularly ADAM10 and ADAM17, mediate the release of soluble ULBP2
To address the role of MPs in ULBP2 release, we used a combination of the broad range MP inhibitors GM6001 and TAPI-1. Cells were cultured for 24 h in the presence or absence of inhibitors at non-toxic concentrations and cell surface expression of ULBP2 was analyzed by flow cytometry. As shown in , broad range inhibition of MPs slightly increased cell surface expression of ULBP2 () and simultaneously decreased the amount of sULBP2 in cell culture supernatants (). In parallel, we inhibited the ADAM family members ADAM10 and ADAM17 with GI254023X (GI, ADAM10-specific) and GW280264X (GW, ADAM10/17-specific) compounds.Citation26 These studies revealed that ADAM10 and ADAM17 have a complementary effect on ULBP2 shedding. Inhibition of ADAM10 almost completely abrogated the shedding of ULBP2 in U-87MG cells but only partially in A-172, T-98G and U-251MG cells. However, in the latter cell lines ULBP2 shedding was effectively prevented upon additional inhibition of ADAM17 (). In line with these results, flow cytometric analysis showed that inhibition of shedding by GI and GW increased the cell surface expression of ULBP2 with cell line-dependent variability (representative histograms are shown in and a quantification of 3–4 experiments in Fig. S2A) but had no effect on surface expression of other NKG2DLs (not shown). Of note, there was no effect of the relevant DMSO control. Together these data indicate that ADAM10 and ADAM17 mediate the proteolytic release of ULBP2 in GBM cells. However, the contribution of ADAM10 and ADAM17 to ULBP2 shedding differs among cell lines. Flow cytometric analysis of ADAM10 and ADAM17 surface expression and Western blot analysis of the active and the proform of ADAM10/17 in GBM cell lysates revealed no major differences among the four cell lines. (not shown).
Figure 2. Metalloprotease inhibition modulates ULBP2 cell surface expression and reduces ULBP2 shedding. Cells were treated for 24 h with the combination of the broad range MP inhibitors GM6001 (5 µM) and TAPI-1 (5 µM), or with GI (3 µM) or GW (3 µM) to inhibit ADAM10 and ADAM10/17, respectively. DMSO was used as a solvent control. (A) Cell surface expression of ULBP2 was determined by flow cytometry after treatment with MP inhibitors (filled histograms), with DMSO solvent control (open black histograms), or without treatment (medium control, open gray histograms). Dotted lines represent the isotype controls. (B) The concentration of sULBP2 in supernatants of medium control, DMSO- and inhibitor-treated GBM cells was determined by ELISA. The medium control was set to 100%. Data are presented as mean values of three to four independent experiments +/− SEM. Statistical significance is displayed as *** for p < 0.001; ** for p < 0.01 and * for p < 0.05.
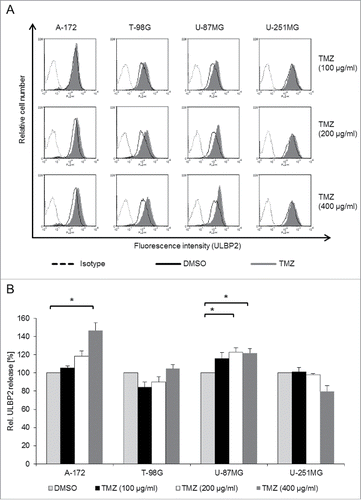
TMZ treatment increases the cell surface expression of ULBP2
Since TMZ has already been shown to enhance NKG2DL expression,Citation16 we investigated the effect of TMZ on cell surface expression and shedding of ULBP2 in the various GBM cell lines. Cells were cultured for 24 h with various concentrations of TMZ or with DMSO as a solvent control. TMZ enhanced the cell surface expression of ULBP2 () and other NKG2DLs (not shown) in A-172, T-98G and U-87MG but only moderately in U-251MG cells (representative histograms are shown in and a quantification of three experiments in Fig. S2B). Shedding of ULBP2, however, was only weakly enhanced in two of the cell lines (A-172, U-87MG; ). Of note, even the highest tested concentration of TMZ (400 µg/mL) had only minor effects on cell viability of T-98G and U-251MG cells as revealed by PI staining. (Fig. S3A).
Figure 3. TMZ increases the cell surface expression and moderately the release of ULBP2. GBM cell lines were treated for 24 h with increasing concentrations of TMZ (100–400 µg/mL) and with DMSO as a control. (A) Cell surface expression of ULBP2 was determined by flow cytometry (filled histograms, TMZ; black open histograms, DMSO controls). Dotted lines represent the matched isotype controls. (B) The concentration of sULBP2 in supernatants of DMSO- and TMZ-treated GBM cells was determined by ELISA. The DMSO control was set to 100%. Data are presented as mean values of three to four independent experiments +/− SEM. Statistical significance is displayed as * for p < 0.05.
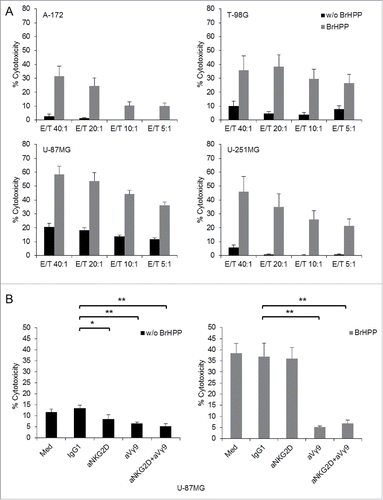
Phosphoantigen-activated γδ T cells efficiently kill GBM cells
γδ T cells have been shown previously to exert cytotoxic activity against GBM cells.Citation23,27 Before investigating the effect of TMZ, we analyzed the capacity of short-term expanded γδ T cells to kill GBM cell lines in a Calcein AM-based cytotoxicity assay in the absence or presence of the phosphoantigen bromohydrin pyrophosphate (BrHPP).Citation28 In the absence of BrHPP, GBM cell lines were largely resistant to Vγ9Vδ2 T cell-mediated cytotoxicity with the exception of U-87MG (). Lysis of all four GBM cells was strongly increased when BrHPP was present during the 4 h cytotoxicity assay () suggesting that TCR-dependent killing plays a major role. In order to investigate the relative contribution of TCR- and NKG2D-dependent killing of GBM cells by Vγ9Vδ2 T cells, we performed the cytotoxicity assay in the absence or presence of monoclonal antibodies directed against the Vγ9 TCR (clone 7A5Citation29) or NKG2D receptor (clone 149810) with U-87MG target cells.Citation22 Vγ9Vδ2 T cells were pre-incubated for 1 h with 10 µg/mL of blocking antibodies and then added to Calcein AM-labeled U-87MG cells, again in the absence (, left-hand panel) or presence of BrHPP (, right-hand panel) to trigger TCR activation. The low level of spontaneous cytotoxic activity of γδ T cells against U-87MG cells was reduced to almost the same extent by blocking of NKG2D or γδ TCR (, left-hand panel). In the presence of BrHPP, however, the contribution of NKG2D receptor to the cytotoxic activity of BrHPP-stimulated Vγ9Vδ2 T cells was negligible, whereas blockade of the γδ TCR almost completely abrogated cytotoxic activity.(, right-hand panel).
Figure 4. Lysis of GBM cell lines by short-term γδ T cell lines. (A) Calcein-AM labeled A-172, T-98G, U-87MG and U-251MG target cells were co-cultured for 4 h at indicated E/T ratios with γδ T cells expanded from four to five healthy donors. Cytotoxicity assays were performed in the absence (black bars) or presence (gray bars) of BrHPP. (B) Involvement of NKG2D and TCR in γδ T cell-mediated killing. Calcein AM-labeled U-87MG cells were co-cultured for 4 h with γδ T cells from five different healthy donors at an E/T ratio of 20:1 with (right-hand panel, gray bars) or without BrHPP (left-hand panel, black bars). Prior to the co-culture, γδ T cells were pre-incubated for 1 h with 10 µg/mL anti-NKG2D and/or anti-Vγ9 TCR anti bodies or with IgG1 as an isotype control. Statistical significance is displayed as ** for p < 0.01 and * for p < 0.05.
TMZ sensitizes GBM cells toward γδ T-cell cytotoxicity
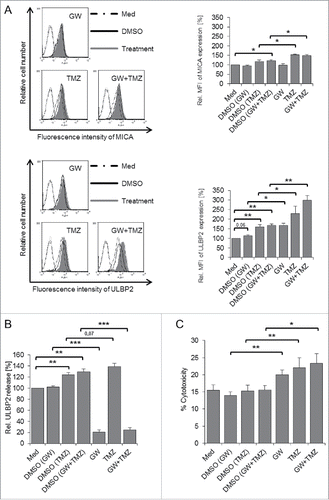
To judge functional implications of the TMZ-induced upregulation of NKG2DLs, we initially used TMZ-treated A-172 as target cells in a CFSE-based cytotoxicity assay. CFSE-labeled A-172 cells were treated with TMZ or with DMSO and incubated for 4 h at various effector/target (E/T) ratios with short-term Vγ9Vδ2 T-cell lines in the presence of BrHPP (TMZ was present in the co-culture at concentration of 100 µg/mL). Thereafter, co-cultures were stained with PI and analyzed for the percentage of CFSE-labeled dead target cells. Non-toxic concentrations of TMZ (, right-hand panel) stabilized the cell surface expression of NKG2DLs (, left-hand panel) and sensitized A-172 cells to increased γδ T cell-mediated cytotoxicity (). This result was further validated in the Calcein AM-based cytotoxicity assay, where DMSO- and TMZ-treated A-172 cells were labeled with Calcein AM and used as targets for Vγ9Vδ2 effector T cells at an E/T ratio of 10:1, again in the absence or presence of BrHPP. As shown in (right-hand panel), TMZ-pretreated A-172 cells displayed enhanced susceptibility to BrHPP activated Vγ9Vδ2 T cell-mediated killing. However, in the absence of BrHPP, there was only minimal lysis of A-172 cells (, left-hand panel). Next, we investigated if TMZ could sensitize other GBM cell lines toward killing by Vγ9Vδ2 T cells in the absence of an additional stimulus. Short-term γδ T cell lines from several different donors were analyzed in the Calcein AM cytotoxicity assay. The cytotoxic potential of γδ T cells varied considerably between donors. In the absence of BrHPP, GBM cell lines were largely resistant to Vγ9Vδ2 T cell-mediated cytotoxicity with the exception of U-87MG cells (Fig. S4). Interestingly, TMZ pre-treatment sensitized A-172, T-98G (p = 0.055) and U-87MG (p < 0.05) but not U-251G cells to γδ T cell-mediated lysis ( for U-87MG, for other cell lines). Comparable results were obtained with CFSE-based cytotoxicity assay (not shown). In extension of the results presented in with untreated target cells, we performed the cytotoxicity assay with TMZ pre-treated U-87MG target cells in the absence or presence of anti-Vγ9 TCR or anti-NKG2D receptor antibodies as above. As presented in , the spontaneous cytotoxic activity of γδ T cells was reduced to the same level by anti-NKG2D and anti-TCR antibodies. The effects of NKG2D- and Vγ9 TCR-blocking antibodies on killing of TMZ-sensitized U-87MG target cells varied considerably among the nine donors displayed in . While blocking of the NKG2D receptor abrogated lysis of TMZ-pre-treated target cells by γδ T cells from some donors, the γδ TCR appeared to be more involved in other cases. In the presence of BrHPP, the contribution of NKG2D receptor to the cytotoxic activity of BrHPP-stimulated Vγ9Vδ2 T cells against TMZ-pretreated U-87MG target cells was moderate, whereas the blockade of the γδ TCR almost completely abrogated the cytotoxic activity (). Interestingly, the moderate sensitizing effect of TMZ pre-treatment on Vγ9Vδ2 T cell-mediated cytotoxicity was still detected even in the presence of both anti-NKG2D and anti-TCR antibodies () suggesting that additional receptor-ligand interactions might be involved. Finally, we also investigated the susceptibility of U-87MG GBM cells pre-treated with TMZ and the MP inhibitor GW. GI and GW, either alone or in combination, did not have a major impact on GBM metabolic activity during the 48 h observation period as revealed by the XTT (sodium 3'-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis (4-methoxy-6-nitro) benzene sulfonic acid hydrate), cell viability assay (Fig. S3B). Combined treatment with GW and TMZ synergistically increased the cell surface expression of ULBP2 on U-87MG (, lower panel) and other GBM cells (Fig. S5A) but had little effect on MICA expression (, upper panel). TMZ slightly increased shedding of ULBP2 from U-87MG cells in comparison to the relevant DMSO control which per se also increased ULBP2 shedding by 20% (). ULBP2 shedding was less or not at all affected in the other GBM lines (Fig. S5B). Moreover, while both GW and TMZ alone sensitized U-87MG cells for killing by γδ T cells in the absence of BrHPP, in combination they further increased the susceptibility. The solvent control DMSO had no effect. ().
Figure 5. TMZ treatment sensitizes A-172 cells to Vγ9Vδ2 T cell-mediated killing. (A) A-172 cells were incubated overnight with 200 μg/mL TMZ or DMSO solvent control. This concentration of TMZ increased cell surface expression of MICA and ULBP2 (left-hand panel) but did not induce cell death as judged by PI staining (right-hand panel). (B) TMZ (gray bars)- or DMSO (black bars)-pre-incubated and CFSE-labeled A-172 cells were co-cultured in duplicates for 4 h with Vγ9Vδ2 T cells at various E/T ratios and stained with PI. BrHPP was present in the co-culture. PI/CFSE double-positive cells were considered as killed A-172 target cells. (C) TMZ (gray bars)- and DMSO (black bars)-pre-incubated A-172 cells were labeled with Calcein AM and co-cultured for 4 h with γδ T cells at E/T ratio 10:1 with (right-hand panel) or without BrHPP (left-hand panel). Data are presented as mean values of experiments with short-term γδ T-cell lines from 5 (B) and 4 (C) donors +/− SEM. Statistical significance is displayed as * for p < 0.05.
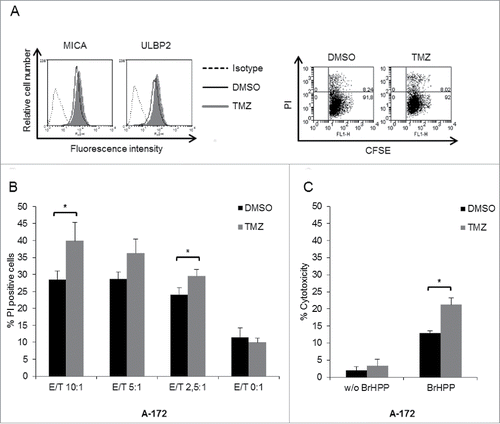
Figure 6. Involvement of NKG2D and TCR in γδ T cell-mediated lysis of GBM cells. U-87MG cells were pre-treated for 20 h with TMZ or DMSO solvent control. (A) Calcein AM-labeled U-87MG target cells were co-cultured for 4 h with γδ T cells at an E/T ratio of 20:1 in the absence of BrHPP. Prior to the co-culture, γδ T cells were pre-incubated for 1 h with 10 µg/mL with blocking antibodies against NKG2D and/or Vγ9 TCR or with IgG1 as an isotype control. γδ T cells were expanded from nine different healthy donors. (B) Similar set-up as in (A) but cytotoxicity assay performed in the presence of BrHPP. Statistical significance is displayed as *** for p < 0.001; ** for p < 0.01 and * for p < 0.05.
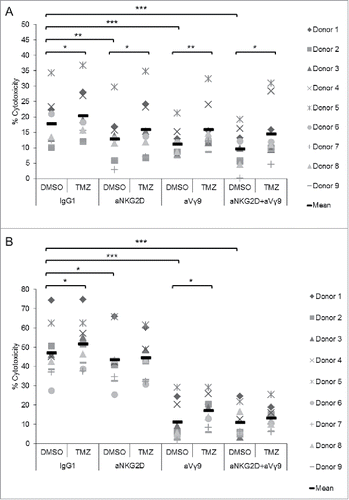
Figure 7. Combination of TMZ and GW treatment synergistically increases expression and shedding of ULBP2 and sensitizes U-87MG cells to Vγ9Vδ2 T cell-mediated killing. (A) U-87MG cells were incubated for 24 h with GW (3 μM), TMZ (200 μg/mL) or a combination of both. Cell surface expression of MICA and ULBP2 was determined by flow cytometry. Representative histograms are depicted in the left-hand panels (dotted histograms: medium control; black open histograms: DMSO solvent control; filled histograms: TMZ/GW treatment). A summary of results of four experiments is shown in the right-hand panels. Median fluorescence intensity (MFI) of MICA (upper right-hand panel) and ULBP2 (lower right-hand panel) expression was set to 100% in the medium-only treated cells. (B) ULBP2 shedding from GW and TMZ-treated U-87MG cells. U-87MG cells were pre-treated as in (A), and the concentration of sULBP2 was determined by ELISA in supernatants of medium-, DMSO solvent control-, GW-, TMZ- and GW/TMZ-treated samples. The amount of sULBP2 in medium control sample was set to 100%. (C) Combined treatment with GW and TMZ sensitizes U-87MG cells to Vγ9Vδ2 T cell-mediated killing. Medium-, DMSO-, GW-, TMZ- and GW/TMZ-treated U-87MG cells were labeled with Calcein AM and co-cultured in triplicates for 4 h with γδ T cells at an E/T ratio of 20:1 in the absence of BrHPP. Data are presented as mean values of five independent experiments +/− SEM. Statistical significance is displayed as*** for p < 0.001, ** for p < 0.01 and * for p < 0.05.
Discussion
To obtain more insight into the plasticity of the NKG2DL system in GBM, we analyzed the expression and shedding of NKG2DLs in four different GBM cell lines and studied the effects of MP inhibitors and TMZ on their expression and release. We observed high levels of MICA and ULBP2 expression on GBM cell lines and low to moderate levels of MICB and ULBP1. In line with our data, Wolpert et al. recently also reported high expression of MICA/ULBP2 on GBM initiating stem cells.Citation13 Although the precise reasons for the differential expression of MICA/ULBP2 versus MICB/ULBP1 are unclear, distinct NKG2DLs can be differentially regulated on the post-transcriptional and/or post-translational level by miRNAs, clathrin mediated-endocytosis, intracellular retention, MP/ADAM-mediated proteolytic cleavage or exosomal release.Citation30,31 However, MP/ADAM-mediated shedding cannot explain the low density of MICB or ULBP1 on GBM cells, since we did not detect any soluble MICB or ULBP1 by ELISA in culture supernatants. In fact, only ULBP2 was released as a soluble protein.
The lack of MICA shedding from A-172 and U-87MG cells can be partially explained by the exosomal release of the truncated MICA allele *008.Citation24,32 On the other hand, ULBP2 which can be attached to the cell surface via a GPI-anchor or expressed as a transmembrane protein, is released in association with exosomes or can be shed via the activity of MPs.Citation7,25 In the analyzed GBM cell lines, inhibition of MPs and specifically ADAM10/17 clearly abrogated ULBP2 shedding and increased its cell surface expression. Interestingly, both ADAM10 and ADAM17 contributed to ULBP2 shedding in GBM cells with the exception of U-87MG where the ADAM10-specific inhibitor GI alone was able to block the release of ULBP2 to the same extent as the ADAM10/17 inhibitor GW. Although the exact mechanism of the regulation of ADAM activity is not precisely known, trafficking, glycosylation, phosphorylation and interactions with accessory molecules might play important roles.Citation33,34 ADAMs cleave cell surface proteins in the juxta-membrane stalk region and substrate specificity is not dependent on the distinct amino acid sequence, but rather on the secondary structure of the protein.Citation34 ULBP2 harbors several single nucleotide polymorphisms resulting in amino acid substitutions (see http://www.ncbi.nlm.nih.gov/SNP) that may cause alterations of the secondary structure of the protein and thus affect the substrate recognition by ADAM proteases. This particular aspect might also explain the lack of MICA shedding from T-98G and U-251MG in the presence of proteolytically active MP, altogether indicating that the regulation of shedding of distinct NKG2DLs is highly complex and divergent in different cell lines and primary tumor cells.
Shedding of NKG2DLs is considered as a tumor immune escape mechanism as it may either reduce the immunogenicity of tumors or even directly influence the expression of NKG2D and the functional activity of NK cells and T-cell subsets.Citation35,36 Proposed therapeutic strategies including the removal of immunosuppressive exosomes by affinity plasmapheresis or the removal of sMICA by neutralizing antibodies aim at controlling the serum levels of sNKG2DLs.Citation37,38 However, direct targeting of distinct ADAM proteases is of particular interest, as it may abrogate the shedding of NKG2DLs and simultaneously stabilize their cell surface expression.Citation10,13,39,40 Moreover, ADAM proteases, especially ADAM10 and ADAM17 are frequently over-expressed in cancer cells.Citation41 By the cleavage of various growth factors and their receptors, adhesion molecules or danger signals such as NKG2DLs, ADAM proteases are considered to be promising targets for tumor therapy.Citation42 Multiple inhibitors targeting ADAMs and other MPs were developed for the treatment of various malignancies. In this scenario, MP inhibitors were remarkably successful in experimental model systems.Citation43 However, although we show that inhibition of MP stabilizes ULBP2 surface expression, the clinical applicability of MP inhibitors is hampered by the lack of selectivity and specificity. For instance, in a phase II clinical trial conducted with GBM patients, the combined administration of the broad range MP inhibitor marimastat together with TMZ had minimal beneficial effect compared to TMZ single therapy but was rather associated with additional toxicity.Citation44,45 Therefore, the diverse physiological activities of ADAM proteases seem to preclude blocking strategies as a generally applicable principle in the context of cancer treatment.Citation46 Moreover, Deng et al. recently observed that sNKG2D can actually cause the activation of murine NK cells and thereby facilitate tumor elimination, which completely contradicts the dogma of the tumor-promoting properties of sNKG2D.Citation47 These findings definitely add more complexity to the NKG2D system and may also partially explain the failure of MP inhibitors in clinical trials for the treatment of various malignancies.
Thus, strategies aiming at enhancing cell surface expression rather than preventing shedding of NKG2DLs might be more promising. Drugs inducing cellular stress including histon deacetylase inhibitors or aldosteron antagonists have been used in different model systems to increase the immunogenicity of tumors by stabilizing the cell surface expression of NKG2DLs.Citation10,13,39 We found that TMZ, a standard chemotherapeutic drug for GBM treatment, increased the expression of NKG2DLs while moderately affecting ULBP2 shedding, and facilitated Vγ9Vδ2 T cell-mediated GBM tumor cell killing. Pre-treatment of A-172 cells and U-87MG cells with TMZ increased the susceptibility to BrHPP-activated γδ T cells. As expected, cytotoxicity exerted by BrHPP-stimulated γδ T cells was mainly mediated through the γδ TCR as shown by antibody blocking, with minimal contribution of the NKG2D receptor. Interestingly, Vγ9Vδ2 T-cells still exerted some cytotoxic activity even if both, NKG2D and γδ TCR, were blocked indicating the possible involvement of additional cytotoxic trigger mechanisms. In the absence of BrHPP, antibody blocking of NKG2D also reduced the low levels of spontaneous γδ T-cell cytotoxicity against GBM target cells. Due to the high donor-dependent variability and marginal effects of TMZ pre-treatment on γδ T-cell killing potential, it is difficult to draw any definitive conclusions. Nevertheless, it seems that in line with investigations in other tumor systems,Citation22 both NKG2D and the TCR contribute to γδ T cell-mediated lysis of GBM target cells. More importantly, even though A-172 cells and T-98G cells were completely resistant to spontaneous γδ T-cell killing, these GBM cells were rendered susceptible upon TMZ pre-treatment. However, the same effect was not observed in U-251MG cells, indicating cell type-dependent variability. Together, these data indicate that TMZ pre-treatment increases the immunogenicity of tumors and augments antitumor activity of Vγ9Vδ2 T cells primarily in a TCR- and NKG2D-dependent manner. However, our results also illustrate the considerable heterogeneity between different GBM cell lines which makes it difficult to predict the γδ T cell-sensitizing effect of TMZ in a clinical setting. The interaction between tumor cells and killer cells (NK cells, γδ T cells) is not only regulated by activating receptors such as NKG2D but is also influenced by inhibitory receptors (e.g., NKG2A) and their ligands which we did not specifically investigate in the current study.Citation48 In addition to NKG2DLs or ligands for inhibitory receptors, TMZ might of course influence multiple other molecules important for the execution of γδ T-cell cytotoxic activity, such as cell adhesion moleculesCitation49 or BTN3A1/CD277,Citation17 an issue which we did not address in the current study but indeed requires further attention. For instance, cell adhesion molecule Nectin-like-molecule-5 binds to DNAX accessory molecule-1 (DNAM-1). DNAM1 is another NK cell activating receptor expressed on γδ T cells playing a significant role in Vγ9Vδ2 T-cell cytotoxicity.Citation50 All four GBM lines expressed Nectin-like-molecule-5 which was increased by previous TMZ exposure (preliminary results, not shown). Therefore, DNAM-1 might indeed be involved in lysis of TMZ-treated GBM cells by γδ T cells.
The application of TMZ in GBM patients can negatively influence the proliferation and cytotoxic activity of immune effector cells including γδ T cells.Citation27,51 However, combination treatment with TMZ and adoptively transferred human cytokin-induced killer cells showed synergistic therapeutic efficacy in a xenograft mouse model with transplanted U-87MG cells.Citation52 Such a synergistic therapeutic effect might be also envisaged with γδ T cells.
In conclusion, our results indicate that TMZ increases the immunogenicity of GBM cells and their susceptibility to Vγ9Vδ2 T-cell-mediated killing, suggesting that the combination of TMZ with the adoptive transfer of in vitro expanded Vγ9Vδ2 T cells might be beneficial for treatment of at least some GBM patients. In contrast to MP inhibitors, TMZ is a well-established component of clinical GBM treatment. Upon surgery of the primary tumor, the sensitizing effect of TMZ on γδ T cell killing could be determined in the individual patient. Large-scale in vitro expansion protocols for Vγ9Vδ2 T cells have already been established and several clinical trials documented the overall safety of Vγ9Vδ2 T-cell immunotherapy of various tumor entitiesCitation53-55and the benefit in the treatment of metastatic solid tumors when used in combination with other therapeutic strategies.Citation56
Materials and methods
Cells lines and reagents
The GBM cell lines A-172 (ECACC 880624218), T-98G (ECACC 92090213), U-87MG (ECACC 89081402) and U-251MG (formerly known as U-373MG; ECACC 89081403) were obtained from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK). Vγ9Vδ2 T-cell lines were established from healthy adult donors as previously described.Citation57 These studies have been approved by the appropriate institutional review board (D 405/10). Briefly, Ficoll-Hypaque-separated peripheral blood mononuclear cells were cultured in complete medium supplemented with 50 U/rIL-2 and were stimulated with 2,5 µM zoledronate (kindly provided by Novartis). Vγ9Vδ2 T cell lines were used as effector cells in cytotoxic assays between day 11 and day 15, referred to as short term γδ T-cell lines. Cytotoxicity assays were performed in the absence or presence of 300 nMBr HPP (kindly provided by Innate Pharma, Marseille, France).Citation28 The purity of Vγ9Vδ2 T-cell lines used in the experiments was always >90%. Complete medium was RPMI-1640 (#52400-25, Life Technologies GmbH) supplemented with 10% heat-inactivated FCS, 100 U/mL penicillin and 100 µg/mL streptomycin. All cell lines were kept at 37°C in a humidified atmosphere with 5% CO2.
Where indicated, tumor cells were cultured for 24 h in the presence of the broad range MP inhibitors GM6001 (5 µM, Merck Millipore, #364206) and TAPI-1 (5 µM, Merck Millipore #579053) or with DMSO as a solvent control. For the selective inhibition of ADAM10 and ADAM10/17 GI254023X (GI, 3 µM) and GW280264X (GW, 3 µM)Citation26 were used (kindly provided by Prof. Paul Saftig, Institute of Biochemistry, Christian-Albrechts University, Kiel). Where indicated, tumor cells were cultured for 24 h in the presence of various concentrations of TMZ (#T2577, Sigma-Aldrich Chemie GmbH) or with DMSO as a solvent control.
Flow cytometry
The following mAb were used for cell surface staining: PE-conjugated anti-ADAM17 mAb (#FAB9301P, clone 111633), anti-MICB mAb (# FAB1599P, clone 236511) and anti-ULBP1 mAb (#FAB1380P, clone 170818) were purchased from R&D Systems. Unconjugated anti-MICA (clone AMO1, kindly provided by Prof. Alexander Steinle, Institute of Molecular Medicine, University of Frankfurt) and anti-ULBP2 mAb (#ab89930, clone MM0593-7F33) were obtained from BAMOMAB and Abcam, respectively. Anti-ADAM10 mAb (#857.800.000, clone 11G2) was obtained from Gen-Probe. Tumor cells were detached from culture plates with trypsin/EDTA (0.05%/0.02 w/v; #L2153, Biochrom), washed and blocked with Fc-receptor blocking reagent (#130-059-901, MiltenyiBiotec GmbH) according to the manufacturer's instructions. After an additional washing step, the cells were incubated for 30 min with specific antibodies and matched isotype controls. In case of unconjugated antibodies cells were further incubated with a polyclonal PE-conjugated goat-anti-mouse Ab (Life Technologies). After a washing step cells were fixed in 1% paraformaldehyde. 5,000 live cells were analyzed on a FACS Calibur flow cytometer using CellQuestPro Software (BD Biosciences, Heidelberg, Germany).
ELISA
GBM cells were cultured for 24 h and the cell-free culture supernatants were subjected to ELISA. The amounts of sMICB (#DY1599), sULBP1 (#DY1380) and sULBP2 (#DY1298) were determined with the respective sandwich ELISA kits (R&D Systems) following the manufacturer's instructions. sMICA was detected by sandwich ELISA as described by Salih et al. with slight modifications.Citation58 Briefly, a microtiter plate was coated overnight with anti-MICA mAb (clone AMO1, 2 µg/mL). Anti-MICA/B (#BAMO3-500, clone BAMO3, 5 µg/mL, IgG2a) was used as a detection Ab. After a washing step anti mouse-IgG2a-HRP was added to the plate and developed by using tetramethylbenzidine peroxidase substrate system (#DY999, R&D Systems). The absorbance was measured on a microplate reader. (Tecan Group Ltd, Männedorf, Switzerland) at 450 nm.
CFSE- and Calcein AM-based cytotoxicity assays
GBM cells were incubated in the presence of 10 µM CFSE (#21888, Fluka) at 37°C for 10 min. 10 mL of ice-cold RPMI medium containing 10% FCS were added to the cells for 5 min. After three washing steps, CFSE-labeled cells were incubated overnight with TMZ (200 µg/mL) or with DMSO as solvent control. Vγ9Vδ2 T cells were added to the plate at various E/T ratios in duplicates with or without BrHPP in the presence of 12.5 U/mL IL-2. After 4 h, cells were stained with PI and 5,000 CFSE-labeled target cells were analyzed by flow cytometry to quantify PI fluorescence. In Calcein AM-based cytotoxicity assays,Citation59 TMZ- and DMSO-treated cells were incubated with 5 µM Calcein AM (#C3099, Life Technologies) for 30 min at 37°C. Following two washing steps cells were co-cultured in triplicates in the presence of 12,5 U/mL rIL-2 with Vγ9Vδ2 T cells at various E:T ratios with or without BrHPP. Where indicated, effector cells were pre-incubated for 1 h with 10 µg/mL anti TCR-Vγ9 mAb 7A5,Citation29 anti-NKG2D mAb(#MAB139, clone 149810, R&D systems) or mouse isotype control IgG1 (#18443, clone MOPC-21, Abcam) before the addition of Calcein AM-labeled tumor target cells. 100 µL of supernatant were transferred to non-binding black µCLEAR® 96 well microplates (#147827, Greiner bio-one) and fluorescence intensities were measured using a Tecan microplate reader at 490 nm with reference filter at 530 nm. Fluorescence intensity correlates with the amount of calcein released into the supernatant.Citation59 Percentage cytotoxicity was calculated as follows: (extinctionexperimental – extinctionspontaneous/extinctionmaximum – extinctionspontaneous, where the maximal calcein release was determined in wells containing target cells and 1% triton X-100.
Statistical analysis
The paired, two-tailed Student's t-Test was performed. Statistical significance was set at p < 0.05 and displayed as *** for p < 0.001; ** for p < 0.01 and * for p < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_S_1093276.zip
Download Zip (7 MB)Acknowledgments
We thank Prof. Paul Saftig (Institute of Biochemistry, Christian-Albrechts University Kiel) for the ADAM10/17 inhibitors GI254023X and GW280264X and Prof. Alexander Steinle (Institute of Molecular Medicine, University of Frankfurt) for supplying anti-MICA mAb (AMO1). BrHPP was kindly provided by Innate Pharma (Marseille, France). Many thanks are due to all group members for their support in establishing γδ T-cell lines.The expert technical assistance of Signe Valentin, Sandra Ussat, Thi Thuy Hoa Ly and Ina Martens is greatly appreciated. We also thank Prof. Hubertus Maximilian Mehdorn (Dept of Neurosurgery, UKSH Campus Kiel) for support.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (CRC877 project A7 to D.K. and project B4 to O.J.) and LE2571/3-1 (to M.L.). TW was the recipient of a RISE fellowship from the German Academic Exchange Service (DAAD). This work forms part of the Ph.D. thesis of G.C. G.C. was supported by a grant from the Medical Faculty of Kiel University.
References
- Preusser M, de Ribaupierre S, Wohrer A, Erridge SC, Hegi M, Weller M, Stupp R. Current concepts and management of glioblastoma. Ann Neurol 2011; 70:9-21; PMID:21786296; http://dx.doi.org/10.1002/ana.22425
- Bielamowicz K, Khawja S, Ahmed N. Adoptive cell therapies for glioblastoma. Front Oncol 2013; 3:275; PMID:24273748; http://dx.doi.org/10.3389/fonc.2013.00275
- Jung TY, Choi YD, Kim YH, Lee JJ, Kim HS, Kim JS, Kim SK, Jung S, Cho D. Immunological characterization of glioblastoma cells for immunotherapy. Anticancer Res 2013; 33:2525-33; PMID:23749904
- Ullrich E, Koch J, Cerwenka A, Steinle A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology 2013; 2:e26097; PMID:24353908; http://dx.doi.org/10.4161/onci.26097
- Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev 2010; 235:267-85; PMID:20536569; http://dx.doi.org/10.1111/j.0105-2896.2010.00893.x
- Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Ann Rev Immunol 2013; 31:413-41; PMID:23298206; http://dx.doi.org/10.1146/annurev-immunol-032712-095951
- Fernandez-Messina L, Ashiru O, Aguera-Gonzalez S, Reyburn HT, Vales-Gomez M. The human NKG2D ligand ULBP2 can be expressed at the cell surface with or without a GPI anchor and both forms can activate NK cells. J Cell Sci 2011; 124:321-7; PMID:21224393; http://dx.doi.org/10.1242/jcs.076042
- Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene 2008; 27:5944-58; PMID:18836475; http://dx.doi.org/10.1038/onc.2008.272
- Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-β downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. NeuroOncol 2010; 12:7-13; PMID:20150362; http://dx.doi.org/10.1093/neuonc/nop009
- Leung WH, Vong QP, Lin W, Janke L, Chen T, Leung W. Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXR gamma activation. J Exp Med 2013; 210:2675-92; PMID:24190430; http://dx.doi.org/10.1084/jem.20122292
- Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, Stevanovic S, Rammensee HG, Steinle A. Tumor-associated MICA is shed by ADAM proteases. Cancer Res 2008; 68:6368-76; PMID:18676862; http://dx.doi.org/10.1158/0008-5472.CAN-07-6768
- Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Furst D, Mytilineos J, Kalthoff H, Janssen O, Oberg HH et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int J Cancer 2013; 133:1557-66; PMID:23526433; http://dx.doi.org/10.1002/ijc.28174
- Wolpert F, Tritschler I, Steinle A, Weller M, Eisele G. A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. NeuroOncol 2014; 16:382-91; PMID:24327582; http://dx.doi.org/10.1093/neuonc/not232
- Chitadze G, Bhat J, Lettau M, Janssen O, Kabelitz D. Generation of soluble NKG2D ligands: proteolytic cleavage, exosome secretion and functional implications. Scand J Immunol 2013; 78:120-9; PMID:23679194; http://dx.doi.org/10.1111/sji.12072
- Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. CurrMolPharmacol 2012; 5:102-14; PMID:22122467; http://dx.doi.org/10.2174/1874467211205010102
- Lamb LS, Jr, Bowersock J, Dasgupta A, Gillespie GY, Su Y, Johnson A, Spencer HT. Engineered drug resistant γδ T cells kill glioblastoma cell lines during a chemotherapy challenge: a strategy for combining chemo- and immunotherapy. PloS One 2013; 8:e51805; PMID:23326319; http://dx.doi.org/10.1371/journal.pone.0051805
- Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, Li J, Kuball J, Adams EJ, Netzer S et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood 2012; 120:2269-79; PMID:22767497; http://dx.doi.org/10.1182/blood-2012-05-430470
- Vavassori S, Kumar A, Wan GS, Ramanjaneyulu GS, Cavallari M, El Daker S, Beddoe T, Theodossis A, Williams NK, Gostick E et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat Immunol 2013; 14:908-16; PMID:23872678; http://dx.doi.org/10.1038/ni.2665
- Wang H, Henry O, Distefano MD, Wang YC, Räikkönen J, Mönkkönen J, Tanaka Y, Morita CT. Butyrophilin 3A1 plays an essential role in prenyl pyrophosphate stimulation of human Vγ2Vδ2 T cells. J Immunol 2013; 191:1029-42; PMID:23833237; http://dx.doi.org/10.4049/jimmunol.1300658
- Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G. Human T cell receptor γδ cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med 2003; 197:163-8; PMID:12538656; http://dx.doi.org/10.1084/jem.20021500
- Kabelitz D, Wesch D, He W. Perspectives of γδ T cells in tumor immunology. Cancer Res 2007; 67:5-8; PMID:17210676; http://dx.doi.org/10.1158/0008-5472.CAN-06-3069
- Wrobel P, Shojaei H, Schittek B, Gieseler F, Wollenberg B, Kalthoff H, Kabelitz D, Wesch D. Lysis of a broad range of epithelial tumour cells by human γδ T cells: involvement of NKG2D ligands and T-cell receptor- vs. NKG2D-dependent recognition. Scand J Immunol 2007; 66:320-8; PMID:17635809; http://dx.doi.org/10.1111/j.1365-3083.2007.01963.x
- Bryant NL, Gillespie GY, Lopez RD, Markert JM, Cloud GA, Langford CP, Arnouk H, Su Y, Haines HL, Suarez-Cuervo C et al. Preclinical evaluation of ex vivo expanded/activated γδ T cells for immunotherapy of glioblastoma multiforme. J Neurooncol 2011; 101:179-88; PMID:20532954; http://dx.doi.org/10.1007/s11060-010-0245-2
- Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, Reyburn HT. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res 2010; 70:481-9; PMID:20068167; http://dx.doi.org/10.1158/0008-5472.CAN-09-1688
- Fernandez-Messina L, Ashiru O, Boutet P, Aguera-Gonzalez S, Skepper JN, Reyburn HT, Vales-Gomez M. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J BiolChem 2010; 285:8543-51; PMID:20080967; http://dx.doi.org/10.1074/jbc.M109.045906
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003; 102:1186-95; PMID:12714508; http://dx.doi.org/10.1182/blood-2002-12-3775
- Bryant NL, Suarez-Cuervo C, Gillespie GY, Markert JM, Nabors LB, Meleth S, Lopez RD, Lamb LS, Jr. Characterization and immunotherapeutic potential of γδ T-cells in patients with glioblastoma. NeuroOncol 2009; 11:357-67; PMID:19211933; http://dx.doi.org/10.1215/15228517-2008-111
- Espinosa E, Belmant C, Pont F, Luciani B, Poupot R, Romagné F, Brailly H, Bonneville M, Fournié JJ. Chemical synthesis and biological activity of bromohydrin pyrophosphate, a potent stimulator of human γδ T cells. J BiolChem 2001; 276: 18337-44; PMID:11279081; http://dx.doi.org/10.1074/jbc.M100495200
- Janssen O, Wesselborg S, Heckl-Östreicher B, Pechhold K, Bender A, Schondelmaier S, Moldenhauer G, Kabelitz D. T cell receptor/CD3-signaling induces death by apoptosis in human T cell receptor γδ+ T cells. J Immunol 1991; 146:35-9; PMID:1824593
- Stern-Ginossar N, Mandelboim O. An integrated view of the regulation of NKG2D ligands. Immunology 2009; 128:1-6; PMID:19689730; http://dx.doi.org/10.1111/j.1365-2567.2009.03147.x
- Aguera-Gonzalez S, Boutet P, Reyburn HT, Vales-Gomez M. Brief residence at the plasma membrane of the MHC class I-related chain B is due to clathrin-mediated cholesterol-dependent endocytosis and shedding. J Immunol 2009; 182:4800-8; PMID:19342658; http://dx.doi.org/10.4049/jimmunol.0800713
- Aguera-Gonzalez S, Gross CC, Fernandez-Messina L, Ashiru O, Esteso G, Hang HC, Reyburn HT, Long EO, Vales-Gomez M. Palmitoylation of MICA, a ligand for NKG2D, mediates its recruitment to membrane microdomains and promotes its shedding. Eur J Immunol 2011; 41:3667-76; PMID:21928280; http://dx.doi.org/10.1002/eji.201141645
- Gooz M. ADAM-17: the enzyme that does it all. Crit Rev BiochemMolBiol 2010; 45:146-69; PMID:20184396; http://dx.doi.org/10.3109/10409231003628015
- Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev 2003; 17:7-30; PMID:12514095; http://dx.doi.org/10.1101/gad.1039703
- Hedlund M, Nagaeva O, Kargl D, Baranov V, Mincheva-Nilsson L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PloS One 2011; 6:e16899; PMID:21364924; http://dx.doi.org/10.1371/journal.pone.0016899
- Salih HR, Holdenrieder S, Steinle A. Soluble NKG2D ligands: prevalence, release, and functional impact. Front Biosci 2008; 13:3448-56; PMID:18508446; http://dx.doi.org/10.2741/2939
- Marleau AM, Chen CS, Joyce JA, Tullis RH. Exosome removal as a therapeutic adjuvant in cancer. J Transl Med 2012; 10:134; PMID:22738135; http://dx.doi.org/10.1186/1479-5876-10-134
- Jinushi M, Hodi FS, Dranoff G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Nat AcadSci USA 2006; 103:9190-5; PMID:16754847; http://dx.doi.org/10.1073/pnas.0603503103
- Kato N, Tanaka J, Sugita J, Toubai T, Miura Y, Ibata M, Syono Y, Ota S, Kondo T, Asaka M et al. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia 2007; 21:2103-8; PMID:17625602; http://dx.doi.org/10.1038/sj.leu.2404862
- Huang B, Sikorski R, Sampath P, Thorne SH. Modulation of NKG2D-ligand cell surface expression enhances immune cell therapy of cancer. J Immunother 2011; 34:289-96; PMID:21389869; http://dx.doi.org/10.1097/CJI.0b013e31820e1b0d
- Lendeckel U, Kohl J, Arndt M, Carl-McGrath S, Donat H, Röcken C. Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J Cancer Res ClinOncol 2005; 131:41-8; PMID:15565459; http://dx.doi.org/10.1007/s00432-004-0619-y
- Duffy MJ, McKiernan E, O'Donovan N, McGowan PM. Role of ADAMs in cancer formation and progression. Clin Cancer Res 2009; 15:1140-4; PMID:19228719; http://dx.doi.org/10.1158/1078-0432.CCR-08-1585
- Witters L, Scherle P, Friedman S, Fridman J, Caulder E, Newton R, Lipton A. Synergistic inhibition with a dual epidermal growth factor receptor/HER-2/neu tyrosine kinase inhibitor and a disintegrin and metalloprotease inhibitor. Cancer Res 2008; 68:7083-9; PMID:18757423; http://dx.doi.org/10.1158/0008-5472.CAN-08-0739
- Groves MD, Puduvalli VK, Hess KR, Jaeckle KA, Peterson P, Yung WK, Levin VA. Phase II trial of temozolomide plus the matrix metalloproteinase inhibitor, marimastat, in recurrent and progressive glioblastoma multiforme. J ClinOncol 2002; 20:1383-8; PMID:11870183; http://dx.doi.org/10.1200/JCO.20.5.1383
- Groves MD, Puduvalli VK, Conrad CA, Gilbert MR, Yung WK, Jaeckle K, Liu V, Hess KR, Aldape KD, Levin VA. Phase II trial of temozolomide plus marimastat for recurrent anaplastic gliomas: a relationship among efficacy, joint toxicity and anticonvulsant status. J Neurooncol 2006; 80:83-90; PMID:16639492; http://dx.doi.org/10.1007/s11060-006-9160-y
- Rose-John S. ADAM17, shedding, TACE as therapeutic targets. Pharmacol Res 2013; 71:19-22; PMID:23415892; http://dx.doi.org/10.1016/j.phrs.2013.01.012
- Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. Antitumor immunity.A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015; 348:136-9; PMID:25745066; http://dx.doi.org/10.1126/science.1258867
- Angelini DF, Zambello R, Galandrini R, Diamantini A, Placido R, Micucci F, Poccia F, Semenzato G, Borsellino G, Santoni A et al. NKG2A inhibits NKG2C effector functions of γδ T cells: implications in health and disease. J LeukocBiol 2011; 89: 75-84; PMID:20952657; http://dx.doi.org/10.1189/jlb.0710413
- Shojaei H, Oberg HH, Juricke M, Marischen L, Kunz M, Mundhenke C, Gieseler F, Kabelitz D, Wesch D. Toll-like receptors 3 and 7 agonists enhance tumor cell lysis by human γδ T cells. Cancer Res 2009; 69:8710-7; PMID:19887600; http://dx.doi.org/10.1158/0008-5472.CAN-09-1602
- Toutirais O, Cabillic F, LeFriec G, Salot S, Loyer P, Le Gallo M, Desille M, de LaPintiere CT, Daniel P, Bouet F et al. DNAX accessory molecule-1 (CD226) promotes human hepatocellular carcinoma cell lysis by Vg9Vd2 T cells. Eur J Immunol 2009; 39:1361-8; PMID:19404979; http://dx.doi.org/10.1002/eji.200838409
- Fadul CE, Fisher JL, Gui J, Hampton TH, Cote AL, Ernstoff MS. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. NeuroOncol 2011; 13:393-400; PMID:21339188; http://dx.doi.org/10.1093/neuonc/noq204
- Jin J, Joo KM, Lee SJ, Jo MY, Kim Y, Jin Y, Kim JK, Ahn JM, Yoon MJ, Lim J et al. Synergistic therapeutic effects of cytokine-induced killer cells and temozolomide against glioblastoma. Oncol Rep 2011; 25:33-9; PMID:21109954; http://dx.doi.org/10.3892/or_00001038
- Bennouna J, Bompas E, Neidhardt EM, Rolland F, Philip I, Galea C, Salot S, Saiagh S, Audrain M, Rimbert M et al. Phase-I study of Innacellγδ™, an autologous cell-therapy product highly enriched in γ9δ2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer ImmunolImmunother 2008; 57:1599-609; PMID:18301889; http://dx.doi.org/10.1007/s00262-008-0491-8
- Abe Y, Muto M, Nieda M, Nakagawa Y, Nicol A, Kaneko T, Goto S, Yokokawa K, Suzuki K. Clinical and immunological evaluation of zoledronate-activated Vgamma9gammadelta T-cell-based immunotherapy for patients with multiple myeloma. ExpHematol 2009; 37:956-68; PMID:19409955; http://dx.doi.org/10.1016/j.exphem.2009.04.008
- Kobayashi H, Tanaka Y, Yagi J, Osaka Y, Nakazawa H, Uchiyama T, Minato N, Toma H. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: a pilot study. Cancer ImmunolImmunother 2007; 56:469-76; PMID:16850345; http://dx.doi.org/10.1007/s00262-006-0199-6
- Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, Nieda M. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer 2011; 105:778-86; PMID:21847128; http://dx.doi.org/10.1038/bjc.2011.293
- Oberg HH, Peipp M, Kellner C, Sebens S, Krause S, Petrick D, Adam-Klages S, Röcken C, Becker T, Vogel I et al. Novel bispecific antibodies increase γδ T-cell cytotoxicity against pancreatic cancer cells. Cancer Res 2014; 74:1349-60; PMID:24448235; http://dx.doi.org/10.1158/0008-5472.CAN-13-0675
- Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, Steinle A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 2003; 102:1389-96; PMID:12714493; http://dx.doi.org/10.1182/blood-2003-01-0019
- Neri S, Mariani E, Meneghetti A, Cattini L, Facchini A. Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. ClinDiagnLaboImmunol 2001; 8:1131-5; PMID:11687452; http://dx.doi.org/10.1128/CDLI.8.6.1131–1135.2001