
ABSTRACT
Epidermal growth factor receptor (EGFR) and its mutant form EGFRvIII are overexpressed in a large proportion of glioblastomas (GBM). Immunotherapy with an EGFRvIII-specific vaccine has shown efficacy against GBM in clinical studies. However, immune escape by antigen-loss variants and lack of control of EGFR wild-type positive clones limit the usefulness of this approach. Chimeric antigen receptor (CAR)-engineered natural killer (NK) cells may represent an alternative immunotherapeutic strategy. For targeting to GBM, we generated variants of the clinically applicable human NK cell line NK-92 that express CARs carrying a composite CD28-CD3ζ domain for signaling, and scFv antibody fragments for cell binding either recognizing EGFR, EGFRvIII, or an epitope common to both antigens. In vitro analysis revealed high and specific cytotoxicity of EGFR-targeted NK-92 against established and primary human GBM cells, which was dependent on EGFR expression and CAR signaling. EGFRvIII-targeted NK-92 only lysed EGFRvIII-positive GBM cells, while dual-specific NK cells expressing a cetuximab-based CAR were active against both types of tumor cells. In immunodeficient mice carrying intracranial GBM xenografts either expressing EGFR, EGFRvIII or both receptors, local treatment with dual-specific NK cells was superior to treatment with the corresponding monospecific CAR NK cells. This resulted in a marked extension of survival without inducing rapid immune escape as observed upon therapy with monospecific effectors. Our results demonstrate that dual targeting of CAR NK cells reduces the risk of immune escape and suggest that EGFR/EGFRvIII-targeted dual-specific CAR NK cells may have potential for adoptive immunotherapy of glioblastoma.
Abbreviations
| CAR | = | chimeric antigen receptor |
| EGFR | = | epidermal growth factor receptor |
| EGFRvIII | = | EGFR variant III |
| GBM | = | glioblastoma |
| NSG | = | NOD-SCID IL2R γnull. |
Introduction
Glioblastoma is the most common primary malignant brain tumor in adults and currently incurable. Standard therapy includes maximal safe resection followed by radiotherapy and chemotherapy with temozolomide. Nevertheless, median survival remains at approximately 15 mo.Citation1,2 Recurrence is almost universally observed, emphasizing the need for more effective treatments. EGFR gene amplification and EGFR overexpression can be detected in 40 to 60% of GBM tumors,Citation3 while the receptor is not or only minimally expressed in normal brain tissue.Citation4 However, despite sporadic evidence for clinical activity, GBM therapy with EGFR-targeted agents like monoclonal antibody cetuximab and tyrosine kinase inhibitors has largely failed.Citation5,6 In contrast, specific immunotherapy does not rely on signaling inhibition and may bypass respective resistance mechanisms.Citation5,7 GBM cells with EGFR gene amplification often co-express the EGFR mutant form EGFRvIII, which drives tumorigenicity and mediates radio- and chemoresistance.Citation8,9 EGFRvIII harbors an in-frame deletion of exons 2 to 7 of the wild-type EGFR gene, generating a neo-epitope at the N-terminus of the receptor. Hence, EGFRvIII can be targeted by specific immunotherapy such as the peptide vaccine rindopepimut, which resulted in a survival benefit for GBM patients.Citation10 However, at recurrence the majority of patients' tumors had lost EGFRvIII expression, indicating strong immune-mediated selection and immune escape. This may also limit clinical success of adoptive therapy with T or NK cells genetically engineered to express an EGFRvIII-specific CAR which demonstrated antitumor activity in preclinical models.Citation11,12
To study the consequences of CAR cell therapy of glioblastoma on distinct tumor cell subpopulations, we developed GBM models characterized by expression of varying levels of EGFR with or without concurrent EGFRvIII expression. As effector cells, we generated variants of the continuously expanding human NK cell line NK-92 genetically engineered to express CARs that recognize epitopes unique to EGFR or EGFRvIII, or an EGFR domain present in both target receptors. Phase I studies in cancer patients demonstrated safety and clinical activity of unmodified NK-92 cells.Citation13-15 Likewise, CAR-engineered NK-92 cells targeting the EGFR-related tumor-associated antigen ErbB2 (HER2) are under development for clinical applications.Citation16 Here, we investigated in vitro antitumor activity of EGFR- and EGFRvIII-targeted NK cells against established and primary human GBM cells, and dependence of cell killing on CAR signaling and expression of the respective target receptors. For analysis of in vivo activity of mono- and dual-specific CAR NK cells and treatment-induced selection of tumor cell subpopulations, we employed NOD-SCID IL2R γnull mice carrying intracranial GBM xenografts either expressing EGFR or EGFRvIII, or mixed tumors consisting of EGFR-expressing GBM cells, and cells co-expressing EGFR and EGFRvIII.
Results
Generation of CAR NK cells targeting EGFR and EGFRvIII
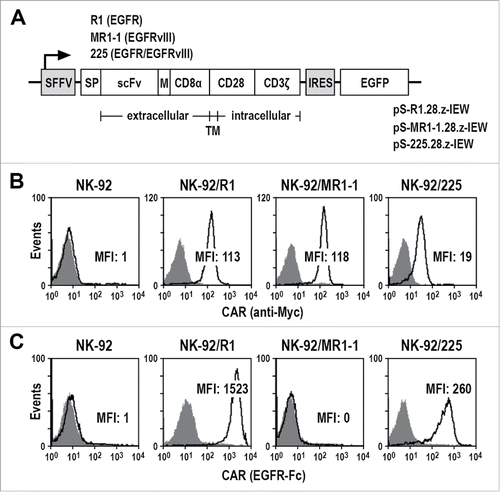
CARs were constructed that contain an immunoglobulin heavy chain signal peptide, scFv(R1), scFv(MR1-1) or scFv(225) antibody fragments which recognize epitopes exclusive for EGFR or EGFRvIII, or an epitope common to both receptors,Citation17-19 a Myc-tag, an optimized CD8α hinge region,Citation16 the CD28 transmembrane and intracellular domains, and the CD3ζ intracellular domain (). Corresponding truncated CARs that lack intracellular signaling domains served as controls (Fig. S1A). Upon transduction of human NK-92 cells with lentiviral CAR vectors, single cell clones displaying high and stable CAR expression were selected ( and Fig. S1B). As expected, EGFR-specific NK-92/R1.28.z (NK-92/R1) and EGFR/EGFRvIII dual-specific NK-92/225.28.z (NK-92/225) cells bound recombinant EGFR-Fc protein, while EGFRvIII-specific NK-92/MR1-1.28.z (NK-92/MR1-1) did not (). Similar results were obtained with NK cells expressing signaling-incompetent CARs (Fig. S1C).
Figure 1. Generation of CAR NK cells. (A) Lentiviral transfer plasmids pS-R1.28.z-IEW, pS-MR1-1.28.z-IEW and pS-225.28.z-IEW encoding under control of the Spleen Focus Forming Virus promoter (SFFV) CARs consisting of an immunoglobulin heavy chain signal peptide (SP), scFv fragments derived from EGFR-specific antibody R1, EGFRvIII-specific MR1-1, or 225 recognizing EGFR and EGFRvIII, followed by a Myc-tag (M), CD8α hinge region (CD8α), transmembrane and intracellular domains of CD28, and the intracellular domain of CD3ζ. Enhanced green fluorescent protein (EGFP) cDNA separated from the CAR sequence by an internal ribosome entry site (IRES) served as a marker. (B) CAR surface expression on NK-92/R1, NK-92/MR1-1 and NK-92/225 single cell clones was determined by flow cytometry with Myc-tag-specific antibody (open areas). Isotype antibody (filled areas) and parental NK-92 cells served as controls. (C) Binding of recombinant EGFR-Fc protein to the surface of CAR NK cells was measured by flow cytometry (open areas). CAR NK cells only treated with secondary antibody (filled areas) and parental NK-92 cells served as controls. MFI: mean fluorescence intensity (geometric mean).
Cytotoxicity of CAR NK cells against established and primary glioblastoma cells
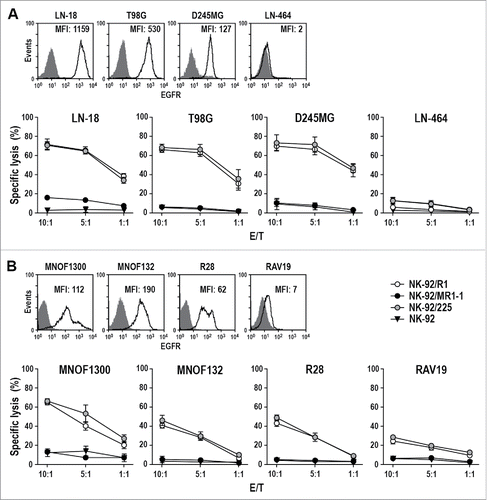
Antitumor activity of the CAR NK cells was first assessed using established human GBM cell lines (). After two hours of co-incubation, NK-92/R1 and NK-92/225 cells efficiently and selectively killed EGFR-positive LN-18, T98G and D245MG cells, which do not express EGFRvIII (Fig. S2A) and were resistant to EGFRvIII-specific NK-92/MR1-1 and untargeted NK-92. None of the NK cell lines showed activity against EGFR-negative LN-464 cells, demonstrating that cell killing was dependent on specific recognition of the target antigen and CAR activation. Accordingly, NK-92/R1.TM and NK-92/225.TM cells expressing signaling-incompetent CARs failed to lyse EGFR-positive targets while retaining CAR-independent natural cytotoxicity toward K562 cells (Fig. S3). Next, we analyzed primary GBM stem cell cultures kept under conditions which maintain most of the characteristics of the original tumors.Citation20 MNOF1300, MNOF132 and R28 cells displayed elevated EGFR levels while being negative for EGFRvIII (Fig. S2B), and similar to established GBM cells were sensitive to NK-92/R1 and NK-92/225 cytotoxicity while showing resistance toward EGFRvIII-specific NK-92/MR1-1 and parental NK-92 (). In concordance with their much lower EGFR expression level, RAV19 primary GBM cells displayed only moderate sensitivity to NK-92/R1 and NK-92/225 cells after 2 h of co-incubation. Nevertheless, upon extended exposure for 16 h EGFR-specific NK-92/R1 and dual-specific NK-92/225 cells killed all of the primary GBM target cells efficiently (Fig. S4), with parental NK-92 and EGFRvIII-specific NK-92/MR1-1 cells under these conditions also showing limited antitumor activity mediated through their natural cytotoxicity mechanisms.
Figure 2. Cytotoxicity of CAR NK cells against GBM cells. (A) Expression of EGFR on the surface of established LN-18, T98G, D245MG and LN-464 GBM cells was determined by flow cytometry with EGFR-specific antibody (open areas). Isotype antibody served as control (filled areas). Cell killing by NK-92/R1, NK-92/MR1-1 and NK-92/225 cells was investigated after co-incubation with the GBM cells for 2 h at different E/T ratios. Parental NK-92 were included for comparison. (B) Expression of EGFR on the surface of primary MNOF1300, MNOF132, R28 and RAV19 GBM cells and cell killing by CAR NK cells were determined as described above. Cytotoxicity data are shown as mean values ± SEM; n = 3. MFI: mean fluorescence intensity (geometric mean).
LNT-229 cells as a model for EGFR and EGFRvIII-positive glioblastoma
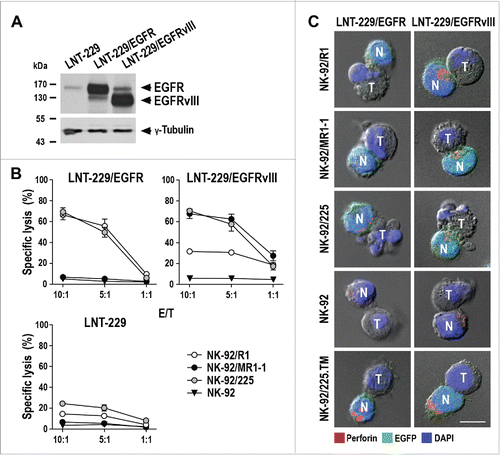
GBM cells frequently lose endogenous EGFRvIII expression during in vitro culture. Therefore, we employed LNT-229 GBM cells ectopically expressing EGFR or EGFRvIII to further investigate antitumor activity of the CAR NK cells.Citation21 Parental LNT-229 are negative for EGFRvIII and have low endogenous levels of EGFR, but overexpress the antigens upon transfection with EGFR or EGFRvIII constructs (, Fig. S5). Interestingly, ectopic EGFRvIII expression also enhanced the level of endogenous EGFR, allowing to use LNT-229/EGFRvIII cells as an EGFR/EGFRvIII double-positive model. Corresponding to endogenous EGFR expression, parental LNT-229 cells showed only minimal sensitivity to EGFR-specific NK-92/R1 and EGFR/EGFRvIII dual-specific NK-92/225 cells, while being resistant to EGFRvIII-specific NK-92/MR1-1 and parental NK-92 (). EGFR-overexpressing LNT-229/EGFR cells were efficiently lysed by NK-92/R1 and NK-92/225 cells, while resistance to NK-92/MR1-1 and NK-92 was retained. Ectopic expression of EGFRvIII allowed efficient killing of LNT-229/EGFRvIII cells by EGFRvIII-specific NK-92/MR1-1 and EGFR/EGFRvIII dual-specific NK-92/225, but the cells were also more effectively killed by EGFR-specific NK-92/R1 than parental LNT-229 corresponding to their enhanced endogenous EGFR expression. Similar experiments with renal cell carcinoma cells ectopically expressing EGFR or EGFRvIII confirmed specificity of the CAR NK cells (Fig. S6). BS-153 tumor cells, which represent one of the few established human GBM cell lines that retain endogenous EGFR and EGFRvIII overexpression in culture,Citation22 were effectively killed by EGFR-specific NK-92/R1, EGFRvIII-specific NK-92/MR1-1 and dual-specific NK-92/225 cells (Fig. S7).
Figure 3. Cytotoxicity of CAR NK cells against EGFR- and EGFRvIII-expressing LNT-229 cells. (A) EGFR (170 kDa) and EGFRvIII (140 kDa) were detected in cell lysates of LNT-229 GBM cells ectopically overexpressing full-length EGFR (LNT-229/EGFR) or mutant EGFRvIII (LNT-229/EGFRvIII) by immunoblotting with an EGFR-specific antibody binding to both receptors. Parental LNT-229 served as control. (B) Cytotoxicity of CAR NK cells against LNT-229/EGFR, LNT-229/EGFRvIII and parental LNT-229 cells was investigated after co-incubation of effector and target cells for 2 h at different E/T ratios. Parental NK-92 were included as control. Mean values ± SEM are shown; n = 3. (C) Conjugate formation between CAR NK cells and LNT-229/EGFR and LNT-229/EGFRvIII cells was investigated by confocal microscopy. Tumor (T) and EGFP-positive CAR NK (N; green) cells were co-incubated for 1 h, fixed, permeabilized and stained for perforin (red) to identify cytotoxic granules. Cell nuclei were labeled with DAPI (blue). Parental NK-92 and NK-92/225.TM cells expressing a CAR without intracellular signaling domains were included as controls. Scale bar: 10 µm.
Conjugate formation between NK and glioblastoma cells
CAR NK cells were co-incubated with LNT-229/EGFR or LNT-229/EGFRvIII cells for 1 h, before the cells were stained with perforin-specific antibody to visualize cytotoxic granules. Irrespective of their specificity, all CAR NK and unmodified NK-92 cells formed contacts with the different GBM cells. However, only conjugate formation between LNT-229/EGFR and NK-92/R1 or NK-92/225 cells, and between LNT-229/EGFRvIII and NK-92/MR1-1 or NK-92/225 cells rapidly triggered concentration of perforin-containing granules at the immunological synapse (; schematically shown in Fig. S8). LNT-229/EGFR and LNT-229/EGFRvIII cells underwent apoptosis within 1 h of incubation with NK-92/R1 or NK-92/225, or NK-92/MR1-1 or NK-92/225 cells, respectively, as indicated by membrane blebbing and disintegration of the target cell nucleus. While EGFR-specific NK-92/R1 cells showed some repolarization of lytic granules also after conjugate formation with LNT-229/EGFRvIII cells, no cell killing was observed. Likewise NK-92/225.TM cells expressing a signaling-incompetent CAR readily formed conjugates with LNT-229/EGFR and LNT-229/EGFRvIII cells, but this was not followed by reorientation of cytotoxic granules and target cell apoptosis. Depending on CAR specificity, the GBM cells also triggered degranulation and IFNγ release by the CAR NK cells, while parental NK-92 only responded marginally (Fig. S9).
Activity of CAR NK cells against orthotopic glioblastoma xenografts
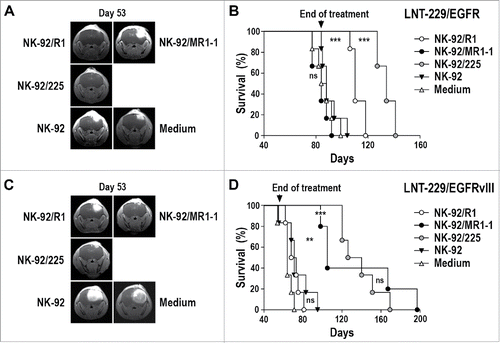
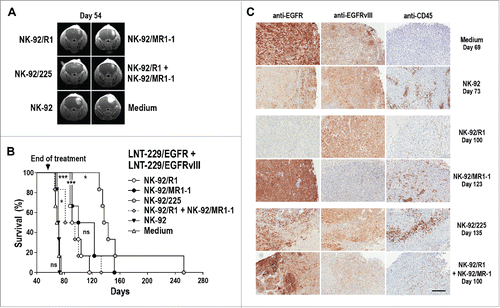
LNT-229/EGFR or LNT-229/EGFRvIII GBM cells were stereotactically injected into the brains of NSG mice. Seven days later, animals were treated by intratumoral injection of NK-92/R1, NK-92/MR1-1, NK-92/225, or parental NK-92 cells once per week for 8 weeks (LNT-229/EGFRvIII model) or 12 weeks (LNT-229/EGFR model). In the case of LNT-229/EGFR tumors, therapy with EGFR-specific NK-92/R1 or dual-specific NK-92/225 cells prevented early tumor outgrowth as assessed by MRI (), while treatment with EGFRvIII-specific NK-92/MR1-1 or parental NK-92 could not suppress tumor growth. This resulted in a marked extension of symptom-free survival of mice treated with NK-92/R1 or NK-92/225 cells (median of 110 and 134 d) in comparison to animals treated with NK-92/MR1-1, untargeted NK-92, or medium (median of 84, 88 and 86 d) (). Importantly, while both, NK-92/R1 and NK-92/225 therapy were effective, dual-specific NK-92/225 cells were more potent in this model enhancing survival even further than NK-92/R1 (p ≤ 0.001). In mice carrying LNT-229/EGFRvIII tumors, therapy with EGFRvIII-specific NK-92/MR1-1 or dual-specific NK-92/225 cells prevented early tumor outgrowth in a similar manner, while tumors developed rapidly in animals treated with EGFR-specific NK-92/R1 or parental NK-92 (). Consequently, symptom-free survival of NK-92/MR1-1- or NK-92/225-treated mice was extended markedly (median of 105 and 133 d) in comparison to animals treated with NK-92/R1, untargeted NK-92, or medium (median of 70.5, 71 and 64 d) ().
Figure 4. Antitumor activity of CAR NK cells against orthotopic LNT-229/EGFR and LNT-229/EGFRvIII GBM xenografts. (A) LNT-229/EGFR cells were stereotactically injected into the right striatum of NSG mice. Seven days later, the animals were treated by intratumoral injection of parental NK-92, EGFR-specific NK-92/R1, EGFRvIII-specific NK-92/MR1-1, or dual-specific NK-92/225 cells once per week for 12 weeks (n = 6). Control mice received injection medium. Tumor growth was monitored by MRI. Tumor development in representative animals from each group at day 53 is shown. (B) Symptom-free survival of the mice from the experiment described in (A). (C) LNT-229/EGFRvIII cells were stereotactically injected into the right striatum of NSG mice. Seven days later, the animals were treated as described above with NK-92 (n = 6), NK-92/R1 (n = 6), NK-92/MR1-1 (n = 5), or NK-92/225 (n = 6) cells once per week for 8 weeks. Control mice received injection medium (n = 6). Tumor development in representative animals from each group at day 53 is shown. (D) Symptom-free survival of the mice from the experiment described in (C). ***p ≤ 0.001; **p ≤ 0.01; ns, p > 0.05.
Therapy-induced selection of tumor cell subpopulations
As a model more closely resembling the heterogeneous tumors of a clinical situation, NSG mice were inoculated intracranially with a 1:1 mixture of LNT-229/EGFR and LNT-229/EGFRvIII cells. Seven days later, animals were treated by intratumoral injection of NK-92/R1, NK-92/MR1-1, NK-92/225, or parental NK-92 cells once per week for 8 weeks. In addition, one group received a 1:1 mixture of NK-92/R1 and NK-92/MR1-1 effector cells. Therapy with EGFR-specific NK-92/R1, EGFRvIII-specific NK-92/MR1-1, dual-specific NK-92/225, or mixed NK-92/R1/NK-92/MR1-1 cells all delayed tumor progression, while parental NK-92 had no effect (). Nevertheless, dual-specific NK-92/225 cells were more potent than monospecific NK-92/R1 and NK-92/MR1-1, with median symptom-free survival extending to 140 versus 95.5 and 111.5 d (). In contrast, median survival of animals treated with parental NK-92 or medium was 71.5 and 70.5 d. Unexpectedly, treatment with mixed NK-92/R1/NK-92/MR1-1 cells was much less effective than treatment with NK-92/225 with survival being similar to that after therapy with monospecific CAR NK cells (median survival of NK-92/R1/NK-92/MR1-1-treated group of 88 d). Tumors from individual animals of each group were analyzed by immunohistochemistry with EGFR- and EGFRvIII-specific antibodies (). While tumors from control mice treated with parental NK-92 or medium still contained both tumor cell subpopulations characterized by high EGFR and absent EGFRvIII expression (LNT-229/EGFR), or high EGFRvIII and moderate EGFR expression (LNT-229/EGFRvIII), therapy with monospecific NK-92/R1 or NK-92/MR1-1 cells led to selective outgrowth of tumors lacking the subpopulation overexpressing the respective target antigen. Accordingly, NK-92/R1-treated tumors consisted almost uniformly of cells overexpressing EGFRvIII and lacking high EGFR levels, while NK-92/MR1-1-treated tumors only contained EGFR-overexpressing but EGFRvIII-negative cells. In contrast, tumors of mice treated with dual-specific NK-92/225 or a mix of NK-92/R1 and NK-92/MR1-1 cells retained heterogeneity with respect to EGFR- and EGFRvIII-overexpression. Staining with an antibody specific for human CD45 revealed the presence of NK cells throughout the tumors, but not in adjacent normal brain tissue ().
Figure 5. Antitumor activity of CAR NK cells against mixed LNT-229/EGFR and LNT-229/EGFRvIII GBM xenografts. (A) LNT-229/EGFR and LNT-229/EGFRvIII cells were mixed at a 1:1 ratio before stereotactic injection of the cells into the right striatum of NSG mice. Seven days later, the animals were treated by intratumoral injection of parental NK-92, EGFR-specific NK-92/R1, EGFRvIII-specific NK-92/MR1-1, dual-specific NK-92/225, or a 1:1 mixture of NK-92/R1 and NK-92/MR1-1 cells once per week for 8 weeks (n = 6). Control mice received injection medium. Tumor development in representative animals from each group at day 54 is shown. (B) Symptom-free survival of the mice from the experiment described in (A). ***p ≤ 0.001; *p ≤ 0.05; ns, p > 0.05. (C) Sections of tumors from individual animals of each treatment group sacrificed at the indicated time point were stained with EGFR- or EGFRvIII-specific antibodies. NK cells present in tumor tissues were detected with CD45-specific antibody. Scale bar: 300 µm.
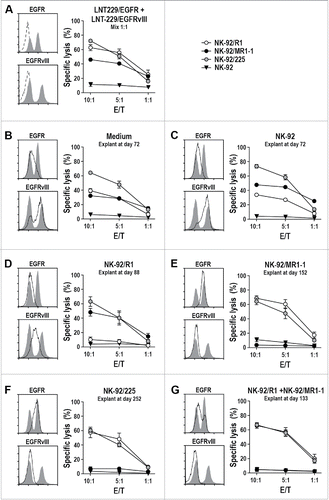
Target antigen expression by explanted tumor cells and sensitivity to CAR NK cells
To investigate treatment-induced selection of tumor cell subpopulations in more detail, tumors from another set of animals of each group were processed and the respective tumor cell suspensions were taken into culture. When compared to a fresh 1:1 mixture of LNT-229/EGFR and LNT-229/EGFRvIII cells displaying similar proportions of cells with high EGFR and absent EGFRvIII expression, or high EGFRvIII and moderate EGFR expression (), tumor cells from animals treated with parental NK-92 or medium contained more cells characterized by high EGFRvIII and moderate EGFR expression, indicating better outgrowth of the more aggressive LNT-229/EGFRvIII cells during in vivo passage in the absence of specific treatment ( and ). Accordingly, in in vitro cytotoxicity assays the explanted tumor cells were less sensitive to EGFR-specific NK-92/R1 than the fresh mixture of LNT-229/EGFR and LNT-229/EGFRvIII, while sensitivity to dual-specific NK-92/225 cells was comparable. Tumor cells recovered from an animal treated with EGFR-specific NK-92/R1 cells had lost the EGFR-overexpressing subpopulation and were no longer sensitive to NK-92/R1 cytotoxicity, while still being effectively killed by EGFRvIII-specific NK-92/MR1-1 and dual-specific NK-92/225 CAR NK cells (). Conversely, cells grown from tumors of animals treated with EGFRvIII-specific NK-92/MR1-1, dual-specific NK-92/225, or the mixture of NK-92/R1 and NK-92/MR1-1 cells did no longer contain an EGFRvIII-positive subpopulation and had lost sensitivity to EGFRvIII-specific NK-92/MR1-1, while sensitivity to EGFR-specific NK-92/R1 and dual-specific NK-92/225 cells was retained (). Hence, dual-specific NK-92/225 cells were the only effector cells still able to kill the different explanted tumor cells, irrespective of the type of treatment and the respective selection pressure present during in vivo tumor growth.
Figure 6. Cytotoxicity of CAR NK cells against ex vivo expanded GBM cells. (A) Cytotoxicity of NK-92/R1, NK-92/MR1-1 and NK-92/225 cells against a fresh 1:1 mixture of LNT-229/EGFR and LNT-229/EGFRvIII cells was investigated after co-incubation of effector and target cells for 2 h at different E/T ratios. Parental NK-92 were included for comparison. Surface expression of EGFR and EGFRvIII by the mixed target cell population was determined by flow cytometry with EGFR- and EGFRvIII-specific antibodies (left panels, gray areas). Control cells were only incubated with secondary antibody (dashed lines). Likewise, EGFR and EGFRvIII expression (solid lines with the initial mixed tumor cells shown as an overlay) and sensitivity to CAR NK cells were determined for GBM cells expanded ex vivo from explanted mixed LNT-229/EGFR and LNT-229/EGFRvIII xenografts of animals treated with: (B) injection medium; (C) parental NK-92; (D) NK-92/R1; (E) NK-92/MR1-1; (F) NK-92/225; (G) mixed NK-92/R1 and NK-92/MR1-1 cells. The tumor cells were recovered from animals from the experiment shown in sacrificed at the indicated time points. Cytotoxicity data are shown as mean values ± SEM; n = 3.
Discussion
In this study, we investigated the antitumor activity of CAR NK cells targeting EGFR, EGFRvIII, or both receptors against glioblastoma in vitro and in vivo. We generated CARs that harbor identical CD28 and CD3ζ signaling domains, but for cell binding carry the different antibody fragments scFv(R1) recognizing an N-terminal epitope unique to EGFRCitation17 (CAR R1.28.z), scFv(MR1-1) specific for the neo-epitope at the deletion breakpoint within EGFRvIIICitation18 (CAR MR1-1.28.z), or scFv(225) which is based on the variable regions of cetuximab and binds to an epitope shared between EGFR and EGFRvIIICitation19,23,24 (CAR 225.28.z). In cell killing experiments, EGFR-specific NK-92/R1 and dual-specific NK-92/225 cells efficiently and to a similar extent lysed EGFR-positive but EGFRvIII-negative established human GBM cells which were resistant to parental NK-92 and EGFRvIII-specific NK-92/MR1-1. This cell killing activity was retained toward primary human GBM cells with stem cell-like characteristics.Citation20 GBM cells ectopically expressing high levels of EGFRvIII together with moderate levels of endogenous EGFR were efficiently killed by dual-specific NK-92/225 and EGFRvIII-specific NK-92/MR1-1 cells, and to a lesser extent by EGFR-specific NK-92/R1 cells, confirming CAR-mediated selectivity. Degranulation of the CAR NK cells upon contact with cognate target cells not only resulted in target cell killing, but was also accompanied by secretion of high levels of IFNγ which may contribute to direct and indirect antitumor effects.
Recurrence of glioblastoma is mainly local,Citation25 suggesting intratumoral treatment as a feasible strategy. In clinical trials for adoptive immunotherapy of GBM, patients were treated by applying lymphokine-activated killer (LAK) cells into the resection cavity.Citation26,27 To model this approach for evaluation of in vivo antitumor activity of the CAR NK cells, we applied weekly stereotactic injection of the cells into the tumor area in orthotopic GBM xenografts in NSG mice. None of the animals carrying LNT-229/EGFR tumors and treated with dual-specific NK-92/225 cells showed visible disease progression during treatment. Similarly, mice treated with EGFR-specific NK-92/R1 had no or only small intracranial tumors at day 53. This resulted in long-term therapeutic benefit for the two treatment groups, with markedly increased symptom-free survival when compared to treatment with untargeted NK-92 or EGFRvIII-specific NK-92/MR1-1 cells, which like injection medium alone had no effect on the course of the disease. Conversely, growth of LNT-229/EGFRvIII tumors was only effectively controlled by treatment with dual-specific NK-92/225 and EGFRvIII-specific NK-92/MR1-1 cells. Hence, the CAR NK cells fully retained selectivity for their target receptors and activity against GBM in vivo.
EGFRvIII-targeted immunotherapy of glioblastoma with a peptide vaccine improved survival in clinical trials.Citation10 However, at recurrence most of patients' tumors had lost EGFRvIII expression, underscoring the strong selective pressure exerted by immunotherapy targeting a single tumor antigen.Citation28 While affecting a much smaller proportion of the treated patients, relapse due to outgrowth of antigen-negative escape variants has also been reported in acute lymphoblastic leukemia after therapy with CD19-specific CAR T cells.Citation29 The feasibility of targeting EGFR- or EGFRvIII-positive GBM cells with CAR NK cells that recognize both target antigens has recently been demonstrated using a CAR based on EGFR-specific antibody 528.Citation30 Nevertheless, so far no data were available addressing antitumor activity of such cells in comparison to monospecific EGFR- or EGFRvIII-targeted NK cells and potential tumor immune escape in an in vivo setting. To investigate treatment-induced selection of tumor subpopulations after CAR NK cell therapy in GBM, we injected NSG mice intracranially with a mixture of LNT-229/EGFR and LNT-229/EGFRvIII cells. These cells are not dependent for growth on overexpression of EGFR or EGFRvIII achieved by stable transfection of the parental LNT-229 GBM cell line. Nevertheless, with respect to the distribution of EGFR and EGFRvIII this model is similar to primary GBM, where not all tumor cells that have amplified copies of the EGFR gene are also EGFRvIII-positive, but those tumor cells that harbor EGFRvIII simultaneously express the wild-type receptor.Citation31 If no specific therapy was applied, explanted tumor cells revealed preferential outgrowth of LNT-229/EGFRvIII, which can be attributed to the enhanced aggressiveness reported for EGFR/EGFRvIII-double positive GBM cells.Citation8,32 Treatment with EGFR-specific NK-92/R1 or EGFRvIII-specific NK-92/MR1-1 cells was both effective resulting in similar extension of survival. Therapy with cetuximab-based dual-specific CAR NK cells, however, was most potent delaying disease development significantly longer than the monospecific effector cells. Surprisingly, a mixture of NK-92/R1 and NK-92/MR1-1 cells was much less effective, with treatment outcome similar to that after monospecific therapy.
While varying in their relative proportions between individual animals, tumors from control groups still contained both, LNT-229/EGFR and LNT-229/EGFRvIII cells. In contrast, treatment with EGFR-specific NK-92/R1 or EGFRvIII-specific NK-92/MR1-1 cells exerted strong selective pressure on the mixed tumors, resulting in immune escape and outgrowth of LNT-229/EGFRvIII or LNT-229/EGFR tumor cell subpopulations lacking the respective target antigen. This was not the case upon simultaneous targeting of EGFR and EGFRvIII with dual-specific NK-92/225 cells or a mixture of monospecific NK-92/R1 and NK-92/MR1-1, with tumors developing after treatment still containing areas contributed by LNT-229/EGFR and LNT-229/EGFRvIII cells. While the tumor cell subpopulations selected by treatment of the animals with either NK-92/R1 or NK-92/MR1-1 cells were no longer sensitive to the respective CAR NK cells during subsequent ex vivo culture, this was not the case after treatment with dual-specific NK-92/225. In this case, explanted tumor cells still expressed at least one of the two targeted antigens and were readily killed by the CAR NK cells, suggesting that disease control by NK-92/225 could have been improved even further by extending frequency or duration of treatment, which could not be tested in this setting due to animal welfare requirements.
Different approaches can be employed for dual targeting of CAR effector cells to increase specificity and reduce the risk of immune escape. These include simultaneous or serial application of CAR cells of different specificities, co-expression of two CAR molecules in the same cells, and the design of bispecific CARs that contain two distinct cell recognition domains.Citation33,34 Employing a CAR based on an antibody like cetuximab that targets a shared epitope in EGFR and EGFRvIII is unique,Citation30,35 since it does not require development of several cell products, multiple transduction of cells, or complex protein design. Nevertheless, unlike EGFRvIII, wild-type EGFR is expressed in many normal tissues, increasing the risk of on-target/off-tumor toxicities if EGFR/EGFRvIII dual-specific CAR effector cells were applied systemically and capable of permanent engraftment and in vivo expansion.Citation36 In our models, we noted persistence of locally injected NK-92 and CAR NK-92 cells for several weeks, but we did not observe obvious expansion of the cells, which stayed within the tumor without invading adjacent healthy brain tissue. Hence, in the case of GBM the risk of adverse effects may be reduced by local application of CAR NK cells, which in contrast to T cells are rather short-lived and have only limited expansion potential in vivo.Citation37
In conclusion, our data demonstrate that cetuximab-based dual-specific CAR NK cells efficiently eliminate EGFR- and/or EGFRvIII-expressing GBM cells without inducing rapid immune escape as observed upon therapy with EGFR- or EGFRvIII-targeted monospecific effectors. Accordingly, such dual-specific CAR NK cells may have potential for development as an adoptive therapy for the treatment of glioblastoma.
Materials and methods
Cells and culture conditions
HEK 293T cells (ATCC) and established GBM cell lines LN-18, LN-464, T98G (kindly provided by Monika Hegi, Lausanne, Switzerland), D245MG (kindly provided by Darell Bigner, Durham, NC) and BS-153 (kindly provided by Katrin Lamszus, Hamburg, Germany) were cultured in DMEM (Lonza) supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin (Life Technologies). LNT-229 cells ectopically expressing EGFR or EGFRvIII were described previously.Citation21 Murine Renca-lacZ, Renca-lacZ/EGFR and Renca-lacZ/EGFRvIII renal cell carcinoma cells were cultured as described.Citation35,38 Human NK-92 cells (kindly provided by NantKwest, Inc., Culver City, CA) were grown in X-VIVO 10 (Lonza) supplemented with 5% heat-inactivated human plasma (German Red Cross Blood Donation Service Baden-Württemberg - Hessen) and 100 IU/mL IL-2 (Proleukin; Novartis Pharma).
Primary human GBM cells MNOF132 and MNOF1300 (kindly provided by Stefan Momma and Julia Tichy, Frankfurt am Main, Germany) as well as R28 and RAV19 (kindly provided by Christoph Beier and Arabel Vollmann-Zwerenz, Regensburg, Germany) were used up to a maximum passage number of 20 and grown in flasks pre-coated with 5 mg/mL laminin (Sigma-Aldrich) in DMEM/F12 medium (Lonza) containing 20 ng/mL each of EGF and bFGF2 (ReliaTech), and 20% BIT Admixture Supplement (Pelo Biotech).Citation20
Generation of CAR-expressing NK-92 cells
CARs R1.28.z, 225.28.z and MR1-1.28.z were designed by in silico assembly of an immunoglobulin heavy chain signal peptide, the antibody fragment scFv(R1),Citation17 scFv(225),Citation19,39 or scFv(MR1-1),Citation18 a Myc-tag, a CD8α hinge region, the CD28 transmembrane and intracellular domains, and the CD3ζ intracellular domain as previously described.Citation16,40 Codon-optimized fusion genes were synthesized (GeneArt) and inserted into lentiviral transfer plasmid pHR'SIN-cPPT-SIEW (pSIEW)Citation41 upstream of IRES and EGFP sequences. Sequences encoding truncated CARs R1.TM, 225.TM and MR1-1.TM were generated by restriction digest of the full-length CAR constructs pS-R1.28.z-IEW, pS-225.28.z-IEW, and pS-MR1-1.28.z-IEW with NaeI, removal of the DNA fragment encoding the CAR signaling domains and religation, resulting in the introduction of a premature stop codon following the transmembrane domain of CD28 in constructs pS-R1.TM-IEW, pS-225.TM-IEW, and pS-MR1-1.TM-IEW. VSV-G pseudotyped vector particles were generated and NK-92 cells were transduced as described.Citation40 NK-92/R1, NK-92/225, and NK-92/MR1-1 single cell clones were generated by flow cytometric cell sorting with a FACSAria cell sorter (BD Biosciences) using Myc-tag specific Alexa Fluor 647-coupled antibody 9E10 (Santa Cruz Biotechnology).
EGFP and CAR expression were monitored by direct flow cytometry or flow cytometry with Myc-tag specific antibody using a FACSCalibur cytometer and CellQuest Pro software (BD Biosciences). CAR-binding to EGFR was determined by flow cytometric analysis of NK cells with recombinant EGFR-Fc fusion protein (R&D Systems) followed by APC-coupled anti-human IgG secondary antibody.
Analysis of target antigen expression
Expression of EGFR and EGFRvIII on the surface of target cells was determined by flow cytometric analysis with Alexa Fluor 647-coupled EGFR-specific antibody R-1, or EGFRvIII-specific antibody DH8.3Citation42 followed by APC-coupled secondary antibody, using FACSCalibur or FACSCanto II flow cytometers (BD Biosciences). Data were analyzed using CellQuest Pro or FACSDiva software (BD Biosciences). For analysis of total cellular EGFR and EGFRvIII, tumor cells were lysed in RIPA buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% NP-40, 0.5% sodium-deoxycholate and cOmplete Mini protease inhibitor cocktail (Roche Diagnostics) for 20 min on ice, followed by sonication and centrifugation to remove cell debris. Proteins from lysates were separated by SDS-PAGE and immobilized on PVDF membranes (Merck Millipore). Membranes were analyzed using antibody D38B1 XP (Cell Signaling) specific for the C-terminus of human EGFR, species-specific HRP-conjugated secondary antibody (Sigma-Aldrich), and chemiluminescent detection.
Cytotoxicity assays
Cell killing activity of NK cells was analyzed in FACS-based assays as described.Citation40 Briefly, target cells were labeled with calcein violet AM (CV; Life Technologies), washed, and co-cultured with effector cells at different E/T ratios for 2 h at 37°C. After co-culture, cells were washed, and 150 µL of a 1 µg/mL propidium iodide solution were added to each sample immediately before flow cytometric analysis in a FACSCanto II flow cytometer (BD Biosciences). Dead target cells were determined as CV and PI double positive. Spontaneous target cell lysis in the absence of effector cells was subtracted to calculate specific cytotoxicity. Data were analyzed using FACSDiva software (BD Biosciences). To investigate survival of tumor cells after longer exposure to NK cells, target cells were seeded in 24-well plates at a density of 7.5 × 104 cells/well and incubated for 1 to 2 d to reach sub-confluency. Then 1 × 105 NK cells were added to each well. After co-incubation for 16 h at 37°C, medium and NK cells were removed; adherent tumor cells were washed and fixed with 200 µL of 4% paraformaldehyde in PBS for 30 min at room temperature. Then cells were incubated for 5 min with 400 µL/well of 0.5% crystal violet (AppliChem) in 20% methanol, followed by extensive washing with dH2O. Cell-bound crystal violet complexes were eluted with Sorenson's buffer (0.1 M sodium citrate, 50% EtOH, pH 4.2) for 30 min at room temperature. Absorption at 540 nm in comparison to tumor cells incubated without NK cells was determined as a measure for the relative number of viable cells using a SpectraMax 340 microplate reader with SoftMax Pro software (Molecular Devices).
Degranulation assay and conjugate analysis
Induction of degranulation in NK cells upon co-incubation with target cells at a 1:1 ratio for 5 h at 37°C was investigated as described,Citation43 measuring surface expression of lysosomal-associated membrane protein LAMP-1 (CD107a) using BD FastImmune CD107a APC detection antibody (BD Biosciences) according to the manufacturer's instructions. NK cells stimulated with 1 µg/mL phorbol 12-myristate 13-acetate (PMA) and 1 µg/mL ionomycin (Sigma-Aldrich), or kept without target cells served as controls.
For analysis of conjugate formation and redistribution of cytotoxic granules by confocal laser scanning microscopy, NK cells (2.5 × 105) and target cells were mixed at a 1:1 ratio and incubated on poly-L-lysine-coated cover slips (Life Technologies) for 1 h at 37°C, fixed for 10 min with phosphate-buffered 4% formaldehyde solution, and permeabilized for 5 min with 0.1% Triton X-100 in PBS. Cells were washed and unspecific binding sites were blocked for 30 min with 10% FBS in PBS. Then samples were incubated for 75 min with perforin-specific antibody δG9 (Santa Cruz Biotechnology), followed by Alexa Fluor 594-coupled anti-mouse antibody (Life Technologies). All antibodies and reagents were added in blocking buffer at room temperature. Cell nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole) (Life Technologies). Cells were washed twice with PBS, embedded using Mowiol 4-88 (Roth) and analyzed with a Leica SP5 confocal laser scanning microscope (Leica Microsystems).
In vivo glioblastoma models
Six to 12 week old female NSG mice were used in all in vivo experiments. Mice were anesthetized with ketamine and xylazine, immobilized in a stereotaxic fixation device (Stoelting) and injected through a burr hole in the skull with 1 × 105 LNT-229/EGFR, LNT-229/EGFRvIII, or a 1:1 mixture of both GBM cell lines in 2 µL PBS using a 10 µL Hamilton syringe (Hamilton) and a Quintessential Stereotaxic Injector (Stoelting). Cells were injected at a speed of 0.5 µL/min into the right striatum with a depth of 3 mm to the skull. Seven days after tumor inoculation, mice were treated by stereotactic injection of 2 × 106 NK-92/R1, NK-92/225, NK-92/MR1-1 or parental NK-92 cells in 3 µL of NK-92 medium. Treatment was repeated weekly for 8 to 12 weeks. Tumor growth was monitored by MRI performed in prone position using a MAGNETOM Trio scanner (Siemens) and a surface coil array for 1H imaging of small animals (RAPID Biomedical). Mice were anesthetized and injected intraperitoneally with 150 µL of 1 mmol/mL gadolinium-diethylenetriamine pentaacetic acid (Gadovist; Bayer Vital). Standard T2 weighted and T1 weighted sequences were acquired. Mice were sacrificed when they developed neurological symptoms or lost more than 20% of body weight. The in vivo experiments were approved by the responsible government committee (Regierungspräsidium Darmstadt, Darmstadt, Germany) and were conducted according to the applicable guidelines and regulations.
Immunohistochemistry
Brain tissues of sacrificed mice were fixed with phosphate-buffered 4% formaldehyde solution (Roth) and subsequently paraffin-embedded. Sections of 3 µm thickness were prepared, deparaffinized and hydrated. Slides were stained according to a standardized staining protocol (Bond Polymer Refine IHC protocol, IHC-F; Leica Microsystems) using murine primary antibodies specific for human CD45 (clone 2B11 + PD7/26, 1:100 dilution; Dako), EGFR (clone DAK-H1-WT, 1:50 dilution; Dako) and EGFRvIII (DH8.3, 14 µg/mL final concentration), followed by polymeric HRP-conjugated anti-mouse antibody (DAB Polymer Refine Detection Kit; Leica Microsystems). Finally, slides were counterstained with hematoxylin and embedded in mounting medium.
Statistical analysis
Survival was analyzed by Kaplan–Meier plot and log-rank (Mantel–Cox) test. p values <0.05 were considered significant. Statistical calculations were performed using Prism 5 software (GraphPad Software).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1119354_supplementary_material.pdf
Download PDF (1.4 MB)Acknowledgments
The authors thank Kurt Schönfeld for the 225.28.z CAR construct, Barbara Uherek and Thorsten Geyer for technical assistance, Maurice Harth and Bianca Lienerth for help with magnetic resonance imaging, Petra Dinse for help with immunohistochemical analysis, Pranav Oberoi for helpful comments on the manuscript, and the staff at the animal facility of the Georg-Speyer-Haus for their support.
Funding
This work was supported in part by grants from the Deutsche Forschungsgemeinschaft (DFG) (GRK1172), LOEWE Center for Cell and Gene Therapy Frankfurt (CGT) (HMWK III L 5-518/17.004 2013), German Federal Ministry of Education and Research (BMBF) (Ci3; FKZ 131A009A, 131A009C), the German Cancer Consortium (DKTK), the Alfons und Gertrud Kassel-Stiftung, and institutional funds of the Georg-Speyer-Haus. The Georg-Speyer-Haus is funded jointly by the German Federal Ministry of Health and the Ministry of Higher Education, Research and the Arts of the State of Hessen (HMWK).
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987-96; PMID:15758009; http://dx.doi.org/10.1056/NEJMoa043330
- Bähr O, Herrlinger U, Weller M, Steinbach JP. Very late relapses in glioblastoma long-term survivors. J Neurol 2009; 256:1756-8; PMID:19434438; http://dx.doi.org/10.1007/s00415-009-5167-6
- Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, Bigner DD. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res 1990; 50:8017-22; PMID:2253244
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995; 19:183-232; PMID:7612182; http://dx.doi.org/10.1016/1040-8428(94)00144-I
- Hasselbalch B, Lassen U, Poulsen HS, Stockhausen MT. Cetuximab insufficiently inhibits glioma cell growth due to persistent EGFR downstream signaling. Cancer Invest 2010; 28:775-87; PMID:20504227; http://dx.doi.org/10.3109/07357907.2010.483506
- Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, Easaw J, Belanger K, Forsyth P, McIntosh L et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol 2010; 65:353-61; PMID:19499221; http://dx.doi.org/10.1007/s00280-009-1041-6
- Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets 2012; 12:197-209; PMID:22268382; http://dx.doi.org/10.2174/156800912799277557
- Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A 1994; 91:7727-31; PMID:8052651; http://dx.doi.org/10.1073/pnas.91.16.7727
- Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJ. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc Natl Acad Sci U S A 1998; 95:5724-9; PMID:9576951; http://dx.doi.org/10.1073/pnas.95.10.5724
- Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE 2nd, McLendon RE, Mitchell DA et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010; 28:4722-9; PMID:20921459; http://dx.doi.org/10.1200/JCO.2010.28.6963
- Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res 2014; 20:972-84; PMID:24352643; http://dx.doi.org/10.1158/1078-0432.CCR-13-0709
- Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, Schmitz M, Schackert G, Pastan I, Temme A. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J Immunother 2015; 38:197-210; PMID:25962108; http://dx.doi.org/10.1097/CJI.0000000000000082
- Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res 2001; 10:535-44; PMID:11522236; http://dx.doi.org/10.1089/15258160152509145
- Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy 2008; 10:625-32; PMID:18836917; http://dx.doi.org/10.1080/14653240802301872
- Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, Suttorp M, Seifried E, Ottmann OG, Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013; 15:1563-70; PMID:24094496; http://dx.doi.org/10.1016/j.jcyt.2013.06.017
- Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bönig H, Köhl U, Kloess S et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther 2015; 23:330-8; PMID:25373520; http://dx.doi.org/10.1038/mt.2014.219
- Jannot CB, Beerli RR, Mason S, Gullick WJ, Hynes NE. Intracellular expression of a single-chain antibody directed to the EGFR leads to growth inhibition of tumor cells. Oncogene 1996; 13:275-82; PMID:8710366
- Beers R, Chowdhury P, Bigner D, Pastan I. Immunotoxins with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin Cancer Res 2000; 6:2835-43; PMID:10914732
- Wels W, Beerli R, Hellmann P, Schmidt M, Marte BM, Kornilova ES, Hekele A, Mendelsohn J, Groner B, Hynes NE. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int J Cancer 1995; 60:137-44; PMID:7814146; http://dx.doi.org/10.1002/ijc.2910600120
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006; 9:391-403; PMID:16697959; http://dx.doi.org/10.1016/j.ccr.2006.03.030
- Steinbach JP, Klumpp A, Wolburg H, Weller M. Inhibition of epidermal growth factor receptor signaling protects human malignant glioma cells from hypoxia-induced cell death. Cancer Res 2004; 64:1575-8; PMID:14996711; http://dx.doi.org/10.1158/0008-5472.CAN-03-3775
- Schulte A, Liffers K, Kathagen A, Riethdorf S, Zapf S, Merlo A, Kolbe K, Westphal M, Lamszus K. Erlotinib resistance in EGFR-amplified glioblastoma cells is associated with upregulation of EGFRvIII and PI3Kp110delta. Neuro Oncol 2013; 15:1289-301; PMID:23877316; http://dx.doi.org/10.1093/neuonc/not093
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7:301-11; PMID:15837620; http://dx.doi.org/10.1016/j.ccr.2005.03.003
- Hartmann C, Müller N, Blaukat A, Koch J, Benhar I, Wels WS. Peptide mimotopes recognized by antibodies cetuximab and matuzumab induce a functionally equivalent anti-EGFR immune response. Oncogene 2010; 29:4517-27; PMID:20514015; http://dx.doi.org/10.1038/onc.2010.195
- Chamberlain MC. Radiographic patterns of relapse in glioblastoma. J Neurooncol 2011; 101:319-23; PMID:21052776; http://dx.doi.org/10.1007/s11060-010-0251-4
- Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Hochster HS, Moore EJ, Pierz DM, Chen DK, Budzilovich GN, Ransohoff J. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer 1995; 76:840-52; PMID:8625188; http://dx.doi.org/10.1002/1097-0142(19950901)76:5%3c840::AID-CNCR2820760519%3e3.0.CO;2-R
- Dillman RO, Duma CM, Schiltz PM, DePriest C, Ellis RA, Okamoto K, Beutel LD, De Leon C, Chico S. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother 2004; 27:398-404; PMID:15314549; http://dx.doi.org/10.1097/00002371-200409000-00009
- Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer 2015; 15:302-10; PMID:25855404; http://dx.doi.org/10.1038/nrc3918
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507-17; PMID:25317870; http://dx.doi.org/10.1056/NEJMoa1407222
- Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, Victor A, Meisen WH, Kim SH, Grandi P et al. CAR-Engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep 2015; 5:11483; PMID:26155832; http://dx.doi.org/10.1038/srep11483
- Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000; 60:1383-7; PMID:10728703
- Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, Chen J, Lau J, Knobbe-Thomsen C, Weller M et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013; 24:438-49; PMID:24135280; http://dx.doi.org/10.1016/j.ccr.2013.09.004
- Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther 2013; 21:2087-101; PMID:23939024; http://dx.doi.org/10.1038/mt.2013.185
- Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schönfeld K, Koch J, Dotti G et al. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids 2013; 2:e105; PMID:23839099; http://dx.doi.org/10.1038/mtna.2013.32
- Schmidt M, Maurer-Gebhard M, Groner B, Köhler G, Brochmann-Santos G, Wels W. Suppression of metastasis formation by a recombinant single chain antibody-toxin targeted to full-length and oncogenic variant EGF receptors. Oncogene 1999; 18:1711-21; PMID:10208432; http://dx.doi.org/10.1038/sj.onc.1202489
- Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 2015; 7:275ra22; PMID:25696001; http://dx.doi.org/10.1126/scitranslmed.aaa4963
- Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014; 3:e28147; PMID:25340009; http://dx.doi.org/10.4161/onci.28147
- Maurer-Gebhard M, Schmidt M, Azemar M, Altenschmidt U, Stöcklin E, Wels W, Groner B. Systemic treatment with a recombinant erbB-2 receptor-specific tumor toxin efficiently reduces pulmonary metastases in mice injected with genetically modified carcinoma cells. Cancer Res 1998; 58:2661-6; PMID:9635594
- Müller N, Hartmann C, Genssler S, Koch J, Kinner A, Grez M, Wels WS. A bispecific transmembrane antibody simultaneously targeting intra- and extracellular epitopes of the epidermal growth factor receptor inhibits receptor activation and tumor cell growth. Int J Cancer 2014; 134:2547-59; PMID:24243620; http://dx.doi.org/10.1002/ijc.28585
- Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother 2012; 61:1451-61; PMID:22310931; http://dx.doi.org/10.1007/s00262-012-1212-x
- Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, Kinnon C, Grez M, Thrasher AJ. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther 2002; 13:803-13; PMID:11975847; http://dx.doi.org/10.1089/10430340252898984
- Hills D, Rowlinson-Busza G, Gullick WJ. Specific targeting of a mutant, activated EGF receptor found in glioblastoma using a monoclonal antibody. Int J Cancer 1995; 63:537-43; PMID:7591264; http://dx.doi.org/10.1002/ijc.2910630414
- Oberoi P, Jabulowsky RA, Bähr-Mahmud H, Wels WS. EGFR-targeted granzyme B expressed in NK cells enhances natural cytotoxicity and mediates specific killing of tumor cells. PLoS One 2013; 8:e61267; PMID:23573299; http://dx.doi.org/10.1371/journal.pone.0061267