
ABSTRACT
Natural Killer (NK) cells are innate effector cells that are able to recognize and eliminate tumor cells through engagement of their surface receptors. NKp30 is a potent activating NK cell receptor that elicits efficient NK cell-mediated target cell killing. Recently, B7-H6 was identified as tumor cell surface expressed ligand for NKp30. Enhanced B7-H6 mRNA levels are frequently detected in tumor compared to healthy tissues. To gain insight in the regulation of expression of B7-H6 in tumors, we investigated transcriptional mechanisms driving B7-H6 expression by promoter analyses. Using luciferase reporter assays and chromatin immunoprecipitation we mapped a functional binding site for Myc, a proto-oncogene overexpressed in certain tumors, in the B7-H6 promoter. Pharmacological inhibition or siRNA/shRNA-mediated knock-down of c-Myc or N-Myc significantly decreased B7-H6 expression on a variety of tumor cells including melanoma, pancreatic carcinoma and neuroblastoma cell lines. In tumor cell lines from different origin and primary tumor tissues of hepatocellular carcinoma (HCC), lymphoma and neuroblastoma, mRNA levels of c-Myc positively correlated with B7-H6 expression. Most importantly, upon inhibition or knock-down of c-Myc in tumor cells impaired NKp30-mediated degranulation of NK cells was observed. Thus, our data imply that Myc driven tumors could be targets for cancer immunotherapy exploiting the NKp30/B7-H6 axis.
Introduction
Natural Killer (NK) cells are innate immune cells that are able to sense and lyse tumor cells through their specialized set of activating and inhibitory surface receptors. Inhibitory receptors of the killer immunoglobulin-like receptor (KIR) family recognize self-MHC class I molecules. Activating NK cell receptors, such as the family of the natural cytotoxicity receptors (NCRs) recognize virus-, stress or transformation-induced ligandsCitation1-3. The family of NCRs comprises three members, NKp30, NKp44 and NKp46 all of which are involved in the killing of tumor cells.Citation4, 5 NKp30 belongs to the CD28 family and is expressed on the majority of NK cells.Citation2 It is not only involved in tumor cell lysis but also in the interaction with dendritic cells.Citation6, 7 NKp30 possesses alternatively spliced isoforms, which are of prognostic value for patients with gastrointestinal stromal tumorsCitation8 and for patients with high risk neuroblastoma responding to induction chemotherapy.Citation9 Intriguingly, the initially described ligands for NKp30, the human cytomegalovirus pp65 tegument proteinCitation10 and the HLA-B-associated transcript 3 (BAT3) proteinCitation11, were both intracellular proteins. B7-H6 (Natural cytotoxicity triggering receptor 3 ligand 1, NCR3LG1) was the first NKp30-ligand identified being a transmembrane protein expressed on tumor cell surfaces.Citation12
B7-H6 belongs to the B7 family and the interaction of B7-H6 on tumor cells with NKp30 on NK cells leads to efficient NK cell activation.Citation12 Neither NKp30 nor B7-H6 orthologues exist in mice. B7-H6 is expressed on the cell surface of many tumor cell lines of different origin and certain tumors in situ but not on hematopoietic cells of healthy individuals or primary keratinocytes.Citation12, 13 In monocytes and neutrophils, inflammatory stimuli were described to lead to an up-regulation of cell surface expression and exosomal secretion of B7-H6Citation14. On tumor cells, B7-H6 expression is decreased after inhibition of histone deacetylation.Citation13 Furthermore, B7-H6 is released from the surface of tumor cells by metalloproteases.Citation15 Mechanisms that determine the expression of B7-H6 on tumors at the transcriptional level are so far unknown.
In mammals, the Myc family of transcription factors (TFs) consists of four members: c-Myc, N-Myc, L-Myc and S-Myc. In humans, the family members c-Myc, N-Myc and L-Myc have all been associated with tumorigenesis.Citation16 In complex with Myc-associated factor X (Max), Myc proteins bind to defined DNA sequences, termed enhancer-boxes (E-boxes) and activate transcription of certain target genes. Max can also bind together with Mad1 to E-boxes. In contrast to the Myc/Max heterodimer, binding of the Max/Mad1 heterodimer leads to transcriptional repression.Citation17 Myc controls the transcription of up to 15% of all human genes and promotes cell growth, proliferation, angiogenesis and invasiveness and inhibits differentiation and senescenceCitation16. An overexpression of Myc is commonly found in human cancersCitation18. For instance, translocation of the MYC gene that leads to its constitutively enhanced expression is found in virtually all cases of Burkitt's lymphoma.Citation19, 20 In neuroblastoma, amplified N-Myc oncogene identifies a highly aggressive subtype that is associated with high N-Myc mRNA and protein levels Citation21.
Our study reveals a novel mechanism of transcriptional regulation of B7-H6 in tumor cells. By B7-H6 promoter analysis, we identified the proto-oncogene Myc as a functional regulator of B7-H6 expression in tumor cells enhancing NKp30-mediated tumor cell recognition by NK cells. Thus, Myc-driven tumors could be promising targets for NK cell-based cancer immunotherapy.
Results
The B7-H6 gene contains a functional binding site for the transcription factor Myc
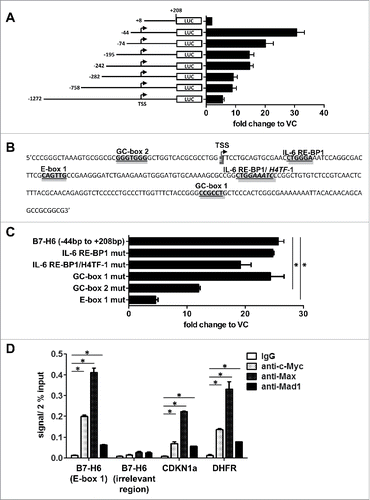
To analyze the transcriptional regulation of the B7-H6 gene the indicated fragments of the B7-H6 gene promoter were cloned from human genomic DNA by PCR. In luciferase reporter assays in HeLa cells, the transfection of sequential deletion constructs of the B7-H6 promoter resulted in the highest luciferase activity when a fragment ranging from −44 bp to +208 bp was used (). Similar results were obtained when HEK cells were transfected with these constructs (data not shown). Sequence analysis of potential transcription factor (TF) binding sites using the Transcription Element Search System (TESS: http://www.cbil.upenn.edu/cgi-bin/tess/tess) revealed two potential binding sites for Sp1 (GC-boxes), one binding site for Myc (E-box), two IL-6 responsive element-binding protein 1 (IL-6 RE-BP1) sites and one histone H4 gene transcription factor 1 (H4TF-1) binding site overlapping with one of the IL-6 RE-BP1 sites in the −44 bp to +208 bp fragment (). We prepared plasmids with specific mutations in each of these sites impairing transcription factor binding and performed luciferase reporter assays in HeLa cells. We observed a significant reduction in luciferase activity when GC-box 2 or E-box 1 was mutated (). Mutating GC-box 1, IL-6 RE-BP1 or H4TF-1 binding sites did not significantly alter luciferase activity (). Since Myc is known as a proto-oncogene overexpressed in many tumors and the reduction of luciferase activity was strongest when E-box 1 was mutated, we focused our further analysis on the role of Myc. Myc binds as a heterodimer with Max to E-box sequences and activates transcription, whereas heterodimers of Max with Mad1 repress transcription.Citation16 We used chromatin immunoprecipitation (ChIP) to determine whether c-Myc, Max or Mad1 bind to the E-box of the B7-H6 promoter in HeLa cells (). As positive controls ChIPs of CDKN1a, coding for the cell cycle inhibitor p21Citation22, and dihydrofolate reductase (DHFR)Citation23 that are c-Myc target genes were included. Of note, we detected even higher levels of binding of c-Myc and Max to the B7-H6 gene as compared to the positive controls. Binding of Mad1 was less pronounced. No binding to an irrelevant gene region of B7-H6 was observed. Moreover, we did not detect binding of c-Myc and Max to the B7-H6 gene promoter in the B7-H6-negative leukemic cell line JVM (data not shown). In conclusion, our data indicate binding of c-Myc and Max to the B7-H6 promoter.
Figure 1. Identification of functional transcription factor (TF) binding sites in the B7-H6 gene promoter. (A) Luciferase reporter assays after transfection of HeLa cells with different B7-H6 gene promoter deletion constructs are shown. HeLa cells were co-transfected with Gaussia luciferase reporter plasmids containing the different B7-H6 promoter fragments and an SV40 promoter-driven Cypridina luciferase control plasmid. 24 h after transfection, both luciferase activities were measured in the supernatant of the cells. Gaussia luciferase activity was normalized to Cypridina luciferase activity and fold change was calculated relative to VC (set as 1). (B) Scheme of the B7-H6 gene promoter fragment ranging from −44 bp to +208 bp depicting potential TF binding sites. (C) Potential TF binding sites were mutated in the B7-H6 promoter fragment ranging from −44 bp to +208 bp and transfected into HeLa cells as described in (A). (D) Binding of c-Myc, Max and Mad1 to Ebox 1 of the B7-H6 gene was analyzed by ChIP in HeLa cells. The amount of immuno-precipitated chromatin bound by either isotype control or specific antibodies was quantified by qRT-PCR. Oligos amplifying CDKN1a and DHFR were used as positive controls for Myc ChIP. Oligos for an irrelevant gene region of B7-H6 were used to control for unspecific binding of antibodies. Specific signals were set relative to signals obtained for the 2% input chromatin (set as 1). (A, C) The mean +/− SD (n = 4) from two independently performed experiments is shown. (D) The mean of duplicates +/− SD is indicated. One representative experiment out of three independently performed experiments is depicted. VC = vector control. TSS = transcription start site. *p <0.05 determined by Student's t-test.
B7-H6 expression is regulated by Myc proteins
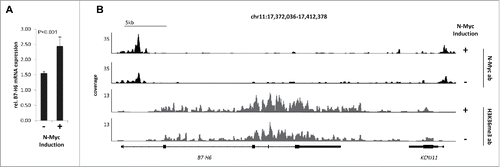
To investigate whether knock-down of c-Myc by siRNAs would affect B7-H6 expression, we transfected two different siRNAs targeting c-Myc into HeLa cells. Protein expression of c-Myc was efficiently reduced by each siRNA by itself, but both siRNAs together had the strongest effect (). 48 h after knock-down of c-Myc, B7-H6 surface expression was significantly reduced (). In parallel, surface expression of the NK cell ligand CD155 was not affected (). Of note, 48 h after knock-down of c-Myc no significant effect on cell viability as measured by the percentage of 7-AAD negative cells was observed (data not shown). Next, we analyzed, whether B7-H6 was down-regulated at the transcriptional level by qRT-PCR. Indeed, relative B7-H6 mRNA expression was strongly reduced upon siRNA mediated knock-down of c-Myc in HeLa cells compared to control treated Hela cells (). To investigate whether knock-down of c-Myc affected the stability of B7-H6 mRNA, we treated HeLa cells 24 h after c-Myc knock-down with the transcription inhibitor Actinomycin D (ActD) and relative B7-H6 expression was analyzed by qRT-PCR at different time points (). Inhibition of transcription diminished B7-H6 mRNA expression levels to a similar extent in control siRNA-treated and c-Myc siRNA-treated HeLa cells (). These data indicate that the decrease of B7-H6 mRNA levels was not due to decreased B7-H6 mRNA stability. B7-H6 cell surface and mRNA expression was also reduced after c-Myc knock-down in the pancreatic carcinoma cell line Capan-1 and the melanoma cell line A375 (). Additionally, shRNA-mediated knock-down of N-Myc in the neuroblastoma cell line IMR5-75-shMYCN led to reduction of B7-H6 protein and mRNA levels (). In these cell lines, other cell surface molecules relevant for NK cell recognition, e.g. MHC-I or CD155, were not affected (). To investigate whether Myc protein overexpression affects B7-H6 expression, we employed the N-Myc-inducible neuroblastoma cell line SY5Y-MYCN. B7-H6 mRNA expression was significantly induced upon N-Myc-induction (). Chromatin immunoprecipitation DNA-sequencing (ChIP-seq) revealed increased occupancy of the B7-H6 locus with N-Myc protein and the transcriptional elongation-associated chromatin mark H3K36me3 (). Together, our data indicate that Myc proteins regulate B7-H6 expression in tumor cells of different origin.
Figure 2. Transient knock-down of c-Mycin HeLa cells leads to reduced B7-H6 expression. HeLa cells were transiently transfected with two different c-Myc siRNAs or negative control (NC) siRNA. (A) Left: Western blot analysis of c-Myc and β-actin expression 48 h after siRNA transfection. Numbers below the blots indicate the % Myc expression relative to NC siRNA-treated cells (set as 100%). All signals were quantified relative to actin (set as 1) (B) Representative histogram overlays show cell surface expression of B7-H6 and CD155 48 h after siRNA transfection. Filled histogram: isotype control, dark gray: NC siRNA, light gray: c-Myc I+II siRNA; open solid histogram: NC siRNA-treated cells; open dashed histogram: c-Myc siRNA I+II-treated cells. Dead cells were excluded by gating on 7-AAD negative cells. (C) Cell surface expression of B7-H6 (upper panel) or CD155 (lower panel) 48 h after siRNA transfection is shown. The mean +/− SD (n = 6) from three independently performed experiments is shown. (D) Upper panel: relative B7-H6 mRNA expression to GAPDH (set as 1) was determined by qRT-PCR at the indicated time points after siRNA transfection. Lower panel: 24 h after siRNA transfection, HeLa cells were treated with Actinomycin D (ActD) and relative B7-H6 mRNA expression to GAPDH (set as 1) was measured by qRT-PCR at the indicated time points after addition of ActD. For each siRNA treatment, the values before ActD treatment were set as 100%. The mean +/− SD (n = 4) from two independently performed experiments is shown. *p <0.05 determined by Student's t-test.
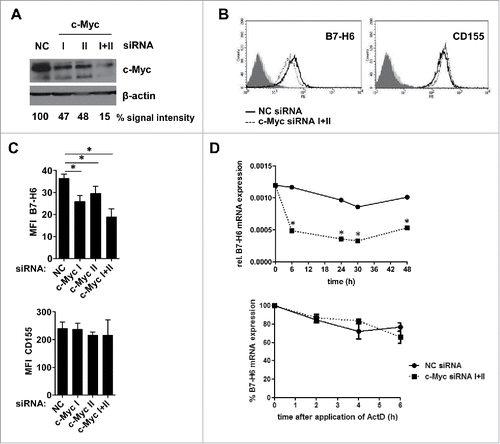
Figure 3. Transient knock-down of c-Myc or N-Myc leads to reduced B7-H6 expression in different tumor cell lines. (A-C) Capan-1 and A375 cells were transiently transfected with a 1:1 mix of two different c-Myc-specific siRNAs or negative control (NC) siRNA. (A) Western blot analysis of c-Myc and actin expression 48 h after siRNA transfection. Numbers below the blots indicate the % Myc expression relative to NC siRNA-treated cells (set as 100%). All signals were quantified relative to actin (set as 1) (B) Representative histogram overlays show cell surface expression of B7-H6 and MHC-I 48 h after siRNA transfection. Filled histogram: isotype control; open solid histogram: NC siRNA-treated cells; open dashed histogram: c-Myc siRNA-treated cells. Dead cells were excluded by gating on 7-AAD negative cells. (C) Quantification of (B). (D-F) Expression of N-Myc-specific shRNA was induced in IMR5–75-shMYCN cells by treatment with doxycyclin (dox) for 24 h. (D) Western blot analysis of N-Myc and actin expression after +/− dox treatment is depicted. Numbers below the blots indicate the % Myc expression relative to untreated cells (set as 100%). All signals were quantified relative to actin (set as 1) (E) Representative histogram overlays show cell surface expression of B7-H6 and CD155 24h after +/− dox treatment. Filled histogram: isotype control; open solid histogram: untreated cells; open dashed histogram: dox-treated cells. Dead cells were excluded by gating on 7-AAD negative cells. (F) Quantification of (E). (G) Relative B7-H6 mRNA expression to GAPDH (set as 1) was determined by qRT-PCR 24 h after siRNA transfection or +/− dox treatment. (C, F) MFI was calculated as geometrical mean of specific staining minus isotype control staining. % relative expression was calculated by setting the MFI of NC siRNA-treated cells (C) or untreated cells (F) as 100%. The mean +/− SD (n = 4) from two independently performed experiments is shown. *p <0 .05 determined by Student's t-test.
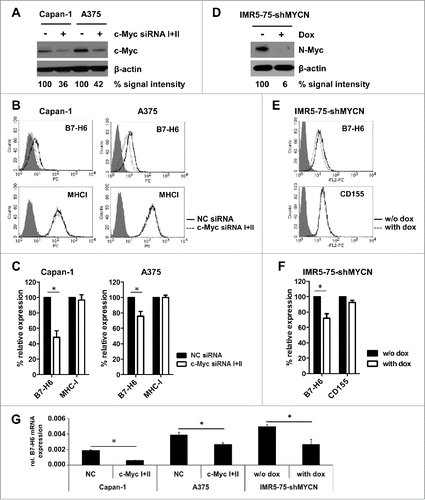
Figure 4. N-Myc overexpression induces B7-H6 mRNA transcription in neuroblastoma cells. Transcriptional activity of B7-H6 upon tetracycline-induced overexpression of N-Myc in the neuroblastoma cell line SH-SY5Y-MYCN (48 h) was monitored by (A) RNA-seq expression analysis and (B) ChIP-seq analysis of the B7-H6 locus. Coverage profiles illustrate occupancy by N-Myc and the transcriptional elongation-associated H3K36me3 mark. Profiles within the neighboring KCNJ11 locus serve as a control. N-Myc profiles were obtained using ChIP with at least 10 million cells, whereas H3K4me3 and H3K36me3 profiles were generated by ChIP with at least 1 million cells. Signals are normalized to input and an average 1x coverage.
Pharmacological inhibition of c-Myc reduces B7-H6 expression in lymphoma and leukemia cell lines
Overexpression or amplification of Myc is often found in hematological malignancies and its inactivation results in tumor regression in lymphoma and leukemia models. Thus, it is regarded as an attractive target for the development of new anti-leukemia or lymphoma therapies.Citation24 Here, we analyzed whether pharmacological inhibition of c-Myc by disrupting the Myc/Max interaction reduces B7-H6 expression in cell lines derived from Burkitt's lymphoma (Raji) or promyelocytic leukemia cells (HL-60). show that after 24h upon pharmacological inhibition of c-Myc with 100µM c-Myc inhibitor, cell surface expression of B7-H6 was reduced in both cell lines. In comparison, surface expression of other NK cell ligands was only slightly modulated. Accordingly, 8 h after c-Myc inhibition relative B7-H6 mRNA expression was diminished in both cell lines (). Titration of the c-Myc inhibitor revealed a dose dependent effect on surface B7-H6 levels after 24h in both Raji and HL-60 cells ().
Figure 5. Pharmacological inhibition of c-Myc reduces B7-H6 expression in lymphoma and leukemia cell lines. (A, B) Raji and HL-60 cells were treated for 24 h with 100 µM c-Myc inhibitor or DMSO as solvent control. (A) Representative histogram overlays show cell surface expression of B7-H6 and CD54/CD155 24 h after treatment. Filled histogram: isotype control; open solid histogram: DMSO-treated cells; open dashed histogram: c-Myc inhibitor-treated cells. Dead cells were excluded by gating on 7-AAD negative cells. (B) Quantification of (A); MFI was calculated as geometrical mean of specific staining minus isotype control staining. % relative expression was calculated by setting the MFI of DMSO-treated cells as 100%. The mean +/− SD (n = 4) from two independently performed experiments is shown. (C) Relative B7-H6 mRNA expression in the indicated tumor cell lines was quantified by qRT-PCR 8 h after c-Myc inhibition. The mean (expression of B7-H6 relative to GAPDH) +/− SD is depicted. The mean of duplicates +/− SD is indicated. One representative experiment out of at least three independently performed experiments is depicted. (D) Raji and HL-60 cells were treated for 24 h with the indicated amounts of c-Myc inhibitor or DMSO as solvent control. The mean of duplicates +/− SD is indicated. One representative experiment out of at least two independently performed experiments is depicted. *p <0.05 determined by Student's t-test
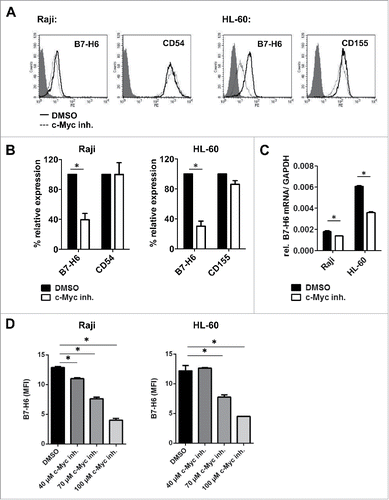
Thus, also pharmacological inhibition of c-Myc downregulates B7-H6 expression in leukemia and lymphoma cell lines.
Relative B7-H6 and Myc mRNA expression positively correlate in tumor cell lines and samples of primary tumors
Next, we asked whether c-Myc expression correlates with B7-H6 expression in different tumor cell lines and we analyzed mRNA expression of c-Myc and B7-H6 in different cell lines derived from solid tumors (n = 11), lymphomas (n = 7) and leukemias (n = 9) (). In these cell lines, relative B7-H6 and c-Myc mRNA expression significantly correlated. To address the correlation of B7-H6 and Myc expression in primary tumor tissues, we analyzed 27 hepatocellular carcinomas (HCC), 42 lymphomas and 493 neuroblastomas (). B7-H6 expression and c-Myc expression was significantly upregulated in a subset of samples of HCC compared to samples of normal liver (, left, middle panel) and relative B7-H6 and c-Myc mRNA expression significantly correlated (, right panel). In samples of lymphoma only a small subset of tumors showed elevated B7-H6 mRNA expression compared to healthy controls (). Lymphoma samples (DLBCL) from two different patients (labeled as #1, #2) showed an elevated relative B7-H6 mRNA expression compared to healthy controls. Importantly, these two samples also showed the highest relative c-Myc mRNA expression (, middle panel). Also in all tested samples of primary lymphoma, relative B7-H6 and c-Myc mRNA expression significantly correlated (, right panel). RNA-seq analysis of 493 neuroblastoma patients, of whom 92 had an N-Myc amplification, showed that patients with amplified N-Myc had also highest B7-H6 expression. Moreover, among all patients B7-H6 and N-Myc levels significantly correlated ().
Figure 6. Relative mRNA expression of c-Myc/ N-Myc and B7-H6 correlates in different tumor cell lines and samples of primary tumors. Relative B7-H6 and c-Myc/N-myc mRNA expression in tumor cell lines (A) and samples of normal and tumor tissues (B, C, D). (A–C) Expression as quantified by qRT-PCR. The mean (expression of B7-H6 relative to GAPDH and c-Myc relative to actin) +/− SEM is depicted. The statistical significance determined by Mann–Whitney test with *p <0.05 is indicated. Right panels: correlations between relative B7-H6 and c-Myc mRNA expression of samples analyzed in the left and middle panels are depicted. Mean (log10-transformed expression relative to GAPDH or β-actin) is shown for each sample. The statistical significance and r squared as determined by Pearson correlation is shown. (A) Tumor cell lines: solid tumors (n = 11), lymphoma (n = 7) and leukemia (n = 9) (B) Hepatocellular carcinoma (HCC): normal liver (n = 3) and HCC (n = 27) (C) Lymphoma: normal lymph nodes (n = 3) and spleens (n = 3), diffuse large B-cell lymphoma (DLBCL, n = 16), follicular lymphoma (FL, n = 10), mantle cell lymphoma (MCL, n = 4), marginal zone lymphoma (MZL, n = 3), lymphoplasmacytic lymphoma (LPL, n = 2) and small lymphocytic lymphoma (SLL, n = 2). #1 = sample of patient 1, #2 = sample of patient 2. (D) Neuroblastoma: patients with N-Myc amplification (black squares, n = 92), patients without N-Myc amplification (gray squares, n = 401). Relative expression as derived from RNA-seq analysis. The statistical significance and r squared as determined by Pearson correlation is shown.
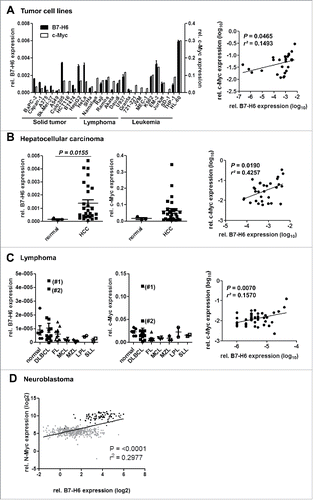
In conclusion, our data indicate that relative B7-H6 and Myc mRNA expression positively correlate in tumor cell lines and samples of primary tumors.
Decreased B7-H6 expression after c-Myc knock-down or pharmacological inhibition leads to reduced NK cell degranulation
Finally, we investigated the effect of c-Myc silencing/inhibition in tumor cells on the degranulation by NK cells. shows that c-Myc knock-down or pharmacological inhibition led to a similar reduction of staining with an anti-B7-H6 mAb or an NKp30-Fc fusion protein suggesting that B7-H6 is most likely the only NKp30 ligand on the cell surface of Raji, HeLa and A375 cells. Of note, staining with an NKG2D-Fc fusion protein was not altered upon treatment (). Importantly, and C reveal that degranulation by NK92-CI cells and primary o/n IL-2 activated NK cells was significantly reduced when co-cultured with c-Myc siRNA or c-Myc inhibitor-treated tumor cells compared to control-treated cells. Degranulation by NK92-CI cells was significantly abrogated by addition of a blocking mAb directed against NKp30. The effect of NKp30 blockade on primary NK cells was significant but less pronounced (, C).
Figure 7. Inhibition or knock-down of c-Myc in tumor cells leads to reduced NK cell degranulation via NKp30. Raji cells were treated for 24 h with 80 µM c-Myc inhibitor or DMSO as solvent control. HeLa or A375 cells were transiently transfected with a 1:1 mixture of two c-Myc-specific siRNAs or negative control (NC) siRNA. 24 h after c-Myc inhibition or 48 h after transfection expression of NK cell ligands was monitored on the cell surface of target cells and co-cultures with NK cells were set up. (A) Expression of B7-H6, NKp30- and NKG2D-ligands on the cell surface of HeLa, A375 and Raji cells after treatment. MFI was calculated as geometrical mean of specific staining minus isotype control staining. Dead cells were excluded by gating on 7-AAD negative cells. % relative expression was calculated by setting the MFI of DMSO- or NC siRNA-treated cells, respectively, as 100%. The mean +/− SD (n = 6) from three independently performed experiments is shown. (B, C) Degranulation of NK-92CI cells (B) or primary O/N IL-2-activated NK cells (C) after 4 h co-culture with treated HeLa, A375 or Raji cells was analyzed (left, middle panel). α-NKp30 mAb or isotype control were added to co-cultures at a final concentration of 10 µg/mL (right panels). Black bars: isotype control; white bars = α-NKp30 mAb (B, C) One representative experiment out of at least two independently performed experiments is shown. The mean of triplicates +/− SD is indicated. *p <0.05 determined by Student's t-test.
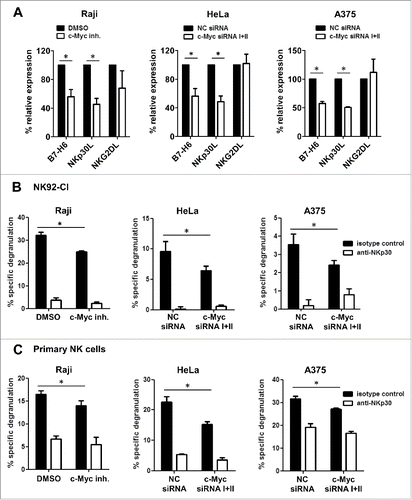
Taken together, our data indicate that c-Myc inhibition or siRNA mediated knock-down leads to reduced levels of B7-H6 in tumor cells resulting in impaired degranulation by NK cells in an NKp30-dependent manner.
Discussion
B7-H6 is the first described tumor cell surface-expressed ligand of NKp30.Citation12 B7-H6 is expressed on the cell surface of many different tumor cell lines and on certain tumor cells in situ but not on hematopoietic cells from healthy individuals or primary keratinocytes.Citation12, 15, 25 So far, mechanisms that drive the expression of B7-H6 on tumors have not been elucidated. Several studies suggested that expression of activating NK cell ligands on tumors may be directly induced by oncogenes.Citation26 For instance, in mice ligands for the activating receptor NKG2D were described to be induced by the oncogenes c-MycCitation27 or H-RasV12.Citation28 Additionally, H-RasV12Citation28, BCR-ABLCitation29, HER2/HER3Citation30 and c-MycCitation31 have been reported to up-regulate expression of human NKG2D ligands.
Here, we analyzed the transcriptional regulation of B7-H6 by cloning sequential 5′-deletion sequences of the B7-H6 gene promoter in front of a luciferase reporter gene and transfected these constructs into HeLa cells (). Unexpectedly, reduction of the length of the B7-H6 gene promoter from a (−282 bp to +208 bp) fragment to a (−44bp to +208bp) fragment resulted in increasing relative luciferase activities (). This indicates the presence of activating TF binding sites in the region from −44 bp to +208 bp and in addition the presence of potentially inhibitory transcription factor binding sites in the upstream region ranging from −282 bp to −44 bp. We identified functional activating binding sites for Sp1 and Myc in the region from −44 bp to +208 bp (). As predicted by TESS, the region from −282 bp to −44 bp contained potential TF binding sites for Sp1, AP-2α, USF1, c-Ets-2 and Myc. The role of these TFs as negative regulators of B7-H6 transcription still needs to be determined.
Using chromatin immunoprecipitation, we detected high levels of binding of c-Myc and Max to the B7-H6 gene promoter, whereas binding of Mad1 and Sp1 was less pronounced ( and data not shown). In neuroblastoma cells, N-Myc induction led to increased B7-H6 mRNA expression, which was accompanied by an elevated coverage of the B7-H6 locus with N-Myc and the transcription-associated chromatin mark H3K36me3 (). Of note, overexpression of B7-H6 in normal human dermal fibroblasts (HDFa) did not lead to an induction of B7-H6 expression (data not shown) indicating that also other factors operating in transformed cells might contribute to drive B7-H6 expression. Together, our data suggest that Myc proteins can activate transcription of the B7-H6 gene in transformed cells.
Knock-down of c-Myc by siRNAs diminished B7-H6 expression at protein and mRNA level in HeLa cells (). The downregulation of relative B7-H6 mRNA expression was rapid, visible already after 6 h, and no effect on B7-H6 mRNA stability was detected suggesting that c-Myc knock-down directly affected transcription of the B7-H6 gene. Of note, knock-down of c-Myc only partially reduced B7-H6 expression indicating that additional factors regulate B7-H6 expression. Accordingly, we observed that siRNA mediated knock-down of both c-Myc and Sp1 further, but still incompletely, diminished B7-H6 expression in HeLa cells compared to knock-down of c-Myc by itself (data not shown). We confirmed that siRNA-mediated c-Myc knock-down down-regulated B7-H6 protein and mRNA expression in additional tumor cell lines derived from solid tumors (). However, we did not succeed to efficiently transfect the lymphoma or leukemia cell lines used in with c-Myc siRNAs (data not shown).
We found a positive correlation of B7-H6 and Myc mRNA in tumor cell lines and samples of primary hepatocellular carcinoma (HCC), primary lymphoma and primary neuroblastoma (). We observed elevated relative B7-H6 mRNA expression in a subset of cDNA samples of primary hepatocellular carcinomas and in a small subset of cDNA samples of primary lymphomas compared to healthy controls (). Recently, B7-H6 was also described in metastatic neuroblastoma samples.Citation9 We confirmed B7-H6 expression in primary neuroblastoma and neuroblastoma cell lines and show a correlation of B7-H6 and N-Myc transcripts in patients (, 6D). It is intriguing that B7-H6 expressing tumors are not eradicated by NK cells. Solid tumors use multiple mechanisms to escape from NK cell immunosurveillance. Many studies demonstrate low levels of NK cell infiltration in solid tumors.Citation32, 33 Importantly, NK cells in solid tumors are frequently functionally impaired and in certain conditions soluble B7-H6 might contribute to downregulation of the NKp30 receptor. Moreover, alternatively spliced NKp30 isoforms affecting the prognosis of GISTCitation8 and neuroblastoma patientsCitation9 were described. Thus, despite expression of B7-H6, c-Myc expressing tumors might be able to escape from NK cell mediated attack. Of note, we could not detect elevated B7-H6 mRNA or protein expression in samples of primary leukemia compared to healthy PBMCs (data not shown), suggesting a high selective pressure against B7-H6-positive tumor cells in blood malignancies.
Pharmacological inhibition of c-Myc reduced B7-H6 expression in lymphoma and leukemia cell lines () resulting in reduced NKp30-dependent degranulation by NK cells (). Of note, we detected similar NKG2D ligand expression after c-Myc inhibition or knock-down in the cell lines tested (). In contrast to our study, Nanbakhsh et al.Citation31 observed down-regulation of NKG2D ligands on Cytarabine-resistant cell lines upon treatment with Myc inhibitors. In our study only non-Cytarabine-resistant cell lines were tested and it is possible that differences in the cell lines used might account for the deviating observations. Nevertheless, under certain circumstances c-Myc might drive expression of both NKG2D and NKp30-ligands alerting tumor cells for NK cell attack.
Overexpression of Myc is frequently found in different tumor entities and its inactivation results in tumor regression and differentiation.Citation24, 34 Thus, Myc is regarded as an attractive target for the development of new anti-cancer therapies.Citation24 So far, Myc inhibitors that for example disrupt the Myc/Max interaction were not successful in vivo probably due to their rapid metabolism and low concentration in tumors.Citation35 In contrast, small molecule inhibitors of the bromodomain and extra-terminal (BET) domain family of chromatin adaptors efficiently reduce c-Myc expression and delay tumor growth in vivo.Citation36, 37 BET inhibitors are currently tested in phase I and II clinical trials for the treatment of different tumor entities. Since our data show downregulation of B7-H6 expression upon Myc inhibition and reduced NK cell-mediated degranulation, patients treated with such Myc inhibitors might not benefit from combinations with NK cell-based therapies. Nevertheless, targeting B7-H6 by itself for immunotherapy may help eradicating c-Myc-driven tumors that express enhanced levels of B7-H6 on their cell surface.
Material and methods
Cells, reagents and treatments
The following cell lines were used: HeLa, A375, SkMel-37, Capan-1, Raji, HL-60 (all from American Type Culture Collection) and NK-92CI.Citation38 For modulation of N-Myc levels, stable neuroblastoma cell models were used where N-Myc can be either up- or downregulated upon addition of tetracycline [SH-SY5Y-MYCN and IMR5-75-shMYCN, respectivelyCitation39, 40. Cell lines were grown in RPMI-1640 containing 2 mM L-glutamine and 10% fetal calf serum (FCS, all from Invitrogen). NK92-CI cells were grown in alpha-minimal essential medium containing 12.5% FCS, 12.5% horse serum (Invitrogen), 2 mM L-glutamine and 50 µM beta-mercaptoethanol (both Sigma-Aldrich). NK cell isolation from healthy donors was performed as previously described.Citation25 NK cells were cultured overnight in CellGro Stem Cell Growth Medium (CellGenix) with 10% human AB serum (PAA) and 200 U/ml recombinant human interleukin 2 (IL-2, NIH). All media were supplemented with 100 U/ml Penicillin and 100 µg/ml Streptomycin (Sigma-Aldrich) and cell lines were routinely tested for contamination by the Multiplex cell Contamination Testing service at the German Cancer Research Center. Cell lines were treated as indicated with c-Myc inhibitor (Calbiochem) or DMSO (Sigma-Aldrich) as solvent control for 24 h. For inhibition of transcription, Actinomycin D (10 µg/ml, Sigma-Aldrich) was added to cell cultures 24 h after siRNA treatment.
Site-directed mutagenesis
Site-directed mutagenesis was performed using a two-stage PCR protocol as described by Wang et al.Citation41 E-box 1 was mutated by replacing CACTTG by TACTTA, GC-box 1 by replacing CCGCCT by CCGTTT, GC-box 2 by replacing GGGTGGG by GTTTGGG and IL-6 RE-BP1 sites as described by Hocke et al.Citation42 Disruption of sites was confirmed by analyzing the mutated sequences in silico with TESS. Sequences of oligonucleotide primers are depicted in Table S1.
Luciferase reporter assay
B7-H6 reporter constructs were generated by cloning the indicated fragments of the B7-H6 5′-untranslated region (UTR, NCBI Reference Sequence NM_001202439.1) into the pGLuc-basic vector (New England Biolabs). Cells were co-transfected in 96-well plates with the B7-H6 reporter construct or an empty vector control (50 ng each) and the pSV40-Cluc vector (10 ng, New England Biolabs) using Lipofectamine 2000 (Invitrogen). Luciferase activity was measured 24 h after transfection in cell culture supernatants using an Orion L luminometer (Titertek Berthold). Normalization was performed by dividing the relative light unit (RLU) values obtained after transfection with the pGLuc-basic vector with the RLU values obtained with the pSV40-Cluc vector.
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) according to the manufacturer's instructions. Chromatin was isolated from HeLa cells and 10 µg was used for IP. Chromatin was immunoprecipitated with 1 µg rabbit anti-c-Myc (N-262), 0.5 µg rabbit anti-Max (C-17), 1 µg rabbit anti-Mad1 (C-19) (all from Santa Cruz) or polyclonal rabbit IgG as negative control (Cell Signaling Technology). The precipitated chromatin was analyzed by qRT-PCR using primers that amplify a 100 bp region including the E-box 1 (−24 to +76 bp) in the B7-H6 gene. As a control for unspecific binding of the antibodies an irrelevant region of the B7-H6 gene was amplified. Oligonucleotide sequences were as follows: B7-H6 (including E-box 1) (5′-GGGTGGGGCTGGTCAC-3′/5′-TTGCACATCCCACTTCTTCA-3′) and B7-H6 (irrelevant region) (5′-GACCTGGAGCCATTGTGTCT-3′ / 5′-AAGCTGGACTGTTCCCTGTG-3′).
ChIP-seq and deep mRNA-sequencing (mRNA-seq)
ChIP-seq methodology and mRNA-seq in N-Myc-modulated cell lines SH-SY5Y-MYCN and IMR5-75-shMYCN were as previously describedCitation43, 44. B7-H6 and N-Myc expression profiles in primary neuroblastoma were derived from global RNA-seq analyses of 498 neuroblastoma patients.Citation45
Flow cytometry
Stainings for flow cytometry were performed as described previouslyCitation13. Adherent cells were harvested using PBS with 5% EDTA (Sigma-Aldrich). Cells were acquired by gating on 7-AAD− cells to exclude dead cells. Flow cytometry was performed using a FACS Calibur® and analyzed by CellQuest Pro® software (both BD Biosciences, San Jose, CA, USA). For quantification, the geometrical mean (GeoMean) of the isotype control staining was subtracted from the GeoMean of the specific staining.
Western blot
Whole cell lysate preparation, SDS-PAGE and Western Blotting was performed as previously describedCitation25. Polyclonal rabbit antibodies specific for c-Myc or N-Myc (both Cell Signaling Technology, Danvers, MA, USA) were added at a 1:1000 final dilution in PBS/ 0.1% Tween 20/ 5% BSA for an overnight incubation. Equal loading was controlled by reprobing the membranes with a mAb specific for actin (clone C4, MP Biomedicals, Solon, Ohio, USA) at a final dilution of 1:10000 in PBS/ 0.1% Tween 20/ 5% milk for 1 h at room temperature. Western Blot signals were quantified using ImageJ (rsbweb.nih.gov/ij/).
Quantitative real-time PCR (qRT-PCR)
Total RNA isolation, reverse transcription and SYBR Green qRT-PCR was performed as described previously.Citation25 cDNA from hepatocellular carcinoma (HCC) samples and normal livers was obtained from K. Breuhahn (University Hospital Heidelberg, Germany). All HCC samples were surgically resected at the University Hospital of Heidelberg and histologically classified according to established criteria by an experienced pathologist (N.W.). The study was approved by the institutional ethics committee (application no. 206/05) of the Medical Faculty of Heidelberg University. The TissueScan™ RT Lymphoma panel II (LYRT302) was purchased from OriGene. Oligonucleotide sequences were as follows: B7-H6 (5′-GACCTGGAGCCATTGTGTCT-3′ / 5′-AAGCTGGACTGTTCCCTGTG-3′), c-Myc (5′-TCAAGAGGCGAACACCAAC-3′ / 5′-GGCCTTTTCATTGTTTTCCA-3′), GAPDH (5′- GCAAATTCCATGGCACCGT-3′ / 5′ TCGCCCCACTTGATTTTGG-3′) and β-Actin (5′-AGAAAATCTGGCACCACACC-3′/ 5′- AGAGGCGTACAGGGATAGCA-3′).
siRNA mediated knock-down experiments
For transient knockdown of c-Myc small interfering RNA (siRNA) was used. SignalSilence® c-Myc siRNA I/ II and unconjugated SignalSilence® Control siRNA were purchased from Cell Signaling Technology and used at a final concentration of 100 nM. Cells were transfected using HiPerfect Transfection Reagent (Qiagen) according to the manufacturer's instructions.
NK cell degranulation assay
Target cells were pre-treated with c-Myc/control siRNAs or with c-Myc inhibitor/DMSO solvent control and washed twice before co-culture with effector cells. NK-92CI or primary NK cells isolated from buffy coats activated overnight with IL-2 were used as effectors after blocking of NKp30 with a final concentration of 10 µg/ml mouse anti-NKp30 (clone 210845, R&D systems) or addition of an isotype control mIgG2a (R&D systems). Targets and effectors were incubated at a 1:1 ratio as previously described.Citation25 The effectors were analyzed for expression of CD107a by flow cytometry by gating on CD56+ CD3- 7-AAD- cells. The % specific degranulation was calculated by subtracting the % CD107a+ NK cells in samples with NK cells by themselves from the % CD107a+ NK cells cultured with targets.
Statistical analysis
Statistical analyses were performed by calculating the mean +/− SD. Unless stated otherwise, statistical significances were determined using a two-tailed Student's t-test. Correlation analysis was performed using two-tailed Pearson correlation of log10- or log-2 transformed values.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1116674_s02.xlsx
Download MS Excel (10.1 KB)Acknowledgments
The authors thank collaboration partners at Bayer Health Care. Tissue samples were provided by the tissue bank of the National Center of Tumor Diseases (NCT, Heidelberg, Germany) in accordance with the regulations of the tissue bank and the approval of the ethics committee of the Medical Faculty of Heidelberg University.
Funding
This work was supported by the Deutsche José Carreras Leukämie-Stiftung e.V. (A. Cerwenka; R11/06) and Israel's Ministry of Science and Technology (MOST; to A. Cerwenka).
References
- Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol 2001; 1:41-9; PMID:11905813; http://dx.doi.org/10.1038/35095564
- Kaifu T, Escaliere B, Gastinel LN, Vivier E, Baratin M. B7-H6/NKp30 interaction: a mechanism of alerting NK cells against tumors. Cell Mol Life Sci 2011; 68:3531-9; PMID:21877119; http://dx.doi.org/10.1007/s00018-011-0802-7
- Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol 2009; 9:568-80; PMID:19629084; http://dx.doi.org/10.1038/nri2604
- Byrd A, Hoffmann SC, Jarahian M, Momburg F, Watzl C. Expression analysis of the ligands for the Natural Killer cell receptors NKp30 and NKp44. PLoS One 2007; 2:e1339; PMID:18092004; http://dx.doi.org/10.1371/journal.pone.0001339
- Pende D, Parolini S, Pessino A, Sivori S, Augugliaro R, Morelli L, Marcenaro E, Accame L, Malaspina A, Biassoni R et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med 1999; 190:1505-16; PMID:10562324; http://dx.doi.org/10.1084/jem.190.10.1505
- Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Münz C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med 2002; 195:343-51; PMID:11828009; http://dx.doi.org/10.1084/jem.20011149
- Vitale M, Della Chiesa M, Carlomagno S, Pende D, Aric∫ M, Moretta L, Moretta A. NK-dependent DC maturation is mediated by TNFalpha and IFNgamma released upon engagement of the NKp30 triggering receptor. Blood 2005; 106:566-71; PMID:15784725; http://dx.doi.org/10.1182/blood-2004-10-4035
- Delahaye NF, Rusakiewicz S, Martins I, Ménard C, Roux S, Lyonnet L, Paul P, Sarabi M, Chaput N, Semeraro M, Minard-Colin V et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med 2011; 17:700-7; PMID:21552268; http://dx.doi.org/10.1038/nm.2366
- Semeraro M, Rusakiewicz S, Minard-Colin V, Delahaye NF, Enot D, Vély F, Marabelle A, Papoular B, Piperoglou C, Ponzoni M et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci Transl Med 2015; 7:283ra55; PMID:25877893; http://dx.doi.org/10.1126/scitranslmed.aaa2327
- Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, Gazit R, Gonen-Gross T, Hanna J, Nahari E et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol 2005; 6:515-23; PMID:15821739; http://dx.doi.org/10.1038/ni1190
- Pogge von Strandmann, E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, Rothe A, Böll B, Simhadri VL, Borchmann P et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007; 27:965-74; PMID:18055229; http://dx.doi.org/10.1016/j.immuni.2007.10.010
- Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med 2009; 206:1495-503; PMID:19528259; http://dx.doi.org/10.1084/jem.20090681
- Fiegler N, Textor S, Arnold A, Rölle A, Oehme I, Breuhahn K, Moldenhauer G, Witzens-Harig M, Cerwenka A. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood 2013; 122:684-93; PMID:23801635; http://dx.doi.org/10.1182/blood-2013-02-482513
- Matta J, Baratin M, Chiche L, Forel JM, Cognet C, Thomas G, Farnarier C, Piperoglou C, Papazian L, Chaussabel D et al. Induction of B7-H6, a ligand for the Natural Killer cell activating receptor NKp30, in inflammatory conditions. Blood 122:394-404; PMID:23687088; http://dx.doi.org/10.1182/blood-2013-01-481705
- Schlecker, E, Fiegler N, Arnold A, Altevogt P, Rose-John S, Moldenhauer G, Sucker A, Paschen A, von Strandmann EP, Textor S et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res 2014; 74:3429-40; PMID:24780758; http://dx.doi.org/10.1158/0008-5472.CAN-13-3017
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 2005; 6:635-45; PMID:16064138; http://dx.doi.org/10.1038/nrm1703
- Lüscher B. MAD1 and its life as a MYC antagonist: An update. Euro J Cell Biol 2012; 91:506-514; PMID:21917351; http://dx.doi.org/10.1016/j.ejcb.2011.07.005
- Albihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res 2010; 107:163-224; PMID:20399964; http://dx.doi.org/10.1016/S0065-230X(10)07006-5
- Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A 1982; 79:7824-7; PMID:6961453; http://dx.doi.org/10.1073/pnas.79.24.7824
- Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A 1982; 79:7837-41; PMID:6818551; http://dx.doi.org/10.1073/pnas.79.24.7837
- Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 1985; 313:1111-6; PMID:4047115; http://dx.doi.org/10.1056/NEJM198510313131802
- Wong PP, Miranda F, Chan KV, Berlato C, Hurst HC, Scibetta AG. Histone demethylase KDM5B collaborates with TFAP2C and Myc to repress the cell cycle inhibitor p21(cip) (CDKN1A). Mol Cell Biol 2012; 32:1633-44; PMID:22371483; http://dx.doi.org/10.1128/MCB.06373-11
- Boyd KE, Wells J, Gutman J, Bartley SM, Farnham PJ. c-Myc target gene specificity is determined by a post-DNAbinding mechanism. Proc Natl Acad Sci U S A 1998; 95:13887-92; PMID:9811896; http://dx.doi.org/10.1073/pnas.95.23.13887
- Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol 2006; 16:318-30; PMID:16934487; http://dx.doi.org/10.1016/j.semcancer.2006.07.015
- Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res 71, 5998-6009; PMID:21764762; http://dx.doi.org/10.1158/0008-5472.CAN-10-3211
- Huergo-Zapico L, Acebes-Huerta A, López-Soto A, Villa-Álvarez M, Gonzalez-Rodriguez AP, Gonzalez S. Molecular Bases for the Regulation of NKG2D Ligands in Cancer. Front Immunol 2014; 5:106; PMID:24711808; http://dx.doi.org/10.3389/fimmu.2014.00106
- Unni AM, Bondar T, Medzhitov R. Intrinsic sensor of oncogenic transformation induces a signal for innate immunosurveillance. Proc Natl Acad Sci U S A 2008; 105, 1686-91; PMID:18223157; http://dx.doi.org/10.1073/pnas.0701675105
- Liu XV, Ho SS, Tan JJ, Kamran N, Gasser S. Ras activation induces expression of Raet1 family NK receptor ligands. J Immunol 2012; 189:1826-34; PMID:22798674; http://dx.doi.org/10.4049/jimmunol.1200965
- Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, Rousselot P, Tamouza R, Le Bouteiller P, Mahon FX et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol 2006; 176:5108-16; PMID:16585609; http://dx.doi.org/10.4049/jimmunol.176.8.5108
- Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, Seliger B, Lundqvist A, Mimura K, Kiessling R. HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J Immunol 2012; 188:2136-45; PMID:22301547; http://dx.doi.org/10.4049/jimmunol.1102237
- Nanbakhsh A, Pochon C, Mallavialle A, Amsellem S, Bourhis JH, Chouaib S. c-Myc regulates expression of NKG2D ligands ULBP1/2/3 in AML and modulates their susceptibility to NK-mediated lysis. Blood 2014; 123:3585-95; PMID:24677544; http://dx.doi.org/10.1182/blood-2013-11-536219
- Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov 2014; 4:522-6; PMID:24795012; http://dx.doi.org/10.1158/2159-8290.CD-13-0985
- Sconocchia G, Eppenberger S, Spagnoli GC, Tornillo L, Droeser R, Caratelli S, Ferrelli F, Coppola A, Arriga R, Lauro D et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology 2014; 3:e952197; PMID:25610741; http://dx.doi.org/10.4161/21624011.2014.952197
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999; 4:199-207; PMID:10488335; http://dx.doi.org/10.1016/S1097-2765(00)80367-6
- Guo J, Parise RA, Joseph E, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol 2009; 63:615-25; PMID:18509642; http://dx.doi.org/10.1007/s00280-008-0774-y
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146:904-17; PMID:21889194; http://dx.doi.org/10.1016/j.cell.2011.08.017
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 2011; 108:16669-74; PMID:21949397; http://dx.doi.org/10.1073/pnas.1108190108
- Tam YK, Miyagawa B, Ho VC, Klingemann HG. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J Hematother 1999; 8:281-90; PMID:10417052; http://dx.doi.org/10.1089/106161299320316
- Dreidax D, Bannert S, Henrich KO, Schröder C, Bender S, Oakes CC, Lindner S, Schulte JH, Duffy D, Schwarzl T et al. p19-INK4d inhibits neuroblastoma cell growth, induces differentiation and is hypermethylated and downregulated in MYCN-amplified neuroblastomas. Hum Mol Genet 2014; 23:6826-37; PMID:25104850; http://dx.doi.org/10.1093/hmg/ddu406
- Muth D, Ghazaryan S, Eckerle I, Beckett E, Pöhler C, Batzler J, Beisel C, Gogolin S, Fischer M, Henrich KO et al. Transcriptional repression of SKP2 is impaired in MYCN-amplified neuroblastoma. Cancer Res 2010; 70:3791-802; PMID:20424123; http://dx.doi.org/10.1158/0008-5472.CAN-09-1245
- Wang W, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 1999; 26:680-2; PMID:10343905
- Hocke GM, Cui MZ, Ripperger JA, Fey GH. Regulation of the rat alpha 2 macroglobulin gene by interleukin 6 and leukemia inhibitory factor. Folia Histochem Cytobiol 1992; 30:137-9; PMID:1284662
- Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Krämer A, Roncaioli JL, Sand F, Heuckmann JM et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015; 526:700-4; PMID:26466568; http://dx.doi.org/10.1038/nature14980
- Duffy DJ, Krstic A, Schwarzl T, Higgins DG, Kolch W. GSK3 inhibitors regulate MYCN mRNA levels and reduce neuroblastoma cell viability through multiple mechanisms, including p53 and Wnt signaling. Mol Cancer Ther 2014; 13:454-67; PMID:24282277; http://dx.doi.org/10.1158/1535-7163.MCT-13-0560-T
- Zhang W, Yu Y, Hertwig F, Thierry-Mieg J, Zhang W, Thierry-Mieg D, Wang J, Furlanello C, Devanarayan V, Cheng J et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol 2015; 16:133; PMID:26109056; http://dx.doi.org/10.1186/s13059-015-0694-1