
ABSTRACT
Dendritic cells (DC) play a pivotal role in the induction and regulation of immune responses. In cancer, DC-based vaccines have proven to be safe and to elicit protective and therapeutic immunological responses. Recently, we showed that specific mTORC2 (mechanistic target of rapamycin complex 2) deficiency in DC enhances their ability to promote Th1 and Th17 responses after LPS stimulation. In the present study, bone marrow-derived mTORC2-deficient (Rictor−/−) DC were evaluated as a therapeutic modality in the murine B16 melanoma model. Consistent with their pro-inflammatory profile (enhanced IL-12p70 production and low PD-L1 expression versus control DC), intratumoral (i.t.) injection of LPS-activated Rictor−/− DC slowed B16 melanoma growth markedly in WT C57BL/6 recipient mice. This antitumor effect was abrogated when Rictor−/− DC were injected i.t. into B16-bearing Rag−/− mice, and also after selective CD8+ T cell depletion in wild-type hosts in vivo, indicating that CD8+ T cells were the principal regulators of tumor growth after Rictor−/− DC injection. I.t. administration of Rictor−/− DC also reduced the frequency of myeloid-derived suppressor cells within tumors, and enhanced numbers of IFNγ+ and granzyme-B+ cytotoxic CD8+ T cells both in the spleens and tumors of treated animals. These data suggest that selective inhibition of mTORC2 activity in activated DC augments their pro-inflammatory and T cell stimulatory profile, in association with their enhanced capacity to promote protective CD8+ T cell responses in vivo, leading to slowed B16 melanoma progression. These novel findings may contribute to the design of more effective DC-based vaccines for cancer immunotherapy.
Abbreviations
| BM | = | bone marrow |
| DC | = | dendritic cells |
| IFN | = | interferon |
| IL | = | interleukin |
| i.t. | = | intra-tumoral |
| KO | = | knock-out |
| LPS | = | lipopolysaccharide |
| MDSC | = | myeloid-derived suppressor cells |
| mTOR(C) | = | mammalian/mechanistic target of rapamycin (complex) |
| NK | = | natural killer |
| PD-L1 | = | programmed death-ligand 1 |
| RAPA | = | rapamycin |
| TILs | = | tumor-infiltrating lymphocytes |
| TLR | = | Toll-like receptor |
| TME | = | tumor micro-environment |
| WT | = | wild-type |
Introduction
DC are professional antigen-presenting cells that shape immune responses by linking innate and adaptive immunity. Their ability to regulate differentiation, activation, and proliferation of specific T-cell subsets, makes them an attractive target in cancer immunotherapy approaches. Indeed, in a large number of clinical trials performed over the last 20 y, DC-based vaccines have proven to be safe (i.e. low toxicity and no impairment in quality of life), and capable of eliciting tumor-specific immunological responses.Citation1 Recent reviews have focused on the operational discrepancy between the apparent immunogenicity of such vaccines and, thus far, their only limited therapeutic efficacy.Citation1-5 These clinical results suggest that DC-based therapies are biologically active in cancer patients, but that their impact on disease can still be refined and improved upon.
The mammalian/mechanistic target of rapamycin (mTOR) pathway plays a key role in regulating mRNA translation, protein synthesis, glucose metabolism, lipid synthesis and autophagy in various cells, including immune cells.Citation6-8 mTOR performs these functions in two independent complexes: mTORC1 and mTORC2.Citation9 mTORC1 phosphorylates the translational factors S6 kinase-1 (S6K1) and 4E-binding protein-1 (4EBP1),Citation10 regulating different cell processes in response to nutrients and/or growth factors,Citation11 while mTORC2 phosphorylates Akt, protein-kinase Cα and SGK1 (serum and glucocorticoid-regulated kinase 1), leading to regulation of the actin cytoskeleton.Citation10 Inhibitors of mTORC1, including rapamycin and its derivatives everolimus and temsirolimus, have been evaluated previously as antitumor agents,Citation12 with everolimus recently approved for the treatment of breast cancer patients (in combination with exemestane)Citation13 and patients with advanced pancreatic neuroendocrine tumors.Citation14 Novel dual mTORC1/2 inhibitors are currently being studied, both translationally and clinically, as anticancer agents,Citation15 i.e., in hepatocellular carcinoma patients.Citation16
Interestingly, during melanoma progression, a number of alterations have been identified in major components of the mTOR pathway, including PI3K (phosphoinositide 3-kinase) mutation, PTEN (phosphatase and tensin homolog) loss of function, and Akt, S6K1, 4EBP1 and eIF4E (eukaryotic initiation factor 4E) overexpression.Citation17
Recently, Damsky and colleaguesCitation18 have reported that activation of mTORC1 and mTORC2 is required for oncogene-induced senescence evasion in human melanomas with BRAF mutations, leading to malignant transformation. Therefore, mTOR is also an appealing therapeutic target for the development of novel treatment options for patients with melanoma. Indeed, dual mTORC1/2 inhibitors have been reported to improve the antitumor efficacy of PI3K or MEK (mitogen-activated protein kinase) inhibitors in melanoma patients.Citation19-21
We have shown recently that DC deficiency in mTORC2 results in increased production of pro-inflammatory cytokines (mainly IL-12p70 and IL-23) after TLR4 ligation, leading to enhanced T-helper 1 (Th1) and Th17 responses when compared to control DC.Citation22 Given that IL-12p70-producing DC vaccines have been shown to elicit type-1 antigen-specific CD8+ T cell immunity in patients with melanoma, and to correlate positively with time to progression,Citation23 we hypothesized that mTORC2-deficient DC might facilitate more robust therapeutic CD8+ T cell responses capable of inhibiting melanoma growth in vivo.
Herein, we show that i.t. injection of Rictor−/− DC markedly slows B16 melanoma growth in syngeneic WT-recipient mice. This treatment benefit was abrogated in tumor-bearing Rag−/− host mice, and also after selective CD8+ T cell depletion of WT recipient animals in vivo, supporting the critical role of therapy-induced CD8+ T cells as principal regulators of tumor growth. Notably, i.t. administration of Rictor−/− DC also led to reduced frequencies of myeloid-derived suppressor cells (MDSC) within the treated microenvironment, and to enhanced numbers of IFNγ+ and granzyme-B+ CD8+ T cells in the spleens and tumors of Rictor−/− DC-treated animals. Furthermore, in a 2-site disease model, Rictor−/− DC injection into a single tumor, led to the slowed growth of both sites of disease. These data support the translational use of Rictor−/− DC as an immunotherapeutic agent in the setting of advanced-stage cancer, where at least one site of disease is accessible for injection.
Results
Rictor−/− DC exhibit pro-inflammatory properties
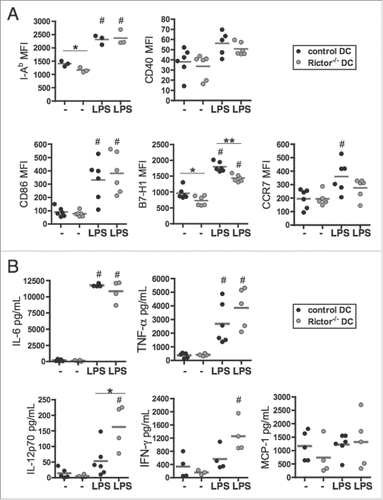
We first evaluated the expression of surface markers and cytokine production by control and Rictor−/− bone marrow-derived DC. When compared with B6 WT control DC, Rictor−/− DC expressed lower levels of MHC class II (I-Ab) when unstimulated and lower levels of B7-H1 (PD-L1) when unstimulated or LPS-activated (). However, there were no significant differences observed in CD40, CD86, or CCR7 expression levels when comparing control and Rictor−/− DC (). Analyses of cytokine secretion profiles supported significantly increased IL-12p70 production by LPS-stimulated Rictor−/− DC vs. control DC, but similar levels of IL-6, TNFα, IFNγ or MCP-1 (CCL2) secretion from both control and Rictor−/− DC (). These results indicate that DC deficient in mTORC2 expression were likely more pro-inflammatory than control DC.
Figure 1. Rictor−/− BMDC display pro-inflammatory properties. (A) CD11c+-gated BMDC were analyzed for cell surface MHC class-II (I-Ab), CD40, CD86, B7-H1 (PD-L1) and CCR7 expression by flow cytometry following 6 d culture in the absence of stimulation (−) or after LPS stimulation (LPS) for the last 18 h of culture. Plots show the means and individual values of n = 3–6 mice. (B) Cytokine levels in supernatants were assessed by cytokine bead array (IL-6, TNFα, MCP-1) or ELISA (IL-12p70 and IFNγ). Data are from n = 4–6 mice. #p < 0.05 compared to corresponding non-stimulated cells; *p < 0.05 and **p < 0.01 between control and Rictor−/− DC.
Intratumoral (i.t.) delivery of LPS-activated rictor−/− DC slows B16 melanoma growth
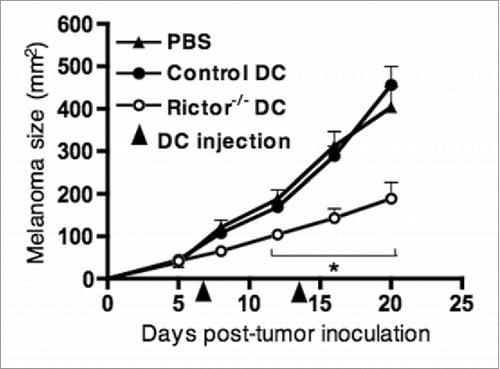
Given that Rictor−/− DC secrete markedly elevated levels of IL-12p70 compared with WT controls, and that this type-1 cytokine plays an important role in promoting protective immune responses within the tumor milieu,Citation24,25 we hypothesized that Rictor−/− DC might regulate cancer growth when delivered into the tumor microenvironment (TME). To test this possibility, 1 × 106 LPS-stimulated control or Rictor−/− DC were injected into established B16 melanomas in WT B6 mice on days 7 (when mean tumor size is approximately 50 mm2) and 14 post-tumor inoculation, and tumor growth monitored subsequently. As shown in , i.t. injection of LPS-activated Rictor−/− DC markedly slowed B16 melanoma growth when compared to tumor-bearing mice treated with control DC or PBS. This therapeutic effect was not observed when the same number of LPS-Rictor−/− DC were instead injected distal to tumors (i.e. intra-peritoneally; data not shown), supporting the need to deliver the treatment within the immediate vicinity of the diseased tissue.
Figure 2. I.t. injection of LPS-activated Rictor−/− DC markedly reduces B16 melanoma growth. C57BL/6 mice bearing day 7 s.c. B16 melanomas were given an i.t. injection of 106 control DC or Rictor−/− DC, that was repeated at day 14 post-tumor inoculation. Tumor growth was monitored every 3–4 d and is shown as mean + SD for five animals per group. p < 0.05 when comparing Rictor−/− DC-injected mice with untreated or control DC-injected mice.
I.t. delivered rictor−/− DC show similar migratory potential to draining lymphoid tissue, but promote reduced frequencies of MDSC within the TME
We next investigated whether slowed tumor growth could be ascribed to superior migratory ability of LPS-Rictor−/− DC (versus control DC) to draining lymph nodes or the spleen, where protective antitumor T cell cross-priming would be expected to occur. To test this possibility, B16 melanoma-bearing mice were injected i.t. with LPS-stimulated and CFSE-labeled control or Rictor−/− DC, and the inguinal lymph nodes, spleens and tumors recovered for analysis on day 3 post-treatment. Our results show that injected LPS-Rictor−/− DC were recovered at lower frequencies () and lower numbers within the tumors when compared to control DC (), suggesting that Rictor−/− DC may have migrated more efficiently to tumor-draining lymph nodes. However, we observed similar numbers of migratory control and Rictor−/− DC in the inguinal lymph nodes of treated animals (), suggesting that Rictor−/− DC did not have a migratory advantage to secondary lymphoid tissues when compared to control DC.
Figure 3. I.t.-delivered rictor−/− DC show similar migration to draining lymphoid tissue, but reduce the frequency of MDSC within the tumor. 5 × 106 CFSE-labeled control DC or Rictor−/− DC were injected i.t. on day 7 post-tumor inoculation in B16-melanoma-bearing B6 mice. After 3 d, tumors, spleens and tumor-draining inguinal lymph nodes were harvested and cells isolated. (A) Plots show the percentages of CFSE+ DC recovered from the tumor. (B, C) Absolute numbers of CFSE+ DC recovered from the tumor (B) and inguinal lymph nodes (C). Box plots show median, 25%- and 75%-quartiles, and both extreme values. (D) Percent CD11c+ and CD11b+Gr1+ cells in the tumor shown as means + SD for three animals per group. *p < 0.05.
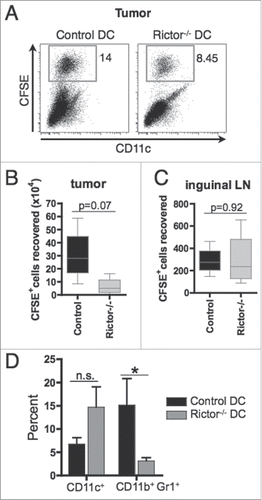
Superior cross-priming of protective T cells or the action of these effector cells in the TME could also occur if the injected LPS-Rictor−/− DC impaired regulatory cell function in vivo. MDSC have been shown to suppress antitumor T cell immune responses in the TME.Citation26,27 We assessed the impact of i.t.-delivered DC on the prevalence of MDSC in the TME. As shown in , the frequency of MDSC (CD11b+Gr1+) in the Rictor−/− DC-treated mice was reduced significantly (> 70 %) when compared to control DC-treated mice, whereas no significant difference was found in overall CD11c+ cell content. This result indicates that the injected Rictor−/− DC may reduce the incidence and regulatory action of MDSC in the TME.
I.t. delivery of rictor−/− DC exerts antitumor activity in a contralateral (distal) tumor site
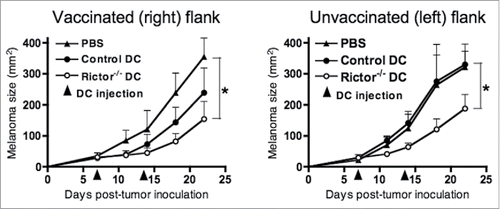
To test whether i.t. Rictor−/− DC could have an effect at a distant tumor site, mice with established tumors on their right and left flanks were injected i.t. on the right with PBS or control DC or Rictor−/− DC on days 7 and 14 post-tumor implantation, while tumors on the left of each animal were left untreated. Tumor sizes were monitored longitudinally every 3–4 d until the termination of the experiment. The results show that Rictor−/− DC had an antitumor effect both in the treated tumor (right side) and the untreated tumor (left side), when compared with both PBS and control DC (). This result suggests that Rictor−/− DC have the potential to reduce the growth of a tumor distant from the site of therapeutic injection.
Figure 4. I.t.-delivery of Rictor−/− DC results in a therapeutic antitumor response in a contralateral tumor site. Mice bearing 7-d B16 tumors in each flank, with similar mean total tumor sizes, were injected with 50 µL of PBS or 50 µL of PBS containing 106 control DC or 106 Rictor−/− DC on the right flanks. Tumors on the left flank of each animal were left untreated. Animals received an identical treatment on day 14 post-tumor implantation. Treated and untreated tumor sizes (in mm2) for each animal were then monitored longitudinally every 3–4 d, and are reported as means + SD for five animals per group. *p < 0.05.
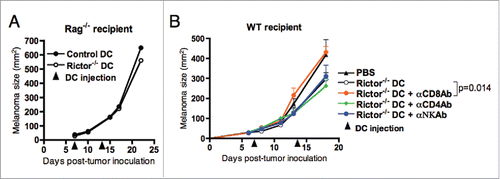
The therapeutic benefit(s) provided by i.t.-delivered Rictor−/− DC is CD8+ T cell-dependent
Given that DC can cross-present antigens acquired from dead/dying cancer cells to T lymphocytes, promoting the development of tumor-specific immune responses,Citation28 we next assessed whether the therapeutic effect of Rictor−/− DC on melanoma growth was mediated by activated T cells. To test this, control or Rictor−/− DC were injected i.t. into B16 melanomas established in Rag−/− mice (deficient in B and T cells), and melanoma growth monitored over 21 d. Our results show no difference in melanoma growth between mice treated with either control DC or Rictor−/− DC (), indicating the importance of B and/or T cells in controlling melanoma growth in Rictor−/− DC-treated recipients. As humoral immune responses have generally been found to be poor at regulating B16 tumor growth,Citation29 we focused further attention on discriminating the roles of T cell subsets in our protective therapy model. WT mice bearing established day-7 B16 melanomas were injected i.t. with Rictor−/− DC in combination with specific monoclonal antibodies to deplete CD8+ or CD4+ T cells, or NK cells in vivo. These studies revealed that depletion of CD8+ T cells, but not CD4+ T cells or NK cells, mitigated the therapeutic benefit associated with i.t.-delivered Rictor−/− DC (). These findings suggest that therapy-induced CD8+ T cells are the primary host immune cell population responsible for regulating tumor growth after treatment with Rictor−/− DC.
Figure 5. Reduction of B16 melanoma growth is dependent mainly on CD8+ T cells. (A) Rag1−/− mice bearing day 7 s.c. B16 melanomas were given i.t. injections of 106 control DC or Rictor−/− DC. DC injection was repeated at day 14 post-tumor inoculation. Tumor growth was monitored every 3–4 d and is shown as means + SD for five animals per group. (B) C57BL/6 mice bearing s.c. B16 melanomas were treated on days 7 and 14 post-tumor inoculation by i.t. injection of 106 Rictor−/− DC. On days 6, 13, and 20 after tumor inoculation, different groups of mice were injected i.p. with control IgG, anti-CD4, anti-CD8+, or anti-NK Ab to specifically deplete these cell populations in vivo. Tumor growth was monitored every 3–4 d and is reported as mean + SD for five animals per group.
Rictor−/− DC administration promotes increased frequencies of cytotoxic CD8+ tumor-infiltrating lymphocytes (TIL) in vivo
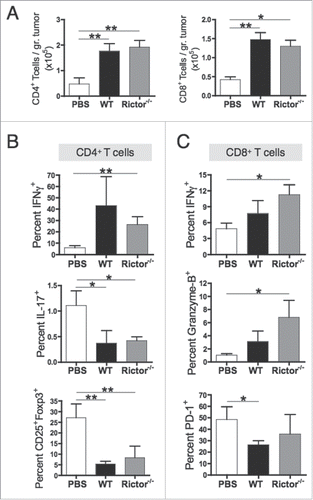
We next analyzed whether i.t.-injected Rictor−/− DC were superior to control DC in priming antitumor T cell responses in vivo. Mice bearing 7-day established B16 melanomas were injected i.t. with 1 × 106 Rictor−/− or control DC, with an identical treatment given 1 week later. Tumors were harvested at day 21 and infiltrating T cells isolated and stained. Although tumors from Rictor−/− DC-treated mice were smaller than those from untreated and control DC-treated mice, the total numbers of CD4+ and CD8+ T cells were similar between control DC- and Rictor−/− DC-treated animals, and higher than in untreated mice (). These results suggest that both control and Rictor−/− DC induce some degree of TIL recruitment, but that only the T cells primed by Rictor−/− DC are able to regulate tumor growth. We next investigated the phenotype of these therapy-induced TIL. Our results show that CD4+ TIL from Rictor−/− DC- or control DC-treated mice displayed similar frequencies of IFNγ+, IL-17+ and regulatory (CD25+Foxp3+) T cell populations (). However, CD8+ TIL from Rictor−/− DC-injected animals displayed higher frequencies of IFNγ+ and granzyme-B+ cytotoxic subpopulations versus CD8+ TIL recovered from controls (). Based on recent evidence that PD-1 plays a key role in modulating immune-mediated regulation of melanoma growth,Citation30 PD-1 expression was evaluated on CD8+ TIL. We observed no significant difference between Rictor−/− DC- and control DC-injected animals in PD-1 expression (). When taken together, these results suggest that i.t.-delivered Rictor−/− DC promote increased recruitment and therapeutic action of cytotoxic CD8+ TIL in vivo.
Figure 6. Rictor−/− DC administration promotes increased frequencies of tumor-infiltrating cytotoxic T cells. C57BL/6 mice bearing s.c. B16 melanomas were injected i.t. at days 7 and 14 post-tumor inoculation with 106 control DC or Rictor−/− DC. At day 21 post-tumor inoculation, tumors were harvested and tumor-infiltrating lymphocytes obtained and stained for the indicated surface markers and intracellular cytokines. (A) Plots show absolute numbers of CD4+ and CD8+ tumor-infiltrating T cells divided by the respective tumor weight. (B) Frequencies of IFNγ+, IL-17+ and CD25+Foxp3+ cells in the total CD4+ T cell population. (C) Frequencies of IFNγ+, granzyme-B+ and PD-1+ cells in the total CD8+ T cell population. Data are shown as means + SD for three to four animals per group, and two independent experiments. *p < 0.05 and **p < 0.01.
I.t.-delivery of Rictor−/− DC promotes improved activation of antitumor CD8+ T cells in the periphery
To evaluate T cell activation in the periphery, spleens were harvested from treated mice at day 21, and isolated CD8+ T cells subsequently evaluated for tumor reactivity. As shown in , splenic CD8+ T cells isolated from Rictor−/− DC-treated mice produced higher levels of IFNγ and granzyme-B in response to stimulation with B16-melanoma cells than splenic CD8+ T cells from untreated or control DC-injected mice. Furthermore, CD8+ T cells from Rictor−/− DC-injected mice proliferated significantly more than control DC-primed T cells in response to B16 antigens (). Quantitation of cytokines in the supernatants of these CD8+ T cell-tumor cell co-cultures also indicated higher secretion levels of IFNγ, IL-6 and TNF-α in those cultures containing Rictor−/− DC-primed CD8+ T cells versus control DC- or no DC-primed CD8+ T cells (). Next, we tested the direct cytotoxicity of these in vivo-primed splenic CD8+ T cells against B16 melanoma cells in vitro. T cells were cultured for 18 h with CFSE-labeled B16 melanoma cells and violet-labeled irrelevant control EL4 thymoma cells at different T cell:target cell ratios. The viability of residual B16 and EL4 cells was then analyzed by flow cytometry (). The results show an increased ability of Rictor−/− DC-primed CD8+ T cells to kill B16 tumor cells compared to control CD8+ T cells (). The percentage killing of irrelevant EL4 target cells was < 4% for all conditions, supporting the B16-specific nature of CD8+ T cell-mediated cytotoxicity. These results support the conclusion that i.t. delivery of Rictor−/− DC improves the cross-priming of cytotoxic, type-1 anti-melanoma CD8+ T cells in the periphery compared to untreated or control DC-treated mice.
Figure 7. Rictor−/− DC administration enhances activation of antitumor CD8+ T cells in the periphery. C57BL/6 mice bearing s.c. B16 melanomas were treated at days 7 and 14 post-tumor inoculation with i.t. injection of 106 control DC or Rictor−/− DC. At day 21 post-tumor inoculation, spleens were harvested and (A, B, C) splenocytes stained with CFSE and then stimulated with irradiated (100 Gy) B16 cells (ratio 10:1 respectively) in the presence of 30 IU/mL recombinant human IL-2 for 5 d in 24-well culture plates. (A, B) Responder T cells were analyzed for CD8+ and intracellular IFNγ and granzyme-B, showing (A) representative plots and (B) means + SD for six animals per group reported from three independent experiments performed. (C) Cell-free supernatants of these cultures were analyzed for IFNγ, IL-6 and TNF-α. Box plots show median, 25%- and 75%-quartiles, and both extreme values. (D, E) Splenic CD8+ T cells isolated from untreated (PBS), control-DC- or Rictor−/− DC-treated mice were cultured with CFSE-labeled B16 melanoma cells and violet-labeled irrelevant control EL4 thymoma cells at ratios of 5:1:1, 2:1:1 or 1:1:1 (where a unit of 1 = 5 × 104 cells) for 18 h. Cells were analyzed by flow cytometry to determine the percentage of viable B16 (green) or EL4 (violet) cells, as shown in (D), versus a control consisting of a 1:1 mixture of each of the labeled tumor cell lines in the absence of T cells. (E) Results are reported as means +/− SD of data obtained from five mice/cohort. Percent killing was determined based on the formula: 100% × [1 – (percentage of viable tumor cells in the presence of T cells/percentage of viable tumor cells in the absence of T cells)]. *p < 0.05, **p < 0.01, ***p < 0.001.
![Figure 7. Rictor−/− DC administration enhances activation of antitumor CD8+ T cells in the periphery. C57BL/6 mice bearing s.c. B16 melanomas were treated at days 7 and 14 post-tumor inoculation with i.t. injection of 106 control DC or Rictor−/− DC. At day 21 post-tumor inoculation, spleens were harvested and (A, B, C) splenocytes stained with CFSE and then stimulated with irradiated (100 Gy) B16 cells (ratio 10:1 respectively) in the presence of 30 IU/mL recombinant human IL-2 for 5 d in 24-well culture plates. (A, B) Responder T cells were analyzed for CD8+ and intracellular IFNγ and granzyme-B, showing (A) representative plots and (B) means + SD for six animals per group reported from three independent experiments performed. (C) Cell-free supernatants of these cultures were analyzed for IFNγ, IL-6 and TNF-α. Box plots show median, 25%- and 75%-quartiles, and both extreme values. (D, E) Splenic CD8+ T cells isolated from untreated (PBS), control-DC- or Rictor−/− DC-treated mice were cultured with CFSE-labeled B16 melanoma cells and violet-labeled irrelevant control EL4 thymoma cells at ratios of 5:1:1, 2:1:1 or 1:1:1 (where a unit of 1 = 5 × 104 cells) for 18 h. Cells were analyzed by flow cytometry to determine the percentage of viable B16 (green) or EL4 (violet) cells, as shown in (D), versus a control consisting of a 1:1 mixture of each of the labeled tumor cell lines in the absence of T cells. (E) Results are reported as means +/− SD of data obtained from five mice/cohort. Percent killing was determined based on the formula: 100% × [1 – (percentage of viable tumor cells in the presence of T cells/percentage of viable tumor cells in the absence of T cells)]. *p < 0.05, **p < 0.01, ***p < 0.001.](/cms/asset/8e59c3af-4a3d-40c8-952d-bfa19769f9af/koni_a_1146841_f0007_b.gif)
Discussion
The main novel findings in this study are as follows: (i) the superior systemic therapeutic efficacy of mTORC2-deficient (vs. control) DC when injected i.t. into mice with 1 or more established B16 melanomas, (ii) the associated enhanced (cross)priming of type-1 cytotoxic (IFNγ+GzB+) CD8+ T cells in secondary lymphoid organs, (iii) treatment-associated enhancement of type-1 cytotoxic CD8+ T cell recruitment into the TME and (iv) treatment-associated reduction in MDSC regulatory populations in the TME. Despite the predicted ability of Rictor−/− DC to also prime and polarize tumor-specific CD4+ T cell responses in treated mice, treatment benefits were CD4+ independent, and the phenotype and functional status of CD4+ T cells in the periphery and the TME of Rictor−/− DC-treated mice was indistinguishable from control treated animals. Our current results are consistent with those reported by Amiel et al. where the injection of rapamycin-treated DC pulsed with LPS and OVA (as a vaccine) in OVA-B16 melanoma-bearing mice delayed tumor growth and increased the frequency of Ag-specific CD8+ TIL.Citation31 Our findings are also in line with the need to promote enhanced frequencies of type-1 tumor-specific CD8+ TIL clinically in order to manifest therapeutic benefit in cancer patients.Citation32-34 Indeed, increased IFNγ secretion from tumor-specific CD8+ T cells has been strongly correlated with prolonged survival in patients with metastatic melanoma post-immunotherapy.Citation35,36
The ability of DC to cross-prime type-1 cytotoxic CD8+ T cell responses has been strongly linked to DC production of IL-12p70.Citation37,38 Furthermore, the clinical efficacy of DC-based vaccines in patients with glioma or melanoma has been strongly associated with the level of IL-12p70 produced by patient-derived DC at the time of their injection.Citation23,39 It has also been reported previously that i.t. DC expression of IL-12 is necessary for the priming of CD8+ T cell responses in patients with breast cancer.Citation40 These reports are consistent with our findings that the superior efficacy of Rictor−/− DC appears to be tied to their differential high levels of IL-12p70 production, associated with the preferential cross-priming of type-1 cytotoxic CD8+ T cells in vivo.
The effective migration of DC to the lymph nodes and subsequent activation of antigen-specific T cells is thought to be important for the success of immunotherapy with DC-based vaccines. Curiously, injected Rictor−/− DC did not display a migratory advantage to tumor-draining secondary lymphoid organs when compared with control DC, which was consistent with their very similar levels of CCR7 expression. Yet the few migratory injected Rictor−/− DC appeared better at activating therapeutic type-1 CD8+ T cells than control DC, pointing to the improved function of Rictor−/− DC once present in secondary lymphoid organs. Our finding of inefficient trafficking of injected DC to lymphoid tissues in vivo is highly consistent with clinical reports. In melanoma patients, only approximately 4% of DC injected intradermally reach the draining lymph nodes, with these few DC still sufficient to induce antigen-specific immunologic responses.Citation41 The fact that most of the injected DC remained at the injection site, in our case the tumor, leads us to hypothesize that the T cell priming may also take place in the TME. Indeed, this has been reported previously for Tbet-engineered DC injected directly into established tumor lesions in mice.Citation42 Recent evidence suggests that extranodal T cell priming in ectopic or tertiary lymphoid organs correlates with better overall survival of cancer-bearing patients.Citation43,44 Therefore, we must be open to the likelihood that it is not necessary for injected DC to migrate to secondary lymph nodes in order to effectively prime tumor-specific T cell responses that yield therapeutic benefit.
Overall, our findings may contribute to the design of more effective DC-based vaccines and cancer immunotherapies. In this regard, transfection with siRNA specific for Rictor has been performed successfully both in mouse BMDCCitation45 and in human monocyte-derived DC.Citation46 Therefore, this strategy could be used to inhibit mTORC2 in patient-derived DC-based cancer immunotherapy approaches. Another strategy to consider would involve the use of dual mTORC1/2 inhibitors, which are currently being studied in translational and clinic studies as potential antitumor agents (ClinicalTrials.gov Identifier: NCT02064608, NCT02403895, NCT02193633).Citation15,47 These agents could be used to inhibit mTORC1/2 in DC ex vivo, with subsequent injection of the conditioned DC. Furthermore, vaccines based on mTORC2-deficient DC could be administered in combination with other anticancer therapies that would be anticipated to improve their effectiveness. For example, they could be combined with agents that help to recruit effector cells into the tumor, as has been shown for tyrosine kinase inhibitors, such as dasatinib.Citation48 Agents that normalize the tumor blood vasculature could also be used in combination with DC-based vaccines. In this regard, drugs targeting vascular endothelial growth factor (VEGF) or its receptor have shown encouraging clinical outcomes in early-phase studies when combined with chemotherapy,Citation49 demonstrating survival extension in patients with metastatic melanoma.Citation50 Immune targeting of the NOTCH antagonist delta-like 1 homolog (DLK1) has also shown promising results in normalizing the vasculature in the TME of renal cell carcinoma mice models.Citation51 Furthermore, while in the present study Rictor−/− DC promoted the loss of MDSC in the TME, the negative impact of Treg on type-1 CD8+ T cell function could likely still serve as a therapeutic impediment. Hence, combined use of Rictor−/− DC with regimens designed to minimize/eliminate Treg numbers/function (i.e., anti-CTLA-4) or to enhance T effector function/survival (i.e. anti-PD1, anti-PD-L1, anti-Tim3, adoptive T cell therapy) would be expected to yield even greater treatment-associated benefits in the cancer setting.Citation52,53
In summary, our data show that i.t. injection of LPS-activated, mTORC2-deficient DC markedly slows established B16 melanoma growth in mice in a CD8+ T-cell-dependent manner. Administration of Rictor−/− DC reduces the frequency of MDSC within the tumors, and enhances numbers of INFγ+ and granzyme-B+ CD8+ T cells both in host spleens and tumors. These data indicate that selective inhibition of mTORC2 in DC can enhance the therapeutic efficacy of DC-based vaccines for cancer immunotherapy.
Materials and methods
Mice
Male C57BL/6J (B6) and B6.129S7-Rag1tm1Mom/J (Rag1−/−) mice were purchased from The Jackson Laboratory. CD11c-specific Rictor−/− mice were madeCitation54 by crossing floxed Rictor miceCitation55 (generously provided by Drs Keunwook Lee and Mark Boothby, Vanderbilt University School of Medicine) with B6 mice expressing CD11c-Cre (The Jackson Laboratory). The genetic background of crossed mice was verified by PCR genotyping and littermates used as negative controls. All studies were performed according to an Institutional Animal Care and Use Committee-approved protocol in accordance with NIH guidelines.
Generation of bone marrow-derived DC
Bone marrow (BM) cells were harvested and cultured to generate DC as described,Citation56 using mouse rGM-CSF and rIL-4 (both 1000 U/mL; R&D Systems, CAA26822 and P07750). On day 7 of culture, DC were purified using anti-CD11c immunomagnetic beads (Miltenyi Biotec, 130-052-001). Where indicated, the TLR4 ligand LPS (100 ng/mL; Salmonella minnesota R595; Alexis Biochemicals, ALX-581-008) was used to stimulate DC for 16–18h.
B16 therapy model
B6 mice received an s.c. injection of 105 B16 melanoma cells in the right flank on day 0. On day 7, mice were randomized into treatment cohorts (five mice each). 106 control DC or Rictor−/− DC were injected intratumorally (i.t.) in 50 μL of PBS on days 7 and 14 post-tumor inoculation. Tumor size was assessed every 3–4 d and recorded in mm2 by determining the product of the largest perpendicular diameters measured by vernier calipers.
For bilateral treatment models, mice were challenged s.c. in each flank with 105 B16 melanoma cells. After 7 d, the animals were randomized into cohorts (five mice/group) with similar mean total tumor sizes, with tumors on the right flanks injected with 50 μL of PBS or 50 μL of PBS containing 106 control DC or 106 Rictor−/− DC. Animals received identical treatment on day 14 post-tumor implantation. Tumors on the left flank of each animal were not treated. Treated and untreated tumor sizes (in mm2) for each animal were then monitored longitudinally every 3–4 d, until the termination of the experiment on day 22 post-tumor inoculation.
Tracking of DC in vivo
Control DC or Rictor−/− DC were stained with CFSE following the manufacturer's instructions (Vibrant CFDASE Cell Tracer Kit; Invitrogen, V12883) and 5 × 106 injected i.t. on day 7 post-tumor inoculation. After 3 d, tumors, spleens and inguinal lymph nodes were harvested and cells isolated and stained for analysis.
In vivo immune cell subset depletion
On days 6, 13, and 20 after tumor inoculation, mice were injected i.p. with purified antibodies (Ab): 50–100 μg rat isotype control IgG (Sigma, I4131), 50 μg anti-CD4+ monoclonal Ab (GK1.5; ATCC®, TIB-207), 100 μg anti-CD8+ monoclonal Ab (ATCC®, TIB-105), or 50 μg anti-asialo GM1 polyclonal Ab (DAKO, 986–10001), as previously described.Citation57 Ab-mediated depletion was 95% effective for the targeted immune cell subset based on flow cytometric analysis of peripheral blood mononuclear cells obtained by tail venipuncture from treated mice 24 h after Ab administration (data not shown).
Tumor-infiltrating leukocyte isolation from B16 tumors
B16 tumors were carefully excised from the mice and weighed. The tumors were then minced and washed using PBS with 5 % fetal bovine serum and 2 % EDTA. Thereafter, they were disrupted mechanically in digestion media [pre-warmed RPMI-1640 with 0.25 mg/mL of Liberase TL (Roche, 05401020001) and 0.02 mg/mL of DNAse I (Sigma, D4527)], and incubated in the same media with agitation for 1 h. The cell suspensions were centrifuged at 1500 rpm for 5 min, several times, until the supernatants were clear. The cells were then filtered through a 40 μm mesh and leukocytes recovered using Lympholyte M (Cedarlane Laboratories Limited, CL5035) gradient centrifugation for 20 min at room temperature. After washing, leukocytes were enumerated using trypan blue (Gibco, 15250–06) in a Neubauer counting chamber.
Evaluation of T cell responses against B16 tumors ex vivo
For ex vivo T cell stimulation, spleens were harvested from mice 21 d after tumor inoculation and pooled splenocytes stimulated with irradiated (100 Gy) B16 cells (ratio 10:1 respectively) in the presence of 30 IU/mL recombinant human IL-2 (30 U/mL; R&D Systems, 202-IL-050) for 5 d in 24-well culture plates. The responder T cells were analyzed for surface markers and intracellular cytokines. Cell-free supernatants were stored at −80°C until cytokine quantitation.
In vitro flow-based cytotoxicity assay
Splenocytes were harvested on the day of euthanasia from bilateral tumor-bearing mice treated by i.t. injection of PBS, control DC or Rictor−/− DC. CD8+ T cells were then isolated from individual, disaggregated spleens by specific MACS (Miltenyi). After trypsinization to obtain single-cell suspensions, B16 melanoma cells and irrelevant control EL4 thymoma (ATCC) cells were labeled for 30 min at 37°C in culture media containing 100 nM carboxyfluorescein succinimidyl ester (CFSE; ThermoFisher) or 1 μM Violet Proliferation Dye 450 (BD Biosciences), respectively. After washing with PBS, T cells and tumor target cells were added to wells of V-bottom (96-well) culture plates (Costar) at T cell:B16 melanoma:EL4 thymoma cell ratios of 5:1:1, 2:1:1 or 1:1:1 (where a unit of 1 = 5 × 104 cells) in a total volume of 200 µL of complete media and further incubated at 37°C for 18 h. Cells were then re-suspended in PBS containing 2% paraformaldehyde and analyzed by flow cytometry to determine the percentage of viable B16 (green) or EL4 (violet) cells versus a control consisting of a 1:1 mixture of each of the labeled tumor cell lines in the absence of T cells. Percent killing was determined based on the formula: 100% × [1 – (percentage of viable tumor cells in the presence of T cells/percentage of viable tumor cells in the absence of T cells)]. Results are reported as means +/– SD of data obtained from five mice/cohort.
Flow cytometric analyses
For assessment of intracellular cytokine expression, T cells were examined after 4–5 h restimulation with PMA (0.5 μg/mL, Sigma-Aldrich, P8139) and ionomycin (1 μg/mL, Sigma-Aldrich, I0634) in the presence of GolgiStop (BD Biosciences, 555029). After extensive washes, cells were stained with appropriate Ab, and fixed/permeabilized (eBioscience, 00-5123-43 and 00-5223-56) prior to intracellular staining. Fluorochrome-conjugated mAbs were purchased from eBioscience, BD Bioscience, Biolegend or Miltenyi Biotec. Appropriately-conjugated, isotype-matched IgGs served as controls. Data were acquired with a LSR II or Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo (TreeStar).
Cytokine quantitation
Cytokines in DC or MLR culture supernatants were quantified by ELISA (eBiosciences) and/or cytometric bead array (CBA; BD Bioscience, 558266) where indicated, following the manufacturer's instructions.
Statistical analyses
Results are expressed as mean ± 1SD. Significant differences between groups were determined using the Student's t test or one-way ANOVA test (GraphPad Prism), with p < 0.05 considered significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Kanishka Mohib for sharing the protocol to process B16 tumors and isolate tumor-infiltrating leukocytes.
Funding
This work was supported by NIH R01 CA169118 (W.J.S.) and NIH P50 CA121973 (R.J.F.). D.R.R. is the recipient of a Thomas E. Starzl Postdoctoral Fellowship in Transplantation Biology. A.R.W. is in receipt of an NIAID T32 institutional research training pre-doctoral fellowship (AI74490).
References
- Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014; 15:e257-e267; PMID:24872109; http://dx.doi.org/10.1016/S1470-2045(13)70585-0.
- Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013; 39:38-48; PMID:23890062; http://dx.doi.org/10.1016/j.immuni.2013.07.004.
- Butterfield LH. Dendritic cells in cancer immunotherapy clinical trials: are we making progress? Front Immunol 2013; 4:454; PMID:24379816; http://dx.doi.org/10.3389/fimmu.2013.00454.
- Galluzzi L, Senovilla L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, Tartour E, Zitvogel L, Kroemer G. Trial watch: Dendritic cell-based interventions for cancer therapy. OncoImmunology 2012; 1:1111-34; PMID:23170259; http://dx.doi.org/10.4161/onci.21494.
- Mac Keon S, Ruiz MS, Gazzaniga S, Wainstok R. Dendritic cell-based vaccination in cancer: therapeutic implications emerging from murine models. Front Immunol 2015; 6:243; PMID:26042126; http://dx.doi.org/10.3389/fimmu.2015.00243.
- Yang Q, Guan K-L. Expanding mTOR signaling. Cell Res 2007; 17:666-81; PMID:17680028; http://dx.doi.org/10.1038/cr.2007.64.
- Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 2009; 9:324-37; PMID:19390566; http://dx.doi.org/10.1038/nri2546.
- Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 2010; 33:301-11; PMID:20870173; http://dx.doi.org/10.1016/j.immuni.2010.09.002.
- Sarbassov DD, Ali SM, Kim D-H, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol CB 2004; 14:1296-302; PMID:15268862; http://dx.doi.org/10.1016/j.cub.2004.06.054.
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal 2009; 2:pe24; PMID:19383975; http://dx.doi.org/10.1126/scisignal.267pe24.
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017.
- Huang S, Houghton PJ. Inhibitors of mammalian target of rapamycin as novel antitumor agents: from bench to clinic. Curr Opin Investig Drugs Lond Engl 2000 2002; 3:295-304; PMID:25231953; http://dx.doi.org/10.1093/annonc/mdu456
- Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, Noguchi S, Perez A, Rugo HS, Deleu I et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol Off J Eur Soc Med Oncol ESMO 2014; 25:2357-62; http://dx.doi.org/10.1093/annonc/mdu456.
- Chan J, Kulke M. Targeting the mTOR signaling pathway in neuroendocrine tumors. Curr Treat Options Oncol 2014; 15:365-79; PMID:25092520; http://dx.doi.org/10.1007/s11864-014-0294-4.
- Sun S-Y. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett 2013; 340:1-8; PMID:23792225; http://dx.doi.org/10.1016/j.canlet.2013.06.017.
- Ashworth RE, Wu J. Mammalian target of rapamycin inhibition in hepatocellular carcinoma. World J Hepatol 2014; 6:776-82; PMID:25429315; http://dx.doi.org/10.4254/wjh.v6.i11.776.
- Pópulo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci 2012; 13:1886-918; http://dx.doi.org/10.3390/ijms13021886.
- Damsky W, Micevic G, Meeth K, Muthusamy V, Curley DP, Santhanakrishnan M, Erdelyi I, Platt JT, Huang L, Theodosakis N et al. mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 2015; 27:41-56; PMID:25584893; http://dx.doi.org/10.1016/j.ccell.2014.11.014.
- Werzowa J, Koehrer S, Strommer S, Cejka D, Fuereder T, Zebedin E, Wacheck V. Vertical inhibition of the mTORC1/mTORC2/PI3K pathway shows synergistic effects against melanoma in vitro and in vivo. J Invest Dermatol 2011; 131:495-503; PMID:21048785; http://dx.doi.org/10.1038/jid.2010.327.
- Ho AL, Musi E, Ambrosini G, Nair JS, Deraje Vasudeva S, de Stanchina E, Schwartz GK. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PloS One 2012; 7:e40439; PMID:22808163; http://dx.doi.org/10.1371/journal.pone.0040439.
- Gopal YNV, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1 and oxidative phosphorylation in melanoma. Cancer Res 2014; 74:7037-47; PMID:25297634; http://dx.doi.org/10.1158/0008-5472.CAN-14-1392.
- Raïch-Regué D, Rosborough BR, Watson AR, McGeachy MJ, Turnquist HR, Thomson AW. mTORC2 deficiency in myeloid dendritic cells enhances their allogeneic Th1 and Th17 stimulatory ability after TLR4 ligation in vitro and in vivo. J Immunol 2015; 194:4767-76; PMID:25840913; http://dx.doi.org/10.4049/jimmunol.1402551.
- Carreno BM, Becker-Hapak M, Huang A, Chan M, Alyasiry A, Lie W-R, Aft RL, Cornelius LA, Trinkaus KM, Linette GP. IL-12p70-producing patient DC vaccine elicits Tc1-polarized immunity. J Clin Invest 2013; 123:3383-94; PMID:23867552; http://dx.doi.org/10.1172/JCI68395.
- Lasek W, Zagożdżon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother CII 2014; 63:419-35; PMID:24514955; http://dx.doi.org/10.1007/s00262-014-1523-1.
- Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol 2015; 35:S185–S198; PMID:25818339; http://dx.doi.org/10.1016/j.semcancer.2015.03.004.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253-68; PMID:22437938; http://dx.doi.org/10.1038/nri3175.
- Umansky V, Sevko A. Melanoma-induced immunosuppression and its neutralization. Semin Cancer Biol 2012; 22:319-26; PMID:22349515; http://dx.doi.org/10.1016/j.semcancer.2012.02.003.
- Spel L, Boelens J-J, Nierkens S, Boes M. Antitumor immune responses mediated by dendritic cells: How signals derived from dying cancer cells drive antigen cross-presentation. Oncoimmunology 2013; 2:e26403; PMID:24482744; http://dx.doi.org/10.4161/onci.26403.
- Yang C, Lee H, Pal S, Jove V, Deng J, Zhang W, Hoon DSB, Wakabayashi M, Forman S, Yu H. B cells promote tumor progression via STAT3 regulated-angiogenesis. PloS One 2013; 8:e64159; PMID:23734190; http://dx.doi.org/10.1371/journal.pone.0064159.
- Mamalis A, Garcha M, Jagdeo J. Targeting the PD-1 pathway: a promising future for the treatment of melanoma. Arch Dermatol Res 2014; 306:511-9; PMID:24615548; http://dx.doi.org/10.1007/s00403-014-1457-7.
- Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, Pearce EJ. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol 2012; 189:2151-8; PMID:22826320; http://dx.doi.org/10.4049/jimmunol.1103741.
- Nordlund JJ, Kirkwood JM, Forget BM, Milton G, Albert DM, Lerner AB. Vitiligo in patients with metastatic melanoma: a good prognostic sign. J Am Acad Dermatol 1983; 9:689-96; PMID:6643767; http://dx.doi.org/10.1016/S0190-9622(83)70182-9.
- Palucka AK, Ueno H, Connolly J, Kerneis-Norvell F, Blanck J-P, Johnston DA, Fay J, Banchereau J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J Immunother 2006; 29:545-57; PMID:16971810; http://dx.doi.org/10.1097/01.cji.0000211309.90621.8b.
- Keilholz U, Weber J, Finke JH, Gabrilovich DI, Kast WM, Disis ML, Kirkwood JM, Scheibenbogen C, Schlom J, Maino VC et al. Immunologic monitoring of cancer vaccine therapy: results of a workshop sponsored by the Society for Biological Therapy. J Immunother 2002; 25:97-138; PMID:12074049; http://dx.doi.org/10.1097/00002371-200203000-00001.
- Kirkwood JM, Lee S, Moschos SJ, Albertini MR, Michalak JC, Sander C, Whiteside T, Butterfield LH, Weiner L. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/-granulocyte-monocyte colony-stimulating factor and/or IFN-alpha2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clin Cancer Res 2009; 15:1443-51; PMID:19228745; http://dx.doi.org/10.1158/1078-0432.CCR-08-1231.
- Schaefer C, Butterfield LH, Lee S, Kim GG, Visus C, Albers A, Kirkwood JM, Whiteside TL. Function but not phenotype of melanoma peptide-specific CD8(+) T cells correlate with survival in a multiepitope peptide vaccine trial (ECOG 1696). Int J Cancer 2012; 131:874-84; PMID:22021080; http://dx.doi.org/10.1002/ijc.26481.
- Gately MK, Wolitzky AG, Quinn PM, Chizzonite R. Regulation of human cytolytic lymphocyte responses by interleukin-12. Cell Immunol 1992; 143:127-42; PMID:1352483; http://dx.doi.org/10.1016/0008-8749(92)90011-D.
- Trinchieri G. Proinflammatory and immunoregulatory functions of interleukin-12. Int Rev Immunol 1998; 16:365-96; PMID:9505196; http://dx.doi.org/10.3109/08830189809043002.
- Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, Mintz AH, Engh JA, Bartlett DL, Brown CK et al. Induction of CD8+ T-Cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with type-1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol 2011; 29:330-6; PMID:21149657; http://dx.doi.org/10.1200/JCO.2010.30.7744.
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014; 26:623-37; PMID:25446896; http://dx.doi.org/10.1016/j.ccell.2014.09.006.
- Verdijk P, Aarntzen EHJG, Lesterhuis WJ, Boullart ACI, Kok E, van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JFM et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res 2009; 15:2531-40; PMID:19318472; http://dx.doi.org/10.1158/1078-0432.CCR-08-2729.
- Chen L, Taylor JL, Sabins NC, Lowe DB, Qu Y, You Z, Storkus WJ. Extranodal induction of therapeutic immunity in the tumor microenvironment after intratumoral delivery of Tbet gene-modified dendritic cells. Cancer Gene Ther 2013; 20:469-77; PMID:23846252; http://dx.doi.org/10.1038/cgt.2013.42.
- Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, Schell MJ, Sondak VK, Weber JS, Mulé JJ. Twelve-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep 2012; 2:765; PMID:23097687; http://dx.doi.org/10.1038/srep00765.
- Chen L, Fabian KL, Taylor JL, Storkus WJ. Therapeutic use of dendritic cells to promote the extranodal priming of anti-tumor immunity. Front Immunol 2013; 4:388; PMID:24348473; http://dx.doi.org/10.3389/fimmu.2013.00388
- Brown J, Wang H, Suttles J, Graves DT, Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J Biol Chem 2011; 286:44295-305; PMID:22045807; http://dx.doi.org/10.1074/jbc.M111.258053.
- Wei W-C, Liu C-P, Yang W-C, Shyur L-F, Sheu J-H, Chen S-S, Yang N-S. Mammalian target of rapamycin complex 2 (mTORC2) regulates LPS-induced expression of IL-12 and IL-23 in human dendritic cells. J Leukoc Biol 2015; 97:1071-80; PMID:25877925; http://dx.doi.org/10.1189/jlb.2A0414-206RR.
- Jiang Q, Weiss JM, Back T, Chan T, Ortaldo JR, Guichard S, Wiltrout RH. mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Res 2011; 71:4074-84; PMID:21540234; http://dx.doi.org/10.1158/0008-5472.CAN-10-3968.
- Lowe DB, Bose A, Taylor JL, Tawbi H, Lin Y, Kirkwood JM, Storkus WJ. Dasatinib promotes the expansion of a therapeutically superior T-cell repertoire in response to dendritic cell vaccination against melanoma. Oncoimmunology 2014; 3:e27589; PMID:24734217; http://dx.doi.org/10.4161/onci.27589.
- Kim KB. Is there a role for targeting vascular endothelial growth factor/receptor axis in the treatment of patients with metastatic melanoma? Cancer 2013; 119:477-80; PMID:22915026; http://dx.doi.org/10.1002/cncr.27756.
- Algazi AP, Cha E, Ortiz-Urda SM, McCalmont T, Bastian BC, Hwang J, Pampaloni MH, Behr S, Chong K, Cortez B et al. The combination of axitinib followed by paclitaxel/carboplatin yields extended survival in advanced BRAF wild-type melanoma: results of a clinical/correlative prospective phase II clinical trial. Br J Cancer 2015; 112:1326-31; PMID:25867272; http://dx.doi.org/10.1038/bjc.2014.541.
- Chi Sabins N, Taylor JL, Fabian KP, Appleman LJ, Maranchie JK, Stolz DB, Storkus WJ. DLK1: A novel target for immunotherapeutic remodeling of the tumor blood vasculature. Mol Ther 2013; 21:1958-68; PMID:23896726; http://dx.doi.org/10.1038/mt.2013.133.
- Perez-Gracia JL, Labiano S, Rodriguez-Ruiz ME, Sanmamed MF, Melero I. Orchestrating immune check-point blockade for cancer immunotherapy in combinations. Curr Opin Immunol 2014; 27:89-97; PMID:24485523; http://dx.doi.org/10.1016/j.coi.2014.01.002.
- Cohen J, Sznol M. Therapeutic combinations of immune-modulating antibodies in melanoma and beyond. Semin Oncol 2015; 42:488-94; PMID:25965368; http://dx.doi.org/10.1053/j.seminoncol.2015.02.014.
- Rosborough BR, Raich-Regue D, Matta BM, Lee K, Gan B, DePinho RA, Hackstein H, Boothby M, Turnquist HR, Thomson AW. Murine dendritic cell rapamycin-resistant and rictor-independent mTOR controls IL-10, B7-H1, and regulatory T-cell induction. Blood 2013; 121:3619-30; PMID:23444404; http://dx.doi.org/10.1182/blood-2012-08-448290.
- Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 2010; 32:743-53; PMID:20620941; http://dx.doi.org/10.1016/j.immuni.2010.06.002.
- Morelli AE, Zahorchak AF, Larregina AT, Colvin BL, Logar AJ, Takayama T, Falo LD, Thomson AW. Cytokine production by mouse myeloid dendritic cells in relation to differentiation and terminal maturation induced by lipopolysaccharide or CD40 ligation. Blood 2001; 98:1512-23; PMID:11520802; http://dx.doi.org/10.1182/blood.V98.5.1512.
- Qu Y, Chen L, Pardee AD, Taylor JL, Wesa AK, Storkus WJ. Intralesional delivery of dendritic cells engineered to express T-bet promotes protective type 1 immunity and the normalization of the tumor microenvironment. J Immunol 2010; 185:2895-902; PMID:20675595; http://dx.doi.org/10.4049/jimmunol.1001294.