
ABSTRACT
Regulatory T cell (Treg) promote IL-17-mediated colon tumorigenesis in multiple intestinal neoplasia (Min) mice colonized with enterotoxigenic Bacteroides fragilis (ETBF). Although they retain their immunosuppressive function, mucosal Treg are instrumental to initiate ETBF-triggered IL-17 colitis by limiting the availability of IL-2 in the local microenvironment. However, the mechanisms that trigger the recruitment of the pro-carcinogenic Treg in ETBF-colonized colon remained to be elucidated.
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths worldwide. Though most cases of CRC are the result of spontaneous mutagenesis, a fraction of CRC cases are epidemiologically linked to chronic inflammation. Indeed, the risk of developing CRC is significantly increased in patients with either ulcerative colitis or Crohn's disease. These inflammatory bowel diseases (IBD) are associated with a disturbance of the intestinal microbiota, or dysbiosis, so identifying possible microbial causes of IBD and CRC is of great public interest.
Our lab previously uncovered the carcinogenic potential of one such microbe, ETBF, a virulent toxin-producing bacterium that profoundly disrupts the colonic microenvironment. Colonization of ApcMin mice with ETBF rapidly promoted colon tumorigenesis that was dependent upon a robust IL-17-dominated immune response to ETBF.Citation1 Because Tregs have been shown to induce tumor regression by suppressing the inflammatory environment in inflammation-associated tumorigenesis,Citation2 we set out to characterize the role of Tregs in the immune response to ETBF, published recently in Cancer Discovery.Citation3
Quickly after initial colonization with ETBF, Tregs accumulated in the colonic lamina propria. In contrast to our expectations, these microbial-triggered Tregs maintained their suppressive function, as illustrated by significantly increased colonic inflammation in their absence. Interestingly, whereas depletion of Tregs triggered a fulminant colitis, early ETBF-induced tumorigenesis was dramatically reduced in the absence of Tregs, highlighting a marked dissociation between inflammation and tumorigenesis. Upon characterizing the inflammatory response to ETBF, we found it shifted from IL-17-dominated in the presence of Tregs to robustly IFNγ-dominated in the absence of Tregs. We further showed that reduced tumorigenesis in the absence of Tregs was not dependent on the potent antitumor cytokine IFNγ. Elucidating the mechanisms underlying Treg-dependent ETBF tumorigenesis, we established that Tregs in the ETBF-induced inflammatory environment limit excess IL-2 that commonly inhibits IL-17 production. Thus, because IL-17-mediated inflammation was pro-tumorigenic and overt IFNγ was not, the importance of the quality of inflammation over its quantity was underscored.
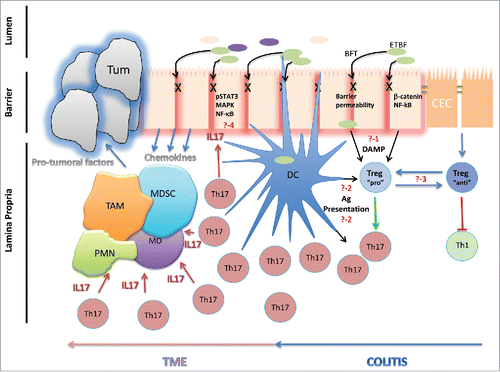
Although we unveiled a novel mechanism whereby microbial-triggered Tregs drive carcinogenic immunity in the colon, several uncertainties remain. First, how does ETBF promote Treg accumulation in the colonic lamina propria (?-1 in )? It is possible, and perhaps likely, that B. fragilis toxin (BFT)-stimulated epithelial cells relay, as yet undetermined, messages that influence lamina propria-resident T cells. The very positioning of colonic epithelium at the interface between the immune-dense lamina propria and microbiota-rich lumen makes these cells the perfect sentinel to relay such messages (e.g., production of IL-33 upon epithelial assault).Citation4 Alternatively, the disruption of the intestinal barrier during ETBF-triggered colitis may allow the direct stimulation of Tregs by ETBF via pattern recognition receptors (e.g., Toll-like receptors).Citation5 Second, what is the antigenic specificity of these Treg and Th17 cells (?-2 in )? Cognate antigen-T cell receptor (TCR) stimulation in the context of TGF-β and IL-2 can induce Treg differentiation and clonal expansion in situ; nonetheless, while direct interactions between ETBF (or BFT) and colonic epithelial cells have been identified,Citation6 no B. fragilis-specific TCRs have been isolated to date. Third, does the immune-suppressive capacity of Tregs that accumulate in response to pathogens toward, which Th17 immunity is most effective, differ from that of Tregs that accumulate during immune responses to pathogens best targeted by Th1 immunity (?-3 in )? That is, perhaps timing of Treg accumulation in anti-viral immunity is delayed so that Th1 responses prevail, but it is possible that the very nature of those Tregs differs from that of Tregs responding to ETBF. For example, some investigators have demonstrated that RORγt+ IL-17-producing Tregs, certainly more prevalent in the storm of Th17 immune responses, are themselves carcinogenic.Citation7 Furthermore, transcription factors (i.e., Gata-3, Tbet or Stat3) expressed by Treg are critical for the suppression of various subsets of T cell subsets (i.e., Th2, Th1 and Th17, respectively). Despite the broadly interesting perspective, it is yet unclear whether Tregs arising from two distinct sets of inflammatory conditions differ in any meaningful way or merely represent a fluid continuum of Treg plasticity that is appropriate for any particular condition. Fourth, how is IL-17 pro-carcinogenic (?-4 in )? While IL-17 is a potent recruiter of neutrophils, which have their own carcinogenic potential, our own [unpublished] investigations have failed to elucidate a prominent role for reactive oxygen and nitrogen species in ETBF-induced tumorigenesis. We are currently studying how ETBF-induced IL-17 directly impacts the colonic epithelium, and other investigators have demonstrated the carcinogenic potential of IL-17, independent of ETBF, directly on the colonic epithelium.Citation8 Thus, it is reasonable to propose a positive-feedback loop, whereby BFT triggers the release of pro-inflammatory Treg/Th17-driving signals by the colonic epithelium that in turn trigger additional pro-inflammatory and survival pathways in the epithelium, ultimately leading to their cancerous transformation.
Figure 1. How do colitogenic Tregs promote microbial-induced colon tumorigenesis? ETBF-derived enterotoxin (B.fragilis toxin or BFT) induced increased barrier permeability via cleavage of E-cadherin of the zonula adherens. The cleavage of E-Cadherin is also responsible for the activation of the β-catenin and Nf-kB signaling pathways in colonic epithelial cells (CEC). The mechanisms by which Treg are recruited and activated in the colon of ETBF colonized mice remained however unclear. Question 1 (?-1): are there damage-associated molecular pattern (DAMP) released by the BFT altered CEC that drives the preferential trafficking of Treg to the distal colon, the preferential site of colon tumorigenesis? Question 2 (?-2): Are the Treg and/or Th17 cells activated via a TCR dependent signal following presentation of ETBF-derived Ag by mucosal dendritic cells (DC)? Question 3 (?-3): Are they two functionally distinct types of Treg which differentially modulate the Th1 and Th17 in accordance to the inflammatory microenvironment? The depletion of Treg in ETBF-colonized Min mice triggered a strong type 1 inflammation, which reinforced that, although they promote Th17 following ETBF colitis, they kept their suppressive activity by restraining Th1 cells. Question 4 (?-4): how does IL-17 promote ETBF-triggered colon carcinogenesis? Myeloid cells and epithelial cells are targets of IL-17 but the mechanisms that sustain the carcinogenic properties of IL-17 remain fairly unknown. IL-17 can promote NF-kB activation in epithelial cells which in return produce chemokines recruiting protumoral myeloid cells including tumor-infiltrating macrophages (TAM) or myeloid derived suppressor cells (MDSC) in the tumor microenvironment (TME). Alternatively, IL-17 can directly activate myeloid cells and drives their local differentiation.
These questions are merely the tip of the iceberg with respect to Tregs, inflammation and microbial cancer biology, and it is without a doubt that the response to these questions will profoundly disarticulate the dogma of Tregs as purely guardians of mucosal immune homeostasis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009; 15(9):1016-22; PMID:19701202; http://dx.doi.org/10.1038/nm.2015
- Erdman SE, Rao VP, Olipitz W, Taylor CL, Jackson EA, Levkovich T, Lee CW, Horwitz BH, Fox JG, Ge Z et al. Unifying roles for regulatory T cells and inflammation in cancer. Int J Cancer 2010; 126(7):1651-65; PMID:19795459; http://dx.doi.org/10.1002/ijc.24923
- Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe JL, Sears CL, Pardoll DM, Housseau F. Regulatory T-cell response to enterotoxigenic bacteroides fragilis colonization triggers IL17-dependent colon carcinogenesis. Cancer Discov 2015; 5(10):1098-109; PMID:26201900; http://dx.doi.org/10.1158/2159-8290.CD-15-0447
- Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014; 513(7519):564-8; PMID:25043027; http://dx.doi.org/10.1038/nature13577
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011; 332(6032):974-7; PMID:21512004; http://dx.doi.org/10.1126/science.1206095
- Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest 2014; 124(10):4166-72; PMID:25105360; http://dx.doi.org/10.1172/JCI72334
- Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, Ham S, Sandall BP, Khan MW, Mahvi DM et al. Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci Transl Med 2012; 4(164):164ra159; PMID:23241743; http://dx.doi.org/10.1126/scitranslmed.3004566
- Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, Zhang W, Zhong Z, Sanchez-Lopez E, Wu LW et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014; 41(6):1052-63; PMID:25526314; http://dx.doi.org/10.1016/j.immuni.2014.11.009