
ABSTRACT
The identification of activation pathways linked to antitumor T-cell polyfunctionality in long surviving patients is of great relevance in the new era of immunotherapy. We have recently reported that dacarbazine (DTIC) injected one day before peptide-vaccination plus IFN-α improves the antitumor lytic activity and enlarges the repertoire of Melan-A-specific T-cell clones, as compared with vaccination alone, impacting the overall survival of melanoma patients. To identify the mechanisms responsible for this improvement of the immune response, we have analyzed the endogenous and treatment-induced antigen (Ag)-specific response in a panel of Melan-A-specific CD8+ T-cell clones in terms of differentiation phenotype, inhibitory receptor profile, polyfunctionality and AKT activation. Here, we show that Melan-A-specific CD8+ T cells isolated from patients treated with chemoimmunotherapy possess a late differentiated phenotype as defined by the absence of CD28 and CD27 co-stimulatory molecules and high levels of LAG-3, TIM-3 and PD-1 inhibitory receptors. Nevertheless, they show higher proliferative potential and an improved antitumor polyfunctional effector profile in terms of co-production of TNF-α, IFNγ and Granzyme-B (GrB) compared with cells derived from patients treated with vaccination alone. Polyfunctionality is dependent on an active AKT signaling related to the engagement of the co-stimulatory molecule ICOS. We suggest that this phenotypic and functional signature is dictated by a fine-tuned balance between TCR triggering, AKT activation, co-stimulatory and inhibitory signals induced by chemoimmunotherapy and may be associated with antitumor T cells able to protect patients from tumor recurrence.
Abbreviations
| Ag | = | antigen |
| APCs | = | antigen presenting cells |
| CTL | = | cytotoxic T lymphocyte |
| DTIC | = | dacarbazine |
| EC50 | = | half-maximal lysis |
| GrB | = | granzyme-B |
| ICOS | = | inducible costimulator |
| ICS | = | intracellular staining |
| IFN | = | interferon |
| iMFI | = | integrated mean fluorescence intensity |
| KLRG1 | = | killer cell lectin-like receptor G1 |
| LAG-3 | = | lymphocyte-activation gene 3 |
| Melan-A | = | melanoma antigen A |
| PBMC | = | peripheral blood mononuclear cells |
| PD-1 | = | Programmed death-1 |
| PD-L1 | = | Programmed death-ligand 1 |
| PHA | = | phytohemagglutinin |
| PI3K | = | phosphatidylinositol 3-kinase |
| pt | = | patient |
| TCR | = | T cell receptor |
| TIM-3 | = | T-cell immunoglobulin and Mucin Domain-3 |
| TNF | = | tumor necrosis factor |
Introduction
Harnessing the T-cell response has proven to be a successful strategy in order to prevent tumor relapse, as recently reported for melanoma patients treated with combined chemoimmunotherapy.Citation1,2 Yet the mechanisms responsible for drug-induced activation of antitumor immune response remain to be elucidated, then understanding the complex regulation of treatment-driven T-cell response is critical for the design of effective combined therapies including cancer vaccines.
T-cell response is a finely regulated process involving the balance between Ag-specific T-cell receptor (TCR) activation and co-stimulatory as well as inhibitory signals.Citation3,4 The co-stimulatory molecule CD28 plays a pivotal role during naive and memory T-cell activation by inducing IL-2 production and promoting proliferation,Citation5,6 while CD27 is critical for the control of TCR activation level and T-cell survival.Citation7,8 The pattern of CD28 and CD27 expression provides a characterization of CD8+ T-cell differentiation stage, with early differentiated cells being CD28+/CD27+ while intermediate and late differentiated T cells displaying a CD28−/CD27+ and CD28−/CD27− phenotype, respectively.Citation9,10 Inducible T-cell co-stimulator (ICOS), another CD28 family member, has been reported to possess partial functional redundancy in CD8+CD28− T cell co-stimulation.Citation11,12 Unlike CD28, which is constitutively expressed on T cells, ICOS is expressed only after activation, and its deficient expression has been associated with extensive T-cell dysfunction,Citation13 a reduced memory T-cell compartment and imbalance between effector and regulatory cells.Citation14-16
As T cells progress toward terminal differentiation, they upregulate the expression of co-inhibitory receptors, such as Lymphocyte-activation gene 3 (LAG-3), T-cell immunoglobulin and Mucin Domain-3 (TIM-3) and Programmed death-1 (PD-1), often in association with an exhausted phenotype and decreased functionality,Citation17-19 both features playing a key function in tumor-induced immune suppression.Citation4 Besides their inhibitory role, the expression of these molecules have recently been observed in highly melanoma-reactive and clonally expanded CD8+ T lymphocytesCitation20 and in activated T cells without impairment of cytokine production.Citation21,22 In particular, PD-1 has been suggested to be linked likely to a T-cell differentiation status rather than to exhaustion.Citation23
Among the mechanisms by which PD-1 has been reported to inhibit T-cell activation, there is the suppression of the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway and the prevention of the CD28-mediated activation.Citation24 PI3K/AKT signaling pathway plays a key role in the survival and activation of T cellsCitation25,26 where it is activated by TCR stimulation in concert with co-stimulatory molecules, such as CD28 and ICOS as well as cytokine receptor engagement.Citation27 As T cells progress toward differentiation and aging, they lose the ability to activate AKT in parallel with the loss of CD28 co-stimulatory molecule.Citation12,28 Notably, the AKT pathway is critical for the control of many aspects of the CD8+ T-cell response including the effector versus memory fate.Citation27,29
A combination of DNA alkylating agent DTIC plus peptide-vaccination and interferon (IFN)-α as adjuvant, has been reported to induce a diversification of the melanoma antigen A (Melan-A)-specific TCR repertoire with potent antitumor activity and significant clinical benefit in the prevention of melanoma relapse.Citation1,2 To identify immune parameters and pathways underlying the clinical advantage observed in patients treated with combined therapy, we have performed an extensive analysis of a panel of Melan-A-specific T-cell clones isolated before and after treatments.
Melan-A-specific CD8+ T cells derived from patients treated with combined therapy showed an improved antitumor polyfunctional profile, a hallmark of protective immunity against viruses and cancer,Citation30-32 compared with those treated with vaccination alone. These polyfunctional highly-reactive Melan-A-specific T cells displayed the highest expression of PD-1 molecule, suggesting that in our settings, this molecule is not associated with T-cell disfunctionality and impairment of cytokine production. This functional effector profile was dependent on an active AKT signaling pathway despite the late differentiated phenotype of T cells, as defined by the absence of CD28 and CD27 co-stimulatory molecules, and was related to the engagement of ICOS. Of clinical relevance, this activation pathway was found only in patients who benefit from chemoimmunotherapy treatment.
Results
Differentiation phenotype of Melan-A-specific CD8+ T cells and improved polyfunctional activity induced by DTIC plus peptide-vaccination
DTIC plus peptide-vaccine combination (Arm2) has been shown to improve the antitumor lytic activity of Melan-A-specific T-cell clones, as compared with vaccination alone (Arm1) and to impact the overall survival of melanoma patients.Citation1,2
First, in order to identify the mechanisms underlying the above functional differences elicited by the chemoimmunotherapy, we evaluated the differentiation phenotype and the multifunctional profile of a number of Melan-A-specific CD8+ T clones isolated from different patients before and after treatment. The phenotypic and functional characterization of CD8+ T cells was performed between the first and fourth round of stimulation with irradiated antigen-presenting cells (APCs) plus phytohemagglutinin (PHA), with overlapping results for each clone. The clinical characteristics of the patients, the phenotype of T-cell clones (n = 66) analyzed in this study and, when available, the TCR clonotypeCitation2 are described in . Naturally occurring Melan-A-specific T cells isolated from patients before therapy (PRE) showed a heterogeneous level of differentiation based on the expression of CD28 and/or CD27, while those isolated after both treatments (POST) were prevalently highly differentiated effector memory (CD28−CD27−CCR7−CD45RA−, ).
Table 1. Patients characteristics, clinical outcome and Melan-A-specific T-cell clones.
Then we investigated the T-cell functional profile in terms of co-production of tumor necrosis factor (TNF)-α, IFNγ and GrB, in response to HLA-A2+ PD-L1+ Melan-A+ (Mel+) melanoma cell lines. shows simultaneous intracellular staining (ICS) for TNF-α, IFNγ and GrB production of representative clones (upper panel) and the quantification of the different possible combinations (lower panel). shows pooled results from Melan-A-specific T cells (n = 14 from Arm1, n = 27 from Arm2), obtained by analyzing both the percentage of positive cells and the integrated mean fluorescence intensity (iMFI), the latter a metric that combines the relative amount of molecule produced (the MFI) with the relative number of cells that make them (the percentage positive cells).Citation33 The analysis of the single molecules evidenced a high production of both TNF-α and IFNγ in clones isolated before treatment (PRE). Of note, after therapy (POST) clones isolated from Arm2 showed a significant increase (p < 0.0001) in the production of both these cytokines as compared with Arm1 T cells (). On the other hand, all clones analyzed showed a high level of GrB production in the absence of specific stimulation, which was maintained after exposure to tumor cells irrespective of the treatment schedule, suggesting that in our settings an increase of this molecule is not critical for the chemoimmunotherapy-induced improvement of antitumor activity (). Stimulation of cells by unrelated HLA/Ag tumor cell lines (Mel-) did not affect the baseline functional profile.
Figure 1. Melan-A-specific CD8+ T cells exhibit an enhanced polyfunctional profile after chemoimmunotherapy. (A) Representative dot plot gating strategy of multicolor intracellular staining for TNF-α (TNF), IFNγ (IFN), and Granzyme-B (GrB) in Melan-A-specific CD8+ T cells stimulated with HLA-A2+ Melan-A-expressing (Mel+) melanoma cell line (top panel). The multicolor histograms in the bottom panel show the quantification of all possible combinations of molecule co-expression, evaluated in the absence of stimulus (−) or after 4 h stimulation with A2+/Melan-A+ (Mel+) or A2+/Melan-A− (Mel-) melanoma cell lines. (B–C) Pooled results from all T-cell clones evaluated (n = 14 from two patients of Arm1, n = 27 from three patients of Arm2), isolated before (PRE) and after therapy (POST), analyzed as percentage of positive cells (%, left) and integrated MFI (iMFI, right) (see Material and Methods for details). Data are displayed singularly (B) and simultaneously (C). Each dot represents the mean value from two to four independent experiments performed with a single T-cell clone. The mean ± SEM of each indicated sample group is shown. Polyfunctionality profile (TNF-α+IFNγ+GrB+) is also shown as box-and-whisker diagram, with 5–95 percentile. */♦p ≤ 0.05, **/♦♦p ≤ 0.01, ***/♦♦♦p ≤ 0.001, ****/♦♦♦♦p ≤ 0.0001, Mann–Whitney two-sample test (*) and two-tail Student’s test (♦), respectively.
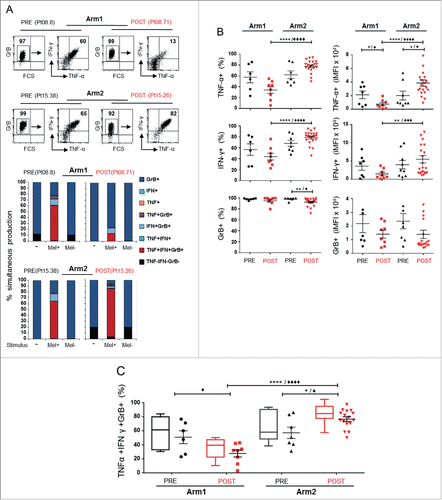
When we analyzed the polyfunctionality of T-cell clones (), a significant increase in molecule co-production was observed in Arm2 patients, while clones from Arm1 showed an impaired functional profile after the treatment. Finally, a strong positive correlation was found by plotting polyfunctionality versus antitumor lytic activity of each T-cell clone, as previously assessedCitation2 (Fig. S1A), proving CD8+ T-cell polyfunctionality as a reliable marker for antitumor activity efficacy. On the other hand, the DTIC-associated functional improvement could not be ascribed to T-cell avidity toward either the natural Melan-A26-35 or the analog peptide used in our vaccine formulation (Melan-AA27L), as defined by half-maximal lysis (EC50) (Figs S1B and C), in agreement with our previous results and other studies.Citation2,34
Combined chemoimmunotherapy activates the AKT pathway in late differentiated T cells
In order to identify signaling pathways potentially activated by DTIC plus vaccination and responsible for the increase in antitumor polyfunctionality of T cells, we focused on AKT signaling, which can favor the induction and maintenance of cytotoxic T lymphocyte (CTL) effector molecules, while its inhibition causes the loss of IFNγ mRNA and prevents IFNγ production.Citation29
In CD8+ T cells, AKT can be activated by TCR activation, cytokines and co-stimulatory molecules.Citation27 Conversely, the loss of AKT enzymatic activity has been associated with the acquisition of a highly differentiated phenotype characterized by the lack of CD28 and CD27, critical molecules for T-cell activation and markers for the classification of T-cell subsets at different stages of differentiation.Citation9,12 We analyzed AKT activation in terms of phosphorylation of Ser473 (pSer473-AKT) in a number of Melan-A specific clones (n = 26). CD8+ T cells isolated before the treatments displayed an early and intermediate level of differentiation and showed pSer473-AKT expression ( and ). After therapy, all clones analyzed lacked CD28 and CD27 molecules, compatible with a late differentiated phenotype. Interestingly, while cells isolated from Arm1 patients showed a negative or low levels of pSer473-AKT, all clones evaluated (n = 11) from Arm2 patients displayed pSer473-AKT () and downstream substrate target activation (i.e. Bclx, pSer2448mTOR, pp70, Fig. S2). In parallel, the status of pSer473 AKT phosphorylation was investigated by flow cytometry in a number of Melan-A-specific CD8+ T cells. A representative staining for T-cell clones isolated PRE and POST therapy from both Arms is shown in Fig. S3 and confirms the results obtained by Western blot analysis.
Figure 2. Melan-A-specific CD8+ T cells isolated after combined chemoimmunotherapy display pSer473-AKT expression unrelated to their differentiation stage. Analysis of AKT activation by Western blot in terms of phosphorylation of Ser473 (pSer473-AKT) and total AKT expression tested on whole cell extracts of 1.5 ×106 viable Melan-A specific clones (n = 26), isolated before (PRE) and after treatment (POST) with vaccination alone (Arm1) or DTIC plus vaccination (Arm2), 18 h following activation by plate-coated anti-CD3 mAb plus rIL-2. Gel loading control was performed analyzing β actin expression. The differentiation status (based on the expression of CD28 and/or CD27 co-stimulatory molecules, see ) for each clone is reported. Cells isolated from patients before the treatments evidence an expression of pSer473-AKT which relates with the differentiation status. Cell clones isolated after therapy (POST) are late differentiated (defined by the lack CD28 and CD27 molecules). Cells isolated from Arm1 (Arm1-POST) do not express pSer473-AKT according to their late differentiated profile, differently clones isolated from Arm2 patients (Arm2-POST) display a strong pSer473-AKT expression. Densitometric quantification (OD) of protein bands was determined by NIH ImageJ software and expressed p-AKT/AKT ratios are expressed either as numbers and columns.
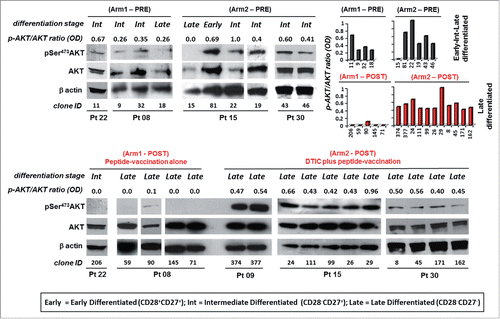
These results indicate that the combination of DTIC plus vaccination activates the AKT signaling pathway in CD28− CD27− late differentiated Melan-A-specific CD8+ T-cell clones with a highly polyfunctional profile.
Since AKT kinase can be activated by IL-2 and IL-15,Citation26 we evaluated a possible correspondence between the AKT activity and the levels of IL-2/IL-15 receptor expression in our clones. We observed a similar expression of the IL-2/IL-15 common receptor β subunit (CD122) in all clones analyzed, while lower levels of IL-2 α subunit (CD25) were observed in cells isolated after therapy, irrespective of the treatment (Fig. S4A). The lack of a critical role of IL-2/IL-2R was proved by the observation that deprivation of IL-2 from culture medium only slightly reduced AKT phosphorylation in T cells isolated from patients after DTIC plus vaccination (Fig. S4B).
In order to evaluate whether this AKT activation was specifically related to Melan-A peptide-mediated stimulation, we also performed biochemical and phenotypical analysis of CD8+ T cells specific for gp100, the other peptide employed in the vaccine formulation. Unlike Melan-A-specific T cells, gp100-specific T cells obtained from Arm2 patient 15, with an intermediate or late differentiation profile, showed a low AKT activation pattern, as shown in Fig. S5 for representative T cell clones. This result strongly suggests that this AKT signaling pathway, identified only in patients treated with DTIC before vaccination, is specifically driven by Melan-A-elicited TCR activation.
DTIC plus peptide-vaccination modulates the pattern of inhibitory molecules in Melan-A-specific CD8+ T cells
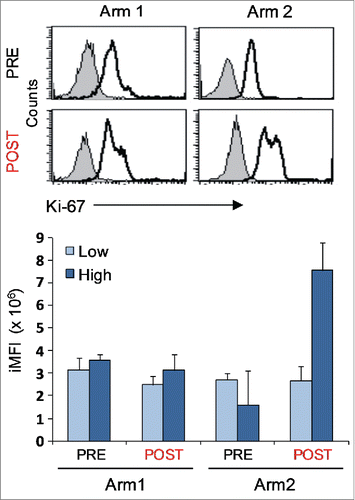
Inhibitory receptors including LAG-3, TIM-3 and PD-1, are generally associated with a chronically stimulated phenotype in highly differentiated T cells,Citation35 although the expression of these receptors “per se” does not indicate that a T cell is exhausted.Citation23 Nevertheless, inhibitory molecules have been shown to control Ser473-AKT phosphorylation in CD8+CD28−CD27− T cells.Citation28,36 To investigate whether the different vaccine-treatments influenced the balance between the inhibitory receptor pattern, we studied the expression of LAG-3, TIM-3 and PD-1 in relation to the AKT pathway in a number of clones, either unstimulated or anti-CD3-activated. Representative plots of multiple staining and the relative gating strategy for molecule co-expression are shown in . In unstimulated T cells, the LAG-3 molecule was undetectable or weakly expressed, whereas TIM-3 was highly expressed in all clones analyzed, either PRE or POST therapy. Differently, the expression of PD-1, absent in T-cells isolated before the treatments, was induced only by the combined therapy (p = 0.001; ). When T cells were stimulated with anti-CD3 mAb, an increase in LAG-3 and PD-1 molecules was observed irrespective of the treatment (). Again, cells isolated after the combined therapy showed the highest level of PD-1, also in terms of co-expression with LAG-3 and TIM-3. Unexpectedly, we found the highest expression of PD-1 along with activated AKT in cells isolated from Arm2 patients, characterized by an improved antitumor activity and polyfunctional profile (, green squares). When we measured PD-L1 in clones isolated from both Arms, we found similar levels of expression independent of the treatment (data not shown). On the other hand, the high expression of inhibitory receptors, and in particular PD-1, did not interfere with the T-cell proliferative potential, as measured by iMFI for the proliferation marker Ki-67, as shown in . Altogether these data suggest that the high expression of inhibitory receptors of Arm2 T-cell clones does not interfere with T-cell proliferative potential and does not affect the chemoimmunotherapy-induced AKT activation.
Figure 3. Melan-A-specific CD8+ T-cell clones isolated after DTIC plus vaccination display the highest level of PD-1 inhibitory receptor expression. (A) Representative flow cytometric analysis of multicolor staining, with the relative gating strategy, for simultaneous LAG-3 (L), TIM-3 (T) and PD-1 (P) expression. The multicolor histograms show the quantification of all possible combinations of molecule co-expression, for representative clones isolated either before (PRE) or at the end (POST) of both treatments. The staining was performed under basal cell culture conditions (−) or after over-night activation by plate-bound anti-CD3 mAb (2 µg/mL) (+). (B) Comparison from pooled data of inhibitory receptor profiles in PRE (black histograms) vs. POST (red histograms) treatment, displayed as single receptor expression, with (+) or without (−) anti-CD3 mAb activation. Each bar represents the mean percentage (± SD) of inhibitory receptor expression of Melan-A-specific T-cell clones isolated from patients of Arm1 (n = 2) and of Arm2 (n = 3), before (PRE, n = 6 for Arm1, n = 7 for Arm2) and after (POST, n = 7-8 for Arm1, n = 7–9 for Arm2) treatments. (C) The percentage of single or simultaneous inhibitory receptor expression in T-cell clones isolated at the end of both treatments (POST), with (■) or without (•) anti-CD3 activation. Each dot represents the mean value out of three–five independent experiments performed on a single T-cell clone. The mean ± SEM of each indicated sample group is shown. Black and green dots, clones negative or positive for pSer473-AKT, respectively, as assessed by Western blot analysis; gray dots, clones not tested for AKT phosphorylation. */♦p ≤ 0.05, **/♦♦p ≤ 0.01, ***/♦♦♦p ≤ 0.001, ****/♦♦♦♦p ≤ 0.0001, Mann–Whitney two-sample (*) test and two-tail Student’s test (♦), respectively.
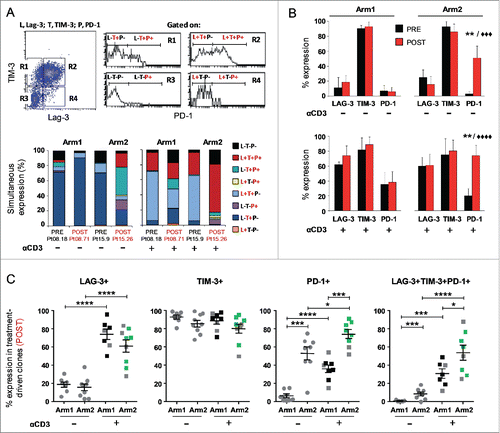
Figure 4. Proliferative potential of Melan-A-specific T-cell clones. Intracellular staining for Ki-67 expression of CTL clones (n = 12) isolated before (PRE) and after (POST) treatments from both Arms, performed 7 d after re-stimulation with allogenic irradiated PBMC, rIL-2 and PHA. Top panel: representative staining for each indicated sample group with anti-Ki-67 (open histograms) or isotype (filled histograms) mAbs, showing the presence of two positive cell populations with diverse amounts of Ki-67 molecule expression (low and high). Bottom panel: magnitude of low and high proliferating cells as measured by integrated mean fluorescence intensity (iMFI), (see Material and Methods for details). Results are shown as mean ± SD.
We then analyzed the expression of the killer cell lectin-like receptor G1 (KLRG1), a marker of terminal differentiation, that is associated with impaired proliferative potentialCitation37 by inhibiting AKT phosphorylation.Citation28 As shown in Fig. S6, KLRG1 expression was not found in any of Melan-A specific CD8+ T cells analyzed.
Inhibition of AKT activation, but not PD-1 blockade, reduces the antitumor activity of Melan-A-specific CD8+ T cells
The unexpected co-presence of high PD-1 levels along with AKT activation in late differentiated polyfunctional T cells isolated after DTIC plus vaccination, prompted us to further explore the involvement of these pathways in T cell antitumor activity.
First, we performed a time course blockade (1, 2 and 15 h) of the AKT signaling pathway in highly polyfunctional CD8+ T-cell clones isolated after combined therapy, before stimulation with Melan-A-expressing tumor cells. Blockade was performed by pre-treatment with non-specific Ly294002 PI3K (PI3Ki) or specific MK-2206 AKT (AKTi) inhibitors and then tumor-induced production of IFNγ was evaluated by ICS. Erk inhibitor FR 180204 was used as internal control.
IFNγ production was inhibited in CD8+ T cells pre-incubated either with LY 294002 (40%) or MK-2206 (70%) for 1 h. Restoration to a basal IFNγ level was observed when cells were pre-treated with the inhibitors for 15 h before tumor recognition (). Blockade with MK-2206 AKT specific inhibitor determined a more durable inhibition of IFNγ production (60% with 2 h blockade) as compared with the non-specific PI3K inhibitor. The inhibition of cytokine production mediated by the AKT inhibitors is not due to a T cell-toxicity event, as indicated by the restoration of IFNγ production after 15 h of T cell treatment. Based on these kinetic settings, blockade of PI3K and AKT activation for 1 h before exposure to Melan-A-expressing cell lines led to the inhibition of both TNF-α and IFNγ production in highly functional CD8+ T-cell clones isolated after combined therapy (, for a representative clone out of three analyzed). GrB, which was expressed also in unstimulated cells, was not affected by the blockade (). shows the mean for the inhibition values obtained in all the clones analyzed. Erk inhibitor FR 180204, used as internal control, did not affect the production either of TNF-α or IFNγ. These results confirm a critical role for AKT activation in the chemoimmunotherapy-induced functional advantage in terms of polyfunctional profiling.
Figure 5. AKT-dependent antitumor activity of highly polyfunctional tumor-specific CD8+ T cells elicited by the combined chemoimmunotherapy. Modulation of TNF-α (TNF), IFNγ (IFN), and Granzyme-B (GrB) co-production by AKT or PD-1 blockade, in clones from Arm2 patient (Pt15). (A) Kinetic of IFNγ production after AKT pathway blockade, with a PI3K (Ly294002, PI3Ki) or AKT inhibitor molecule (MK-2206, AKTi). Figure shows the mean ± SD of three independent experiments performed on a representative Arm2 Melan-A-specific T cell clone (Pt15.29) after specific recognition of Melan-A-expressing cell line (Mel+). Results are shown as a percentage of IFNγ production (top panel) or as a percentage of inhibition with respect to the maximal baseline production (bottom panel). A specific Erk inhibitor (FR 180204) was used as internal control. (B–C) Flow cytometry analysis of multicolor staining from a representative Melan-A-specific clone (Pt15.29), showing the single % of production (B) or the co-production (C) of TNF-α, IFNγ, and GrB in the presence (1 h) or in the absence of inhibitors. TNF-α and IFNγ, but not GrB, are significantly reduced by the AKT blockade. PD-1 blockade, by specific anti-PD1 (α-PD-1) or anti-PD-L1 (α-PD-L1) mAbs (18 h) does not affect the TNF-α, IFNγ, and GrB production capability. (D) Pooled data from three different T-cell clones (Pt15.3, Pt15.26, Pt15.29 ) showing the mean ± SD of the % of inhibition evaluated under different blockade conditions, relative to baseline production.
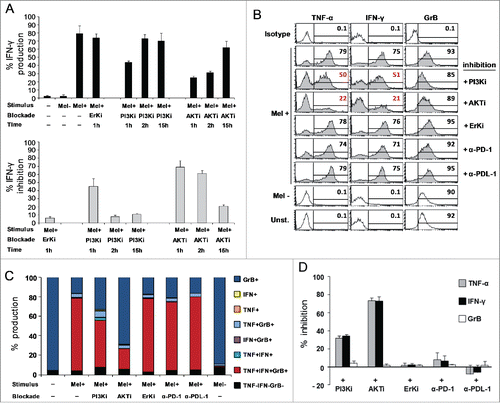
Noteworthy, the production of TNF-α, IFNγ or GrB was not affected by the inhibition of the PD-1 pathway, either by anti-PD-1 or anti-PD-L1 specific mAbs in Melan-A-specific CD8+ T cells isolated after the combined therapy (). On the other hand, blockade of PD-1 pathway slightly enhanced the cytokine production in cells from patients treated with vaccination alone (data not shown).
ICOS is involved in AKT activation induced by chemoimmunotherapy
In order to identify T-cell co-stimulatory molecules potentially responsible for the PI3-K/AKT activation observed in our polyfunctional T cells, we investigated the role of ICOS which shares the CD28 motif of the cytoplasmic tail responsible for the kinase activation. Differently from CD28 which is constitutive, ICOS is induced following TCR stimulationCitation14 and its ligand (ICOS-L/B7H) is expressed by APC and human melanomas.Citation38-40 Flow-cytometric analysis performed following 48 h stimulation by anti-CD3 mAb, evidenced the highest levels of the co-stimulatory molecule in clones isolated from patients treated with the combined therapy (). Of note, a statistically significant correlation was found between high levels of ICOS (cut-off > 30%) and p-AKT expression ().
Figure 6. ICOS is upregulated in Melan-A-specific CD8+ T cells isolated after combined therapy and is involved in chemoimmunotherapy-induced AKT activation. (A) Top panel shows representative staining with anti-ICOS (open histogram) or isotype (filled histogram) mAbs, in two T-cell clones isolated from Arm1 (Pt08.90) and Arm2 (Pt15.26) patients after the treatments (POST). Bottom panel shows pooled data of ICOS expression in Melan-A-specific T-cell clones (n = 15 from two patients of Arm1, n = 18 from three patients of Arm2), isolated before (PRE) and after therapy (POST). Cells were stained after 48 h stimulation with plate-bound anti-CD3 mAb (2 µg/mL) plus rIL-2 and shown in both scatter-plot graph and box-and-whisker diagram, with 5–95 percentile. Each dot represents the mean value out of three–five independent experiments. The mean ± SEM of each group is shown. (B) Correlation between p-AKT and ICOS expression. T-cell clones isolated after both treatments were grouped according to pSerAKT expression levels and the percentage of ICOS+ cells (bottom panel) and shows higher expression of ICOS in AKThigh T cells. Top panel, percentage of clones with ICOS levels > 30% cut-off in p-AKTneg/low versus p-AKThigh groups. p value was calculated with one-tail Fisher’s exact test. */♦p ≤ 0.05, **/♦♦p ≤ 0.01, ***/♦♦♦p ≤ 0.001, Mann–Whitney two-sample test (*) and two-tail Student’s test (♦), respectively. (C) Left panel: effect of ICOS blockade on AKT activation (p-Ser473-AKT) tested in a representative Melan-A specific T-cell clone (Arm2, Pt15.26, POST), following activation by irradiated PBMC-derived APCs plus rIL-2 (−). Blockade of ICOS engagement with anti-ICOS-L (ICOSLi) mAb (10 µg/mL) induced a marked downregulation of p-AKT. Right panel: effect of ICOS engagement on AKT activation (p-Ser473-AKT) tested in a representative Melan-A specific T-cell clone (Arm2, Pt15.111, POST), following activation by anti-CD3 mAb (−). The anti-ICOS-agonist (ICOSa) mAb (12.5 µg/mL) induced upregulation of p-AKT. Gel loading control is represented by total AKT expression. Densitometric quantification of protein bands was determined by NIH ImageJ software. Columns show the mean ± SD of p-AKT/AKT ratios (OD) from two independent experiments. (D) Top panel: bars represent the mean % (± SD) of TNF-α, IFNγ and GrB inhibition by ICOS blockade from two experiments performed on Pt15.26 T cell clone (representative of three different T-cell clones tested). Bottom panel: bars represent the mean % (± SD) of the increased polyfunctional activity induced by ICOS co-stimulation, as calculated with respect to the isotype control mAb.
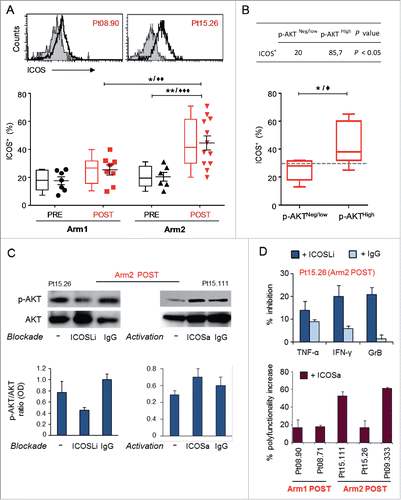
Then, to confirm the contribution of ICOS in AKT activation induced by chemoimmunotherapy, we modulated this pathway either by blocking with an anti-ICOS ligand mAb or activating by an anti-ICOS agonist mAb. For the blockade experiment, whole blood-isolated APC were pre-incubated with anti-ICOS-L/B7H mAb for 15 h, co-cultured with the Melan-A-specific CD8+ T-cell clones expressing p-AKT and analyzed by Western blot. AKT phosphorylation at Ser473 induced by stimulation was reduced by the ICOS blockade of about 50%, as evidenced in the representative immunoblotting of clone Pt15.26, and not affected by the IgG isotype control (, left panel). Of relevance, the blockade of the pathway inhibited the production of TNF-α, IFNγ and GrB, as measured by ICS (, top panel). For the activation experiment, Melan-A-specific CD8+ T-cells were stimulated 24 h with anti-CD3 mAb or anti-CD3 plus anti-ICOS agonist. An increase of AKT phosphorylation and polyfunctional activity was induced by ICOS co-stimulation (, right panel and , bottom panel, respectively). As expected, the increase of polyfunctional activity by ICOS co-stimulation was higher in clones from Arm2 patients, which displayed the highest ICOS expression (). These results suggest a critical role for ICOS in contributing to AKT activation and polyfunctionality in late differentiated CD8+ T cells isolated after chemoimmunotherapy.
Discussion
The identification of activation pathways linked to T-cell polyfunctionality against tumors, associated with clinical benefit, is of great relevance in the new era of immunotherapy and combined chemoimmunotherapy. Recently, we reported that Melan-A-specific CD8+ T cells isolated from long-term survival patients treated with DTIC injected in a tight window before peptide-vaccination plus IFN-α, possess higher tumor reactivity and an enlarged T-cell repertoire, compared to cells isolated after vaccination alone.Citation1,2
Here, we have explored the molecular mechanisms underlying the antitumor functionality of these T cells. We found that Melan-A-specific CD8+ T cells isolated from patients treated with chemoimmunotherapy, despite the late differentiation profile, show the highest antitumor activity in terms of polyfunctionality that is dependent on the activation of the AKT pathway and is associated with the engagement of ICOS, in the presence of LAG-3, TIM-3 and PD-1 inhibitory receptors.
Melan-A-specific T-cell clones derived from patients treated with the combined therapy, revealed a more potent antitumor cytolytic profile in terms of polyfunctionality defined by the co-production of TNF-α, IFNγ and GrB, compared with cells derived from patients treated with vaccination alone. Polyfunctionality reflects the ability of individual T cells to perform multiple effector functions and is crucial for the protective immunity against viruses and cancer.Citation30-32 Polyfunctional CD8+ T cells producing several effector cytokines and molecules are associated with the spontaneous resolution of primary acute HCVCitation41 and have been related to HIV-specific enlarged T-cell repertoire.Citation42,43 Similarly, polyfunctional T cells can be induced in cancer patients after vaccination.Citation44-46 In our study, the long overall survival observed in patients treated with the combined chemoimmunotherapy is in line with the concept that polyfunctional antitumor CD8+ T cells mediate a stronger host antitumor response and may thus prevent disease recurrence. Furthermore, the less effective quality of T-cell responses evidenced in patients treated with vaccination alone suggests that the schedule of combined therapy could have contributed to the induction of protective polyfunctional T cells. Although to our knowledge no data are available about a possible regulation of human T-cell polyfunctionality by combined immunotherapy, preconditioning chemotherapy with the alkylating agent cyclophosphamide has been shown to promote the generation of polyfunctional CD4+ effector T cells in murine models.Citation30
The generation of polyfunctional CD8+ T cells in response to vaccination, although associated with improved protective immunity has not yet been fully linked to a T-cell differentiation phenotype. In our setting, cells isolated after the treatments, in particular those obtained after the combined therapy, display a highly differentiated profile as defined by the CD45RA−CCR7− phenotype and the lack of co-stimulatory molecules CD28 and CD27. Among multiple extracellular pathways, AKT signaling has been reported as a major player in the cell fate decision versus effector CD8+ T cellsCitation29,47 and the magnitude of AKT activation drives the differentiation of T cells into terminal effectors or memory cells.Citation47 On the other hand, it has been shown that AKT activation decreases as CD8+ T cells progress toward differentiation.Citation12,28 Here, we have observed that combined immunotherapy, differently from vaccination alone, results in increased AKT phosphorylation in highly differentiated Melan-A-specific CD28−CD27− T cells. Of note, T-cell polyfunctionality elicited by combined therapy was strictly dependent on AKT activation, as demonstrated by blockade with selective inhibitors, in agreement with previous studies showing the impairment of the IFNγ production induced by AKT inhibition and the induction of an effector phenotype in the presence of an active AKT signaling pathway.Citation29,47 This AKT activation occurred only in Melan-A-specific CD8+ T cells and not in cells specific for gp100, the other peptide used in the vaccine formulation, where the AKT phosphorylation was expressed according to their differentiation profile, suggesting that the structure of the Ag/TCR complex is critical for the activation of this pathway.
It has been recently reported that AKT inhibition induces the generation of long-lived tumor-reactive memory T cells in murine and human models.Citation48-50 The results of Crompton’ and Abu’s studies may differ from our results due to the long AKT inhibition treatment (up to 30 d) performed in gp-100-specific T cells. Similarly, Van der Waart study reports the possibility of expanding CD28+CD27+ CD8+ T memory-like efficient antitumor cells by inhibiting AKT signaling in naive precursors. These apparently differing results can be ascribed to the differentiation status of the CD8+ T cells evaluated, i.e., late differentiated CD28−CD27− in our settings, and early memory-like CD28+CD27+ T cells in the study by van Der Waart. From our data, although the mechanisms regulating the AKT activation are still unclear, we can speculate that combined therapy may favor an effector phenotype, with an improvement of the proliferative potential as defined by Ki-67 expression, in late differentiated Melan-A-specific CD8+ T cells.
In the possible redundancy of co-stimulatory receptor usage, ICOS is known to enhance T-cell activation when CD28 expression is limited or absent.Citation12,51 We found that combination therapy induced the highest expression of ICOS in Melan-A-specific CD8+CD28−CD27− T cells with the phosphorylation of AKT at Ser 473 partially dependent on the engagement of the ICOS/ICOS-L pathway. This was not related to the expression of receptors for IL-2 and IL-15, cytokines known to be involved in the activation of the AKT pathway,Citation52 confirming previous studies showing that ICOS, differently from CD28, impacts on proliferation along with IFNγ, but not IL-2 production.Citation53 Deficiency in ICOS expression has been associated with extensive T-cell dysfunction and impaired memory T-cell compartment.Citation54 Besides, this molecule has proven effective in the enhancement of cytokine-mediated cell toxicity, in part by increasing IFNγ secretion.Citation55 Of note, ICOS has been identified as a crucial player in the anti-melanoma effects of CTLA-4 blockadeCitation56 and critically co-expressed with PD-1 as a potential identifier of tumor-reactive cells.Citation20
Although inhibitory receptors have so far been considered to mark terminally differentiated “exhausted” T cells,Citation35 they have recently been associated with the activation and differentiation profile of T cells.Citation23 PD-1 and KLRG1 have been shown to contribute to the defect of AKT phosphorylation in highly differentiated CD8+ CD28− CD27−T cells.Citation28,36 Differently, the activation of AKT we observed in Melan-A-specific clones isolated after the chemoimmunotherapy was not the result of a reduced expression of inhibitory receptors since high levels of LAG-3, TIM-3 and PD-1 were found in all the clones analyzed, with cells isolated after the combined therapy showing the highest PD-1 expression. This phenotypic profile may be instrumental for limiting the self-tissue damage, as supported by the lack of autoimmune evidences in our patients. On the other hand the co-expression of PD-1 and ICOS may represent a rheostat in the control of highly reactive Ag-stimulated T cells, as recently reported for PD-1.Citation57 Our findings do not raise doubts about the clinical usage of blocking inhibitory checkpoints in the presence of T cells with such phenotype,Citation4,58-60 as the blockade of PD-1 does not impair the AKT-dependent polyfunctionality. Rather these findings highlight the importance of taking into account the status of T-cell activation and differentiation in the selection of patients who can take advantage of the inhibition of these pathways.
About other players potentially involved in this peculiar activation of AKT in T cells pre-exposed to DTIC and sustained by Melan-A-mediated expansion, we can speculate that it can be the consequence of DNA damage events, as reported for Temozolomide, the active DTIC metabolite.Citation61 Further, we cannot exclude that IFN-α, administrated locally in both treatment Arms as adjuvant,Citation62 may have contributed to the generation of polyfunctional Melan-A-specific CD8+ T cells with an activated AKT pathway.
Of clinical relevance patients who received DTIC plus vaccination, although affected by III/IVa stage melanoma, are currently disease-free, after 10 y from the end of the treatment, thus suggesting that AKT-dependent polyfunctional tumor-reactive T cells, endowed with an enlarged Melan-A-specific TCR repertoire,Citation2 may protect from disease relapse.
Although we recognize that the study of a large number of clones only partially resembles the “in vivo” scenario, the present findings represent a critical contribution for the comprehension of the mechanisms underlying the advantages of combined chemoimmunotherapy. We suggest that combination DTIC plus vaccination may induce a phenotypic and functional signature dictated by a fine-tuned balance between quality of Ag/TCR complex, co-stimulatory signals such as ICOS, inhibitory checkpoints and AKT activation, associated with antitumor T cells able to protect patients from tumor recurrence. As far as new methodologies will improve immune monitoring at single cell level, the analysis of the identified activated AKT pathway may represent a new biomarker of T-cell activation and responsiveness in the course of immunomodulating cancer therapies.
Material and Methods
Patients, cell lines and T-cell clones
Treatment of melanoma patients enrolled in Arm1 and Arm2 vaccination protocol has been described.Citation1 Pt.08 and Pt.22 were vaccinated with Melan-A (A27L) and gp100 (210M) peptides plus IFN-α (Arm1); Pt.09, Pt.15 and Pt.30 received the DTIC one day before the same vaccination schedule (Arm2). Patients were followed for disease progression by a total body CT scan periodically. T2 cell lines were purchased from the American Type Culture Collection. Melanoma cell lines Mel1 (HLA-A2−/Melan-A+, not shown), Mel2 (HLA-A2+/Melan-A−, referred as Mel-) and Mel3 and Mel4 (HLA-A2+/Melan-A+, referred as Mel+) were kindly provided by Dr A. Anichini (Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy). Melanoma cell lines were isolated from surgical specimens of tumors of patients admitted to Fondazione IRCCS Istituto Nazionale dei Tumori, Milan. All lesions were histologically confirmed to be cutaneous malignant melanomas. Molecular and biological characterization of these cell lines has been reported previously.Citation63 Melanoma cell lines were periodically checked for the expression of a panel of tumor associated Ags (i.e. BRAF V600E, MITF, Tyrosinase, Melan-A, gp100, MAGE) by intracellular flow cytometry and Western blot analysis. Cell lines were periodically checked (no more than 2 mo from the assays) morphologically, and tested by growth curve analysis by 3H-Thymidine incorporation assay and Mycoplasma detection. Melan-A-specific and gp100-specific T-cell clones were generated by limiting dilution from presensitized Ag-specific T-cell lines as describedCitation2 and periodically restimulated (every 3 weeks) in RPMI 1640 medium supplemented with 10% human serum (noncommercial, prepared from healthy donors), 1 μg/mL PHA (Roches, 11082132001), 25 U/mL rIL-2 (Roches, 11147528001), and 1 × 106/mL irradiated allogenic peripheral blood mononuclear cells (PBMC) as feeder cells. T-cell clones were used 13–15 d after PHA-stimulation.
Antibodies and flow cytometry
Cell surface-staining was performed using various combinations of the following Abs: PE-Cy5-CD27 (Beckman Coulter, 0826607107), FITC-CD45RA (BD Pharmigen, 335039), PE-Cy7-CCR7 (BD Pharmigen, 557648), PE-CD25 (Miltenyi Biotec, 130-091-024), PE-IL-2/IL15Rβ (CD122, BD Pharmigen, 554525), PE-Cy7-PD-1 (BD Pharmigen, 561272), PE-Cy7-PD-L1 (CD274, BD Pharmigen, 558017), PE-TIM3 (R&D Systems, FAB2365P), FITC-LAG3 (Adipogen, Inc., AG-20B-0012F), polyclonal-KLRG1 (Sigma-Aldrich, SAB1400378), PE-ICOS (BD Pharmigen, 557802). The CD28 hybridoma CD28.2 (IgG1) was provided by Dr D. Olive (Inserm UMR). ICS was performed with the following mAbs: PerCP-Cy5.5-TNF-α (BD Pharmigen, 560679), PE-Cy7-IFNγ (BD Pharmigen, 557643), Alexa Fluor647-GrB (BD Pharmigen, 560212) and Ki-67 (Dako, M7240). For CD28, KLRG1 and Ki-67 staining, a secondary PE-labeled anti-mouse antibody (Dako, R0439) was used. Ag-specificity of T-cell clones was periodically tested by the use of FITC-CD8+ (Miltenyi Biotec, 130-080-601) and PE-Melan-A/gp100 tetramers (Beckman Coulter, 082T01008/082T01012) as described.Citation1 The surface staining was performed for 30 min at 4°C. When possible, dead cells were excluded using propidium iodide staining (MP Biomedicals, 195458). When required, stimulation with plate-bound anti-CD3 mAb (2 μg/mL) (CBT3 IgG2a,Citation64) was performed as indicated. For ICS of p-AKT, T-cell clones were cultured in RPMI medium supplemented with 1% HS for 2 h, and stimulated by plate-bound anti-CD3 mAb for 2 h at 37°C. The staining was performed after fixation with 1.6% paraformaldehyde and permeabilization with 80% methanol by the use of Alexa Fluor 647 anti-AKT pS473 (BD Pharmigen, 561670). Cells were immediately acquired on BD FACSCanto II (BD Bioscience) and analyzed using FlowJo 9.2.3 (TreeStar), FACSDiva and CellQuest software (BD). For receptor blocking experiments, the following functional grade purified mAbs purchased from eBioscience were used: anti-PD1 (IgG1, 16-9989), anti-PD-L1 (IgG1, 16-5983), anti-ICOSL (IgG1, 16-5889) and control Isotype (IgG1, 16-4714). The blockade was performed by incubation of T cells and/or target cells overnight with the corresponding blocking antibody (10 µg/mL) or with the control Isotype. Then cells were washed and co-cultured for the indicated time. For ICOS activation experiments, cells were incubated for 24 h with plate-bound functional grade anti-ICOS agonist mAb (IgG1, eBioscience, 16-9948-82 ) (12.5 µg/mL) in the presence of anti-CD3, before polyfunctional analysis. For some experiments, the magnitude of staining was measured as iMFI, a metric that combines the relative amount of molecule produced (the MFI) with the relative number of cells that make them (the percentage positive cells).Citation33 iMFI is computed by multiplying the percentage with the MFI of positive cells and reflects the total functional response of a population.
TNF-α, IFNγ, GrB production and cytotoxicity assay
For ICS production, Melan-A-specific T-cell clones were co-cultured with related (Mel+) or unrelated (Mel-) HLA/Ag melanoma cell lines, for 4–5 h at 37°C in the presence of the protein transport inhibitor GolgiStop (BD, 554724). T-cell clones were collected, washed with PBS, fixed using 2% paraformaldeide (15 min on ice) and then permeabilized by 5% FCS, 0,5% Saponine in PBS (20 min at RT). Cells were then incubated for 30 min at RT with PerCP-Cy5.5-TNF-α, PE-Cy7-IFNγ and Alexa Fluor647-GrB mAbs. The AKT signaling pathway blockade was performed by pre-incubating T-cell clones with 30 µM PI3K inhibitor molecule (Ly294002, Cell Signaling Technology, 9901) or with 5 µM AKT inhibitor molecule (MK-2206, Merck Pipeline, HY-10358) for 30 min, 1 h, 2 h or 15 h, as indicated. An Erk inhibitor (FR 180204, Santa Cruz Biotechnology, SC203945) was used as internal control. PD-1 and ICOS pathway blockade/activation were performed as described above. Lytic activity and fine Ag recognition were assessed in a standard 4 h 51Cr release assay as described.Citation2 Briefly, T2 cells, Mel1, Mel2, Mel3 and Mel4 melanoma cell lines were used as target cells and cytotoxicity assays were performed by incubating 51Cr-labeled target cells with effector cells at an E:T ratios of 20:1. Fine Ag specificity analysis measured pulsing T2 target cells with decreasing concentration of natural (Melan-A26-35), analog (Melan-AA27L) or irrelevant gp100 peptides. The percentage of specific lysis was calculated as follows: 100 × (experimental release−spontaneous release)/(total release−spontaneous release).
Western blot analysis
T-cell clones were activated overnight by plate-bound anti-CD3 mAb, in complete RPMI medium supplemented with 10% HS, with or without 25 U/mL rIL-2. Whole cell extracts were obtained by lysis in 10% glycerol, 0.1% SDS, 0.5% DOC, 1% NP-40 in PBS. Lysates from 1.5 × 106 cells were separated on 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to Hybond-P membranes (Amersham Pharmacia Biotech, Piscataway, NJ). Membranes were then probed with anti p-AKT (Ser 473, Cell Signaling, 4060S), AKT1 (Cell Signaling, 2938S), Bclx (Cell Signaling, 2762), and pSer2448mTOR (Cell Signaling, 2971) Abs, following the instructions provided by the manufacturer. β-actin Ab (Cell Signaling, 3700S) was used as loading control. Analysis was performed using the ECL Prime Western detection kit (Amersham Pharmacia Biotech, RPN 2236). X-ray films were scanned by HP Scanjet 5470 and processed by Corel Photo Paint 12. Densitometric quantification of protein bands was determined by NIH ImageJ software. For blocking experiments, Melan-A-specific CD8+ T-cell clones, previously pre-incubated overnight or not with anti-ICOS-L mAb, were stimulated in the presence of whole blood-APCs, and then analyzed for pSer473-AKT as described above. For ICOS activation experiments, cells were incubated for 24 h with plate-bound functional grade anti-ICOS agonist mAb (12.5 µg/mL) in the presence of anti-CD3, before pSer473-AKT Western blot analysis.
Statistical analysis
Values are reported as mean ± SEM/SD or median (range). As evaluated in a similar study,Citation65 the comparison between groups was performed with a Mann–Whitney two-sample test, unpaired Student’s t-test or Fisher's Exact test, when appropriate. The correlation analysis was performed with Spearman and Pearson test. p value ≤ 0.05 was considered significant. Statistical analyses were carried out using SPSS software (SPSS version 21, SPSS Inc., Chicago, IL, USA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary Files
Download PDF (490.2 KB)Acknowledgments
The authors thank the patients for study participation and blood donation. They are grateful to Dr F. Di Modugno for continuous helpful discussion. M.V. Sarcone, N. Gualtieri, M Panetta are acknowledged for the technical assistance. A particular thanks to A Soriani and M. L. Foddai for their continuous collaboration.
References
- Nisticò P, Capone I, Palermo B, Del Bello D, Ferraresi V, Moschella F, Aricò E, Valentini M, Bracci L, Cognetti F et al. Chemotherapy enhances vaccine-induced antitumor immunity in melanoma patients. Int J Cancer 2009; 124:130-9; PMID:18839429; http://dx.doi.org/10.1002/ijc.23886
- Palermo B, Del Bello D, Sottini A, Serana F, Ghidini C, Gualtieri N, Ferraresi V, Catricalà C, Belardelli F, Proietti E et al. Dacarbazine treatment before peptide vaccination enlarges T-cell repertoire diversity of melan-a-specific, tumor-reactive CTL in melanoma patients. Cancer Res 2010; 70:7084-92; PMID:20823160; http://dx.doi.org/10.1158/0008-5472.CAN-10-1326
- Frauwirth KA, Thompson CB. Activation and inhibition of lymphocytes by costimulation. J Clin Invest 2002; 109:295-299; PMID:11827987; http://dx.doi.org/10.1172/JCI0214941
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/10.1038/nrc3239
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996; 14:233-58; PMID:8717514; http://dx.doi.org/10.1146/annurev.immunol.14.1.233
- Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol 2003; 3:939-51; PMID:14647476; http://dx.doi.org/10.1038/nri1248
- Agematsu K, Kobata T, Sugita K, Freeman GJ, Beckmann MP, Schlossman SF, Morimoto C. Role of CD27 in T cell immune response. Analysis by recombinant soluble CD27. J Immunol 1994; 153:1421-9; PMID:8046222
- Denoeud J1, Moser M. Role of CD27/CD70 pathway of activation in immunity and tolerance. J Leukoc Biol 2011; 89:195-203; PMID:20699361; http://dx.doi.org/10.1189/jlb.0610351
- Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med 2002; 8:379-85; PMID:11927944; http://dx.doi.org/10.1038/nm0402-379
- Plunkett FJ, Franzese O, Belaramani LL, Fletcher JM, Gilmour KC, Sharifi R, Khan N, Hislop AD, Cara A, Salmon M et al. The impact of telomere erosion on memory CD8+ T cells in patients with X-linked lymphoproliferative syndrome. Mech Ageing Dev 2005; 126:855-65; PMID:15992610; http://dx.doi.org/10.1016/j.mad.2005.03.006
- Kopf M, Coyle AJ, Schmitz N, Barner M, Oxenius A, Gallimore A, Gutierrez-Ramos JC, Bachmann MF. Inducible costimulator protein (ICOS) controls T helper cell subset polarization after virus and parasite infection. J Exp Med 2000; 192:53-61; PMID:10880526
- Plunkett FJ, Franzese O, Finney HM, Fletcher JM, Belaramani LL, Salmon M, Dokal I, Webster D, Lawson AD, Akbar AN. The loss of telomerase activity in highly differentiated CD8+CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. J Immunol 2007; 178:7710-9; PMID:17548608; http://dx.doi.org/10.4049/jimmunol.178.12.7710
- Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, Flavell RA. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001; 409:97-101; PMID:11343121; http://dx.doi.org/10.1038/35051100
- Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999; 397:263-6; PMID:9930702; http://dx.doi.org/10.1038/16717
- Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol 2002; 2:116-26; PMID:11910893; http://dx.doi.org/10.1038/nri727
- Takahashi N, Matsumoto K, Saito H, Nanki T, Miyasaka N, Kobata T, Azuma M, Lee SK, Mizutani S, Morio T. Impaired CD4 and CD8 effector function and decreased memory T cell populations in ICOS-deficient patients. J Immunol 2009; 182:5515-27; PMID:19380800; http://dx.doi.org/10.4049/jimmunol.0803256
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007; 27:670-84; PMID:17950003; http://dx.doi.org/10.1016/j.immuni.2007.09.006
- Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol 2007; 19:408-15; PMID:17656078; http://dx.doi.org/10.1016/j.coi.2007.06.004
- McMahan RH, Golden-Mason L, Nishimura MI, McMahon BJ, Kemper M, Allen TM, Gretch DR, Rosen HR. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest 2010; 120:4546-57; PMID:21084749; http://dx.doi.org/10.1172/JCI43127
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME et al. PD-1 identifies the patient-specific CD8 tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124:2246-59; PMID:24667641; http://dx.doi.org/10.1172/JCI73639
- Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, Tata P, Gupta S, Zilliox MJ, Nakaya HI et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol 2011; 186:4200-12; PMID:21383243; http://dx.doi.org/10.4049/jimmunol.1001783
- Baitsch L, Legat A, Barba L, Fuertes Marraco SA, Rivals JP, Baumgaertner P, Christiansen-Jucht C, Bouzourene H, Rimoldi D, Pircher H et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One 2012; 7:30852; PMID:22347406; http://dx.doi.org/10.1371/journal.pone.0030852
- Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory Receptor Expression Depends More Dominantly on Differentiation and Activation than “Exhaustion” of Human CD8 T Cells. Front Immunol 2013; 4:455; PMID:24391639; http://dx.doi.org/10.3389/fimmu.2013.00455
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005; 25:9543-53; PMID:16227604; http://dx.doi.org/10.1128/MCB.25.21.9543-9553.2005
- Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev 2003; 192:7-20; PMID:12670391; http://dx.doi.org/10.1034/j.1600-065X.2003.00008.x
- Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol 2011; 11:109-17; PMID:21233853; http://dx.doi.org/10.1038/nri2888
- Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol 2013; 4:20; PMID:23378844; http://dx.doi.org/10.3389/fimmu.2013.00020
- Henson SM, Franzese O, Macaulay R, Libri V, Azevedo RI, Kiani-Alikhan S, Plunkett FJ, Masters JE, Jackson S, Griffiths SJ et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood 2009; 113:6619-28; PMID:19406987; http://dx.doi.org/10.1182/blood-2009-01-199588
- Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, Feijoo C, Okkenhaug K, Cantrell DA. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 2011; 34:224-36; PMID:21295499; http://dx.doi.org/10.1016/j.immuni.2011.01.012
- Ding ZC, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, Zhou G. Polyfunctional CD4+ T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood 2012; 120:2229-39; PMID:22859605; http://dx.doi.org/10.1182/blood-2011-12-398321
- Graw F, Regoes RR. Predicting the impact of CD8+ T cell polyfunctionality on HIV disease progression. J Virol 2014; 88:10134-45; PMID:24965450; http://dx.doi.org/10.1128/JVI.00647-14
- Chiu YL, Shan L, Huang H, Haupt C, Bessell C, Canaday DH, Zhang H, Ho YC, Powell JD, Oelke M et al. Sprouty-2 regulates HIV-specific T cell polyfunctionality. J Clin Invest 2014; 124:198-208; PMID:24292711; http://dx.doi.org/10.1172/JCI70510
- Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med 2007; 13:843-50; PMID:17558415; http://dx.doi.org/10.1038/nm1592
- McKee MD, Roszkowski JJ, Nishimura MI. T cell avidity and tumor recognition: implications and therapeutic strategies. J Transl Med 2005; 3:35; PMID:16174302; http://dx.doi.org/10.1186/1479-5876-3-35
- Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology 2010; 129:474-81; PMID:20201977; http://dx.doi.org/10.1111/j.1365-2567.2010.03255.x
- Henson SM, Macaulay R, Franzese O, Akbar AN. Reversal of functional defects in highly differentiated young and old CD8 T cells by PDL blockade. Immunology 2012; 135:355-63; PMID:22211948; http://dx.doi.org/10.1111/j.1365-2567.2011.03550.x
- Voehringer D, Koschella M, Pircher H. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood 2002; 100:3698-702; PMID:12393723; http://dx.doi.org/10.1182/blood-2002-02-0657
- Ito T, Yang M, Wang YH, Lande R, Gregorio J, Perng OA, Qin XF, Liu YJ, Gilliet M. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med 2007; 204:105-15; PMID:17200410; http://dx.doi.org/10.1084/jem.20061660
- Zheng J, Chan PL, Liu Y, Qin G, Xiang Z, Lam KT, Lewis DB, Lau YL, Tu W. ICOS regulates the generation and function of human CD4+ Treg in a CTLA-4 dependent manner. PLoS One 2013; 8:82203; PMID:24312642; http://dx.doi.org/10.1371/journal.pone.0082203
- Martin-Orozco N, Li Y, Wang Y, Liu S, Hwu P, Liu YJ, Dong C, Radvanyi L. Melanoma cells express ICOS ligand to promote the activation and expansion of T-regulatory cells. Cancer Res 2010; 70:9581-90; PMID:21098714; http://dx.doi.org/10.1158/0008-5472.CAN-10-1379
- Badr G, Bédard N, Abdel-Hakeem MS, Trautmann L, Willems B, Villeneuve JP, Haddad EK, Sékaly RP, Bruneau J, Shoukry NH. Early interferon therapy for hepatitis C virus infection rescues polyfunctional, long-lived CD8+ memory T cells. J Virol 2008; 820:10017-31; PMID:18667516; http://dx.doi.org/10.1128/JVI.01083-08
- Almeida JR, Price DA, Papagno L, Arkoub ZA, Sauce D, Bornstein E, Asher TE, Samri A, Schnuriger A, Theodorou I et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 2007; 204:2473-85; PMID:17893201; http://dx.doi.org/10.1084/jem.20070784
- Janbazian L, Price DA, Canderan G, Filali-Mouhim A, Asher TE, Ambrozak DR, Scheinberg P, Boulassel MR, Routy JP, Koup RA et al. Clonotype and repertoire changes drive the functional improvement of HIV-specific CD8 T cell populations under conditions of limited antigenic stimulation. J Immunol 2012; 188:1156-67; PMID:22210916; http://dx.doi.org/10.4049/jimmunol.1102610
- Darrah PA1, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med 2007; 13:843-50; PMID:17558415; http://dx.doi.org/10.1038/nm1592
- Yuan J, Gnjatic S, Li H, Powel S, Gallardo HF, Ritter E, Ku GY, Jungbluth AA, Segal NH, Rasalan TS et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A 2008; 105:20410-20415; PMID:19074257; http://dx.doi.org/10.1073/pnas.0810114105
- Ayyoub M, Dojcinovic D, Pignon P, Raimbaud I, Schmidt J, Luescher I, Valmori D. Monitoring of NY-ESO-1 specific CD4+ T cells using molecularly defined MHC class II/His-tag-peptide tetramers. Proc Natl Acad Sci U S A 2010; 107:7437-7442; PMID:20368442; http://dx.doi.org/10.1073/pnas.1001322107
- Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, Suresh M. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol 2012; 188:4305-14; PMID:22467649; http://dx.doi.org/10.4049/jimmunol.1103568
- van der Waart AB, van de Weem NM, Maas F, Kramer CS, Kester MG, Falkenburg JH, Schaap N, Jansen JH, van der Voort R, Gattinoni L et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 2014; 124:3490-500; PMID:25336630; http://dx.doi.org/10.1182/blood-2014-05-578583
- Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, Tran E, Hanada K, Yu Z, Palmer DC, Kerkar SP, Michalek RD et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 2015; 75:296-305; PMID:25432172; http://dx.doi.org/10.1158/0008-5472.CAN-14-2277
- Abu Eid R, Friedman KM, Mkrtichyan M, Walens A, King W, Janik J, Khleif SN. Akt1 and -2 inhibition diminishes terminal differentiation and enhances central memory CD8+ T-cell proliferation and survival. Oncoimmunology 2015; 4:1005448; PMID:26155399; http://dx.doi.org/10.1080/2162402X.2015.1005448
- Bukczynski J, Wen T, Watts TH. Costimulation of human CD28- T cells by 4-1BB ligand. Eur J Immunol 2003; 33:446-54; PMID:12645943; http://dx.doi.org/10.1002/immu.200310020
- Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol 2006; 6:595-601; PMID:16868550; http://dx.doi.org/10.1038/nri1901
- Arimura Y, Kato H, Dianzani U, Okamoto T, Kamekura S, Buonfiglio D, Miyoshi-Akiyama T, Uchiyama T, Yagi J. A co-stimulatory molecule on activated T cells, H4/ICOS, delivers specific signals in T(h) cells and regulates their responses. Int Immunol 2002; 14:555-66; PMID:12039907; http://dx.doi.org/10.1093/intimm/dxf022
- Takahashi N, Matsumoto K, Saito H, Nanki T, Miyasaka N, Kobata T, Azuma M, Lee SK, Mizutani S, Morio T. Impaired CD4 and CD8 effector function and decreased memory T cell populations in ICOS-deficient patients. J Immunol 2009; 182:5515-27; PMID:19380800; http://dx.doi.org/10.4049/jimmunol.0803256
- Wang J, He M, Shi W, Sha H, Feng J, Wang S, Wang Y. Inducible costimulator (ICOS) enhances the cytolytic activity of cytokine-induced killer cells against gallbladder cancer in vitro and in vivo. Cancer Invest 2009; 27:244-50; PMID:19194830; http://dx.doi.org/10.1080/07357900802239124
- Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med 2014; 211:715-25; PMID:24687957; http://dx.doi.org/10.1084/jem.20130590
- Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol 2013; 14:1212-8; PMID:24240160; http://dx.doi.org/10.1038/ni.2762
- Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology 2012; 1:1223-1225; PMID:23243584; http://dx.doi.org/10.4161/onci.21335
- Aranda F, Vacchelli E, Eggermont A, Galon J, Fridman WH, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Immunostimulatory monoclonal antibodies in cancer therapy. Oncoimmunology 2014; 3:27297; http://dx.doi.org/10.4161/onci.27297
- Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32:1020-30; PMID:24590637; http://dx.doi.org/10.1200/JCO.2013.53.0105
- Caporali S, Levati L, Starace G, Ragone G, Bonmassar E, Alvino E, D'Atri S. AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol Pharmacol 2008; 74:173-83; PMID:18413665; http://dx.doi.org/10.1124/mol.107.044743
- Aricò E, Belardelli F. Interferon-α as antiviral and antitumor vaccine adjuvants: mechanisms of action and response signature. J Interferon Cytokine Res 2012; 32:235-47; PMID:22490303; http://dx.doi.org/10.1089/jir.2011.0077
- Daniotti M, Oggionni M, Ranzani T, Vallacchi V, Campi V, Di Stasi D, Torre GD, Perrone F, Luoni C, Suardi S et al. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene 2004; 23:5968-77; PMID:15195137; http://dx.doi.org/10.1038/sj.onc.1207780
- Nisticò P1, Mortarini R, De Monte LB, Mazzocchi A, Mariani M, Malavasi F, Parmiani G, Natali PG, Anichini A. Cell retargeting by bispecific monoclonal antibodies. Evidence of bypass of intratumor susceptibility to cell lysis in human melanoma. J Clin Invest 1992; 90:1093-9; PMID:1387883; http://dx.doi.org/10.1172/JCI1-15925
- Lövgren T, Baumgaertner P, Wieckowski S, Devêvre E, Guillaume P, Luescher I, Rufer N, Speiser DE. Enhanced cytotoxicity and decreased CD8 dependence of human cancer-specific cytotoxic T lymphocytes after vaccination with low peptide dose. Cancer Immunol Immunother 2012; 61:817-26; PMID:22080404; http://dx.doi.org/10.1007/s00262-011-1140-1