
ABSTRACT
Adoptive natural regulatory T cell (nTreg) therapy has improved the outcome for patients suffering from graft-versus-host disease (GVHD) following allogeneic hematopoietic cell transplantation (Allo-HCT). However, fear of broad immune suppression and subsequent dampening of beneficial graft-versus-leukemia (GVL) responses remains a challenge. To address this concern, we generated alloreactive induced Tregs (iTregs) from resting CD4+ or CD8+ T cells and tested their ability to suppress GVH and maintain GVL responses. We utilized major mismatched and haploidentical murine models of HCT with host-derived lymphoma or leukemia cell lines to evaluate GVH and GVL responses simultaneously. Alloreactive CD4+ iTregs were effective in preventing GVHD, but abrogated the GVL effect against aggressive leukemia. Alloreactive CD8+ iTregs moderately attenuated GVHD while sparing the GVL effect. Hence, we reasoned that using a combination of CD4+ and CD8+ iTregs could achieve the optimal goal of Allo-HCT. Indeed, the combinational therapy was superior to CD4+ or CD8+ iTreg singular therapy in GVHD control; importantly, the combinational therapy maintained GVL responses. Cellular analysis uncovered potent suppression of both CD4+ and CD8+ effector T cells by the combinational therapy that resulted in effective prevention of GVHD, which could not be achieved by either singular therapy. Gene expression profiles revealed alloreactive CD8+ iTregs possess elevated expression of multiple cytolytic molecules compared to CD4+ iTregs, which likely contributes to GVL preservation. Our study uncovers unique differences between alloreactive CD4+ and CD8+ iTregs that can be harnessed to create an optimal iTreg therapy for GVHD prevention with maintained GVL responses.
Abbreviations
| Allo-HCT | = | allogeneic hematopoietic cell transplantation |
| DAPK2 | = | death-associated protein kinase 2 |
| DLI | = | donor lymphocyte infusion; |
| GVHD | = | graft-versus-host disease; |
| GVL | = | graft-versus-leukemia; |
| HLA | = | human leukocyte antigen |
| iTreg | = | induced regulatory T cell |
| miHAgs | = | minor histocompatibility antigens |
| Mln | = | mesenteric lymph nodes |
| nTreg, | = | natural regulatory T cell |
| ROI | = | region of signal intensity |
| Teffs | = | effector T cells |
Introduction
Allo-HCT was established to treat patients suffering from hematological malignancies encompassing leukemias and lymphomas. Allo-HCT replaces a patient's diseased hematopoietic system with a healthy donor stem cell source. The degree of human leukocyte antigen (HLA) disparity dictates the beneficial GVL response, where donor T cells recognize and kill residual leukemic cells, and the deleterious GVHD, where donor T cells recognize and damage host epithelial tissues.Citation1 Unfortunately, a therapy that can separate these two responses within allo-HCT recipients has yet to be established. Presently, the primary therapy for GVHD is broad immune suppression, which can lead to relapse of the underlying malignancy or life-threatening infections.Citation2
nTregs and iTregs play key roles in suppressing autoimmunityCitation3 and in maintaining immune homeostasis.Citation4 nTregs arise from the thymus, recognize self-antigen with high affinity, and represent a stable cell lineage.Citation5 iTregs arise in the periphery,Citation6 recognize non-self-antigens, and are less stable due to their propensity to lose Foxp3 expression, the master transcription factorCitation7 associated with Treg suppressive function.
Harnessing Treg's inherent suppressive nature to control GVHD has been an ongoing area of investigation. Initial clinical trials using nTregs have yielded positive results.Citation8-10 However, nTregs constitute a small percentage of circulating T cells and require expansion due to the high number needed for GVHD attenuationCitation10; thus, reaching optimal cell doses for patientsCitation9 remains an obstacle. Likewise, nTregs are non-selective suppressors, which could result in significant impairment of the beneficial GVL response. Conversely, iTregs can be generated in large numbersCitation11 and enriched for antigen specificityCitation12,13 which could result in targeted separation of GVH and GVL responses. However, iTregs as a GVHD therapy is surrounded with controversy, with some investigators citing beneficial effectsCitation14-17 and others a complete lack of efficacy.Citation18-20 The lack of a consensus on iTreg therapy seems to stem from differences in activation agents (polyclonal vs. antigen-specific), polarizing cytokines, and infusion schedules employed across labs. Yet, it is clear that antigen specificity enhances iTreg potency in GVHD attenuation, but these findings used monoclonal antigen specific iTregs that cannot be readily translated to the clinic.Citation12,13 Therefore, to extend antigen-specific iTreg therapy into clinical settings, we tested the efficacy of alloreactive iTregs in attenuating GVHD and preserving GVL responses.
Utilizing multiple well-established murine models of allo-HCT, we demonstrated that alloreactive CD4+ iTregs potently suppress GVHD; yet, GVL is completely lost against aggressive leukemia. Alloreactive CD8+ iTregs are less stable than CD4+ iTregs, but still moderately attenuated GVHD while preserving the GVL effect. To overcome the obstacles of each singular therapy, we established a combination therapy using both CD4+ and CD8+ iTregs, which was superior in GVHD attenuation and preservation of GVL responses. Mechanistic studies demonstrated, even under the same polarization conditions, CD4+ and CD8+ iTregs represent two distinct regulatory populations that suppress GVH or maintain GVL responses. Finally, we uncovered unique gene expression patterns between alloreactive CD4+ and CD8+ iTregs, which provide further understanding of these cell types and rationale to apply a combinational iTreg therapy after Allo-HCT.
Results
Alloreactive CD4+ iTregs suppression of GVHD is antigen specific
Our previous findings illustrate that antigen specificity increases iTregs suppressive potential.Citation13 However, HY-specific monoclonal iTregs have limited potential to be translated to the clinic. Therefore, we generated alloreactive CD4+ iTregs and tested their ability to alleviate GVHD while maintaining GVL activity. We utilized generation conditions previously describedCitation16; as a control, polyclonal CD4+ iTregs were generated.Citation12 iTregs generated with H2d+ DCs (Fig. S1A) were highly suppressive toward CD4+ and CD8+ T-cell responses to H2d+ alloantigens, and more effective than polyclonal iTregs in vitro (Fig. S1B). These alloreactive iTregs were also capable of suppressing allogeneic responses in vivo, reflected by a significant reduction of T-cell proliferation and IFNγ secretion (Fig. S1C and D). Using a Major histocompatibility complex (MHC)-mismatched B6 to BALB/c HCT model, we injected enriched H2d-alloreactive iTregs three days prior to effector T cells (Teffs), and found that these iTregs significantly attenuated GVHD, displayed by improved survival and body weight maintenance () and decreased pathological damage in all tissues.()
Figure 1. Alloreactive CD4+ iTregs attenuate GVHD in high potency. BALB/c mice were lethally irradiated (700cGy) and transplanted with 5 × 106 TCD-BM from B6 donors and 1 × 106 CD25hi H2d specific iTregs. Three days later, 1.0 × 106 CD25 depleted B6 Teffs were injected alone or with a second dose of H2d iTregs (1–0.5 × 106). Recipients were monitored for survival (A) and body weight loss (B) for 80 d, n = 12. In separate experiments, GVHD target organs were excised for pathological scoring (C). Recipient BALB/c mice were lethally irradiated (700cGy) and transplanted with 5 × 106 TCD-BM from B6 donors and 1 × 106 CD25hi H2d or H2k reactive iTregs. Three days later, 1.0 × 106 CD25-depleted B6 Teffs were injected alone or with a second dose of H2d or H2k reactive iTregs (1–0.5 × 106). Recipients were monitored for survival (D) and body weight loss (E) for 80 d, n = 12. *p < .05; **p < .01; ***p < .001. Error bars indicate the mean of standard error. BM stands for (TCD-BM) only; +Teff stands for (BM + Teff); + H2d-iTregs stands for (BM+Teff+H2d-iTregs), same abbreviations used henceforth.
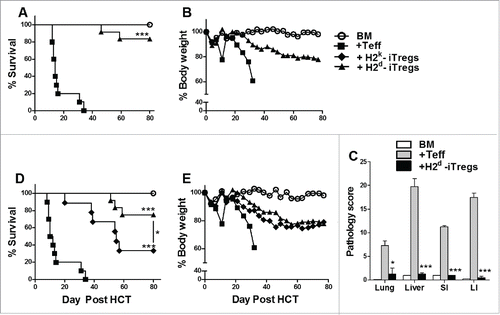
To evaluate the impact of iTreg allo-reactivity in the prevention of GVHD, we generated third party (H2k)-reactive iTregs and compared the ability of H2d- vs. H2k-alloreactive iTregs to suppress GVHD. As expected, host (H2d)-reactive iTregs were superior to third party (H2k)-reactive iTregs in suppressing GVHD (). To understand the mechanism of how iTreg allo-reactivity affects iTreg function, we followed the fate of CD4+ iTregs to observe their effect on Teffs. Host (H2d)-reactive iTregs displayed greater persistence as compared to H2k-iTregs (Fig. S2A and B). Furthermore, host (H2d)-reactive iTregs potently suppressed the expansion of CD4+ Teffs and reduced the expansion of CD8+ Teffs (Fig. S2 A and B). Conversely, the third party (H2k)-reactive iTregs reduced the expansion of CD4+ Teffs but had no effect of CD8+ Teffs, which may account for their ability to moderately attenuate GVHD lethality. Taken together, alloreactive iTregs effectively attenuate GVHD and host-reactive iTregs have greater potency in GVHD prevention.
Alloreactive CD4+ iTregs impair GVL activity against aggressive leukemia
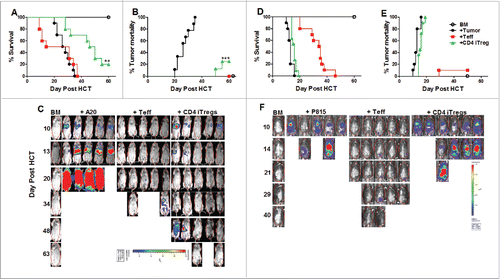
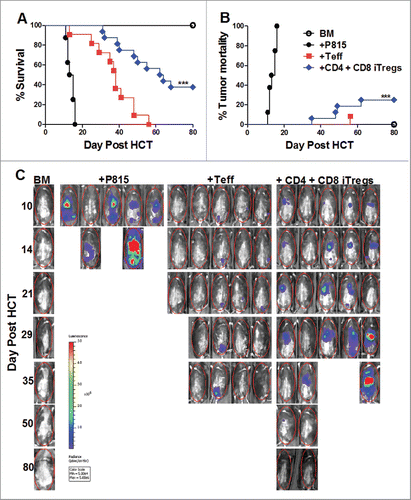
Since GVHD is closely linked to GVL activity, we tested whether alloreactive CD4+ iTregs could suppress GVHD without impairing GVL responses. To further increase iTreg yield and translational potential, we tested non-enriched iTreg's (bulk after culture) pathogenicity following allo-HCT, and found non-enriched alloreactive iTregs (CD4+, CD8+, or in combination) were not pathogenic (Fig. S3A and B). Consequently, we used non-enriched iTregs in all experiments thereafter. Initially, we found that CD4+ iTregs significantly attenuated GVHD () while largely preserving GVL activity against A20 B cell lymphoma (). To ensure the maintenance of GVL function was not tumor specific, we used a haploidentical B6 to BDF1 HCT model with host-original mastocytoma (P815). Surprisingly, the addition of CD4+ iTregs completely abrogated the ability of Teffs to mediate GVL function, reflected by all recipients with CD4+ iTregs succumbing to tumor mortality () with heavy tumor load () similar to those with BM plus tumor alone. To understand the strikingly different outcomes, we encountered between A20 and P815 models, we found A20 cells express significantly higher levels of MHC-II and CD86 compared to P815 (data not shown), suggesting that A20 lymphoma is more immunogenic than P815, possibly contributing to the differential sensitivities we observed. Taken together, while CD4+ alloreactive iTregs are effective in GVHD control, they may impair the GVL activity against aggressive and low immunogenic tumors.
Figure 2. Alloreactive CD4+ iTregs impair GVL activity against aggressive leukemia. BALB/c mice were lethally irradiated and transplanted with 5 × 106 TCD-BM, 1 × 106 CD4+ iTregs, and 1 ×104 luc-A20 cells. Three days later, 0.5 × 106 CD25-depleted Teffs were injected. Recipients were monitored for survival (A), tumor mortality (B) and tumor load (C) for 80 d n = 10/group. BDF1 recipient mice were lethally irradiated (1200cGy) split dose 3 h apart and transplanted with 5 × 106 TCD-BM, 2 × 106 CD4+ iTregs, and 5 × 103 luc-P815 cells. Three days later, 2 × 106 CD25-depleted Teff cells were injected. Recipients were monitor for survival (D) tumor mortality (E) and tumor load (F) for 60 d, n = 15/group. *p < .05; **p < .01; ***p < .001. Error bars indicate the mean of standard error.
CD8+ iTregs moderately attenuate GVHD and fully preserve GVL function
Recent studies showed that CD8+ iTregs arise after allogeneic HCT, which play a significant role in GVHD attenuation.Citation21 Interestingly, a recent report also suggested that human CD8+ iTregs possess direct cytotoxicity against LCL-tumor cell lines.Citation14 We therefore generated alloreactive CD8+ iTregs and evaluated their ability to control GVHD and preserve the GVL response. Using similar polarizing conditions, we were able to generate 30–40% Foxp3+ CD8+ iTregs (Fig. S4A). These CD8+ iTregs displayed significantly higher levels of CD39+CD73+, CTLA-4, and granzyme B compared to nTregs and CD4+ iTregs (Fig. S4B), and were equally suppressive of allogeneic responses as CD4+ iTregs (Fig. S4C). Despite potent suppressive function in vitro, alloreactive CD8+ iTregs only moderately attenuated GVHD (Fig. S5A), but fully preserved the GVL effect against P815 tumor (Fig. S5B and C). Interestingly, CD8+ iTregs themselves (+ CD8 iTregs no Teff) could significantly delay tumor mortality (Fig. S5A–C). In vitro cytotoxic assays confirmed CD8+ iTregs possess direct cytotoxicity against tumor cells (Fig. S5D). These results provide evidence that alloreactive CD4+ and CD8+ iTregs are fundamentally different concerning GVH and GVL responses, with CD4+ iTregs potently suppressing Teffs and CD8+ iTregs contributing cytolytic molecules to GVL maintenance.
Combination of CD4+ and CD8+ iTregs is superior to singular iTreg therapy in GVHD attenuation
We further reasoned that these two types of iTregs could compensate for each other's weaknesses and provide an optimized cellular therapy. We first assessed the potency of a combination therapy (CD4+ + CD8+ iTregs) to suppress GVHD using B6 to BALB/c HCT model. To negate an additive effect within the combinational therapy, the absolute iTreg product injected was equivalent to CD4+ or CD8 iTregs singular therapy. We observed that the inclusion of CD8+ iTregs did not abrogate the ability of CD4+ iTregs to alleviate GVHD, and in fact attenuated GVHD more potently than CD4+ iTreg singular therapy (). In correlation with our previous observations, CD4+ singular therapy significantly improved survival as compared with Teff only recipients (, p = 0.01). By using β-actin luciferase transgenic B6 donors to monitor the expansion of Teffs in vivo, we found that the combinational therapy significantly reduced the expansion of Teffs compared to either CD4+ or CD8+ iTreg singular therapy reflected by significantly decreased bioluminescent signal over time.()
Figure 3. Combinational therapy with CD4+ and CD8+ iTregs is superior to singular iTreg therapy in GVHD attenuation. BALB/c mice were lethally irradiated and transplanted with 5 × 106 TCD-BM, 1 × 106 CD4+ iTregs, 1 × 106 CD8+ iTregs, or 0.5 × 106 CD4+ + 0.5 × 106 CD8+ iTregs. Three days later, 0.5 × 106 CD25-depleted Teffs were injected. Recipients were monitored for survival and body weight loss for 80 d (A), n = 20/group. In separate experiments, 0.25 × 106 luc+Teff were injected and expansion was monitored over the first 40 d post HCT (B). BALB/c mice were lethally irradiated and transplanted as described in (A); however, Teffs were injected from Thy1.1+ reporter mice at 1 × 106/mouse. On days 5, 8, and 11 recipients spleen, liver, and mesenteric lymph nodes harvested and Teff expansion analyzed over time (C) as well as characteristics (D). Absolute numbers of iTregs were also quantified over time in all three organs (E). *p < .05; **p < .01; ***p < .001. Error bars indicate the mean of standard error.
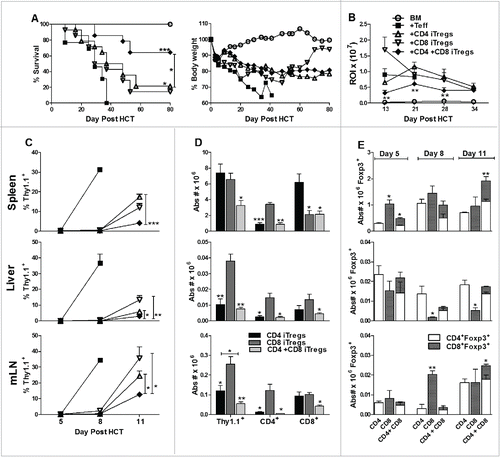
To understand the cellular mechanism of iTreg therapy on GVHD, we performed a time course study to monitor both Teff and iTregs after allo-HCT. Recipient BALB/c's spleen, liver, and mesenteric lymph nodes (mLN) were harvested on day 5, 8, and 11 post HCT. On day 5, Thy1.1+ Teffs were not readily detectable in any of the recipients. On day 8, the expansion of Teffs was significantly suppressed by all iTreg therapies (). On day 11, all the recipients with Teffs alone succumbed to GVHD. Combinational therapy with CD4+ and CD8+ iTregs controlled Teff expansion significantly better than either singular therapy (), which correlated with a less intense bioluminescence signal observed in the combinational therapy (). Interestingly, CD4+ iTregs potently controlled the expansion of CD4+, but not CD8+ Teffs, whereas CD8+ iTregs potently controlled the expansion of CD8+, but not CD4+ Teffs (). Furthermore, the combination of CD4+ and CD8+ iTregs potently controlled the expansion of both CD4+ and CD8+ Teffs (). To evaluate the fate of infused iTregs, we tabulated absolute numbers of residual Foxp3+ infused individual iTregs (Thy1.2+), which reflects iTregs expansion and stability. Overall, equivalent or greater expansion/stability of iTregs was observed when infused in combination as compared to each alone in recipient spleens and mLNs (). While CD8+ iTregs accumulated in mLN, there were significantly fewer in the liver when infused alone versus together with CD4+ iTregs (). To track the maintenance and stability of infused iTregs, we generated iTregs from Ly5.1+ B6 mice, performed HCT as described in , and evaluated Foxp3 expression 17 d post-transplant. Consistently, we found that CD8+ iTregs expanded more rapidly than CD4+ iTregs (Fig. S6A and C), albeit at the loss of Foxp3 expression. Given the injected iTregs were non-enriched (∼60% Foxp3 expression) at the day of transplant, we found CD4+ iTregs had significant retention of Foxp3 expression (∼40%) and were more stable than CD8+ iTregs. (Fig. S6A and B). Taken together, the combinational therapy was more effective in preventing GVHD than either singular therapy, likely because the former was able to control both CD4+ and CD8+ Teffs simultaneously.
Combinational therapy rescues the inhibited GVL response of CD4+ singular therapy
To exclude potential model specific phenomena, we further evaluated the efficacy of combinational therapy in B6 to BDF1 HCT model. We identified an appropriate combined dose of CD4+ and CD8+ iTregs that potently attenuated GVHD significantly better than either iTreg therapy alone. This was supported by pathological analysis of GVHD target organs, including the liver, lung, small intestine and colon (). Taken together with the data presented in , we conclude that the combinational therapy is superior to single iTreg therapy across different HCT models.
Figure 4. Combinational therapy reduces GVHD pathology and tumor burden. BDF1 recipient mice were lethally irradiated (1200cGy) and transplanted with 5 × 106 B6 TCD-BM, and either 2 × 106 CD4+ iTregs, 4 × 106 CD8+ iTregs, or a combination of both CD4+ and CD8+ iTregs, and 5 × 103 luc-P815 cells. Three days later, 2 × 106 Ly5.1+ CD25-depleted Teffs were injected. Fourteen days post BM injection, GVHD target organs were excised for pathological analysis. A representative photomicrograph of each group depicting the average disease score morphology (A) and quantification of pathological damages to organs (B) are shown. Before being euthanized, mice were injected with luciferin, and organs were excised followed by imaging using IVIS 200 imager. (C) A representative mice from each group n = 4. Organ bioluminescent was quantified using Living Imager software (D). *p < .05; **p < .01; ***p < .001. Error bars indicate the mean of standard error.
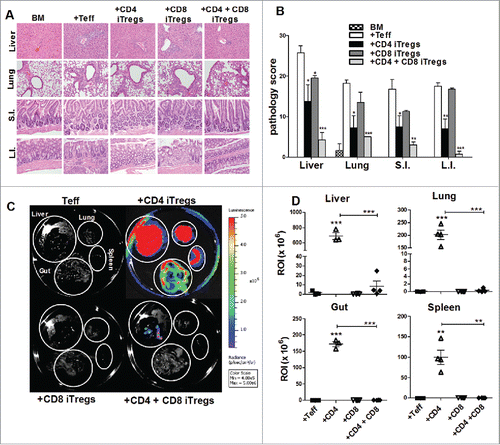
We next addressed the most pertinent question as to whether the combinational therapy can spare the GVL effect against P815. Fourteen days post HCT, we observed that the recipients given CD4+ iTregs alone had significantly increased tumor burden within the liver, lung, spleen, and gut as compared to those that received Teffs alone (). However, the recipients given either CD8+ iTregs alone or combinational therapy had significantly reduced tumor burden as compared to those that received CD4+ iTregs alone within all organs examined (). These results suggest that CD8+ iTregs could overcome the impaired GVL effect mediated by CD4+ iTregs. To extend this observation to long-term survival studies, we simultaneously evaluated the effect of combinational therapy on GVHD and GVL responses against P815. Indeed, the recipients with the combinational therapy had significantly improved long-term survival, resulting from reduced GVHD lethality and tumor mortality (). Taken together with data presented in and Fig. S5, we conclude that the combinational therapy has distinct advantages over either CD4+ or CD8+ iTreg singular therapy resulting in GVHD prevention and GVL preservation.
Figure 5. Combinational therapy preserves the GVL response. BDF1 recipient mice were lethally irradiated (1200cGY) and transplanted with 5 × 106 TCD-BM, 2 × 106 CD4+ iTregs, 4 × 106 CD8+ iTregs, and 5 × 103 luc-P815 cells. Three days later, 2 × 106 B6 CD25-depleted Teffs were injected. Recipients were monitored for survival (A), tumor mortality (B), and tumor load (C) for 80 d, n = 15. *p < .05; **p < .01; ***p < .001.
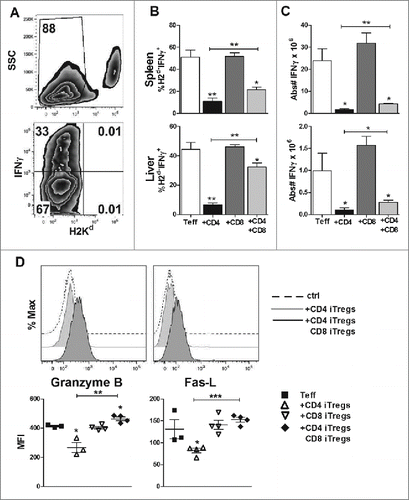
CD8+ iTregs contribute to the preserved GVL effect by maintaining cytolytic functions
To understand why the GVL effect was preserved upon addition of CD8+ iTregs, we analyzed the levels of effector molecules on all donor cells injected (Teffs + CD4+ and/or CD8+ iTregs) to accurately reflect the total cytolytic potential within recipient mice. Importantly, this analysis will not exclude the cytolytic molecules, such as IFNγ, granzyme B, and fas-L, which are secreted or expressed by CD8+ iTregs themselves (Fig. S4). Fourteen days post HCT, CD4+ iTregs alone significantly reduced the percentage of IFNγ+ cells among total donor T cells (H2d−, Teff + iTregs) in recipient spleens and livers (). The percentage of IFNγ+ cells among donor T cells was partially restored with addition of CD8+ iTregs (). To a lesser extent, the absolute number of H2d−IFNγ+ cells was also significantly increased in the recipients given combinational therapy compared to those of CD4+ iTregs alone (). Combinational therapy was also able to restore the expression of granzyme B and fas-L on H2d−CD8+ cells, which was impaired by CD4+ iTregs (). These results indicate that the combinational therapy achieves GVL preservation by maintaining sufficient overall levels of IFNγ secretion and cytolytic molecule expression within recipient mice.
Figure 6. CD8+ iTregs contribute to the preserved GVL effect mediated by combinational therapy. BDF1 mice were transplanted as stated in . Fourteen days post HCT, spleen and liver mononuclear cells were isolated. Representative flow gating strategy (A) displays IFNγ secretion from Teffs and injected iTregs (H2Kd−). Percentages of indicated cell populations, IFNγ production (B), and absolute number are quantified (C). Cells were also stained for granzyme B and Fas-L, with a corresponding isotype control (D) representative histogram analysis of the isotype control, CD4+ singular or combination therapy. Graphical quantification of the mean fluorescence intensity for each molecule is displayed in (D), n = 4. *p < .05; **p < .01; ***p < .001. Error bars indicate the mean of standard error.
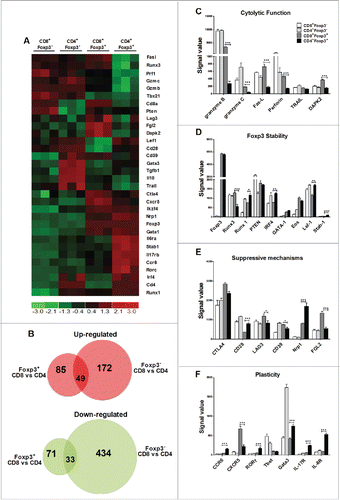
Alloreactive CD4+ and CD8+ iTregs display differential gene expression profiles
Given CD4+ and CD8+ iTregs had distinct effects on GVH and GVL responses; we strove to understand molecular differences between these two types of iTregs and if different molecular signatures could confirm our long-term studies. By using T cells from Foxp3-GFP reporter mice, we generated alloreactive iTregs and sorted pure CD4+Foxp3+ and CD8+Foxp3+ iTregs. The gene expression of alloreactive iTregs (Foxp3+) and Teffs (Foxp3−) was compared at the genomic level. Among 34,356 genes analyzed, even though generated under identical conditions, there were 235 genes differentially expressed between CD4+ and CD8+ Foxp3+ iTregs and 688 genes differentially expressed between CD4+ and CD8+ Foxp3− Teffs. By excluding genes that differ between CD4+ and CD8+ T cells independent of Foxp3 expression, we further identified 85 of the 235 genes uniquely upregulated and 71 of the 235 genes uniquely downregulated in in CD8+Foxp3+ versus CD4+Foxp3+ iTregs; as the expression of those 156 (85 + 71) genes were not significantly different in CD8+Foxp3− versus CD4+Foxp3− Teffs (). To identify the genes or molecules that may contribute to the distinct activity of CD4+ and CD8+ iTregs on GVH/GVL responses, we focused on those that regulate cytolytic function, Foxp3 stability, Treg suppressive mechanisms, and plasticity. 32 representative genes among those were displayed in a heat map (). Consistent with increased cytolytic activity of CD8+ iTregs and hence GVL preservation (Fig. S4 and S5), we found that granzyme B, granzyme C, fas-L, perforin, and DAPK2 (death-associated protein kinase 2) were significantly and uniquely upregulated in CD8+Foxp3+ versus CD4+Foxp3+ iTregs (). Among five transcription factors associated with Foxp3 stability, also known as the “Treg quintet,”Citation22 three of the five genes were significantly downregulated in CD8+Foxp3+ iTregs; reflecting their instability as compared to CD4+ counterparts (), which confirmed our findings in Fig. S6. Among the effector molecules that Tregs utilize to mediate suppression, we found that LAG3, CD39, and FGL2 (fibrinogen-like protein 2) were upregulated, whereas CD28 and Neuropilin-1 were downregulated in CD8+Foxp3+ versus CD4+Foxp3+ iTregs (Fig. 7E), suggesting that CD8+ and CD4+ iTregs may utilize different mechanisms to control the immune response. In addition, CD4+Foxp3+ cells upregulated genes associated with Treg plasticity, such as RORγt, IL-17R, IL-6R and CCR6, as compared with CD8+Foxp3+ cells, suggesting that CD8+ iTregs, although less stable, represent a more terminally differentiated population (). Overall, CD8+ iTregs and CD4+ iTregs are quite distinct on the molecular level, with CD8+ iTregs expressing more cytolytic molecules and less plasticity than CD4+ iTregs.
Figure 7. Alloreactive CD4+ and CD8+ iTregs display differential gene expression profiles. Naive CD4+ or CD8+ B6-Foxp3-GFP T cells were stimulated with BALB/c CD11chi DC with IL-2 only, or with TGFβ and RA three separate times. GFP/Foxp3− CD4+ or CD8+ cells were FACS sorted from the culture with IL-2 alone, and GFP/Foxp3+ CD4+ or CD8+ cells were sorted from the culture with IL-2 plus TGFβ and RA. Total RNA was isolated from 2 × 106 cells, and then labeled and hybridized to microarray chips for gene profiling. 32 representative genes display differential gene expression were shown (A). dChip software using FC > 2 and p < 0.05 identified unique genes that were exclusive either Foxp3 expression (B). Signal values for genes relevant to cytotoxic function (C) Treg stability (D) suppressive mechanisms (E) or Treg plasticity were displayed (F). * p < .05; **p < .01; ***p < .001. Error bars indicate the mean of standard error.
Discussion
In the current study, we demonstrate that host alloreactive iTregs are more potent in GVHD prevention than third-party alloreactive iTregs following allogeneic HCT. This is in accordance with our previous findingsCitation13 that antigen recognition increases iTreg's therapeutic potential. To date, an effective therapy that distinguishes between GVH and GVL responses is highly desirable for patients with hematologic malignancies. Here, we established the first combinational alloreactive iTreg therapy, using both CD4+ and CD8+ iTregs, to provide distinct suppression of GVHD with maintained GVL responses. We uncovered unique roles for alloreactive CD4+ and CD8+ iTregs following allo-HCT. CD4+ iTreg singular therapy potently suppressed GVHD but significantly impaired GVL responses. Conversely, CD8+ iTreg singular therapy had a lesser impact on GVHD but fully preserved GVL activity. Although the current study utilizes iTregs reactive to major (MHC) antigen, we have validated that the generation protocols can be applied to minor antigen (miHAg) settings (data not shown); suggesting miHAgs-reactive iTregs could be utilized to prevent GVHD induction from miHAg disparity. Together, the combinational therapy using these two types of iTregs had complementary roles: CD4+ iTregs reduced the expansion of Teffs to attenuate GVHD, and CD8+ iTregs maintained optimal activation and cytolytic potential of Teffs, thus preserving GVL function.
Although controversy surrounds iTreg therapy for GVHD attenuation, a critical look into the variability among generation conditions clearly demonstrates why differential results exist. First, the activating agent (polyclonal vs. physiological) results in noticeably different outcomes for GVHD, as polyclonal activated iTregs are unable to mitigate diseaseCitation19,20 while physiological activated iTregs significantly attenuate GVHD.Citation12,13,16 It has been found that physiologic activation imparts iTregs with more homing characteristics, like chemokine receptors, than polyclonal activated iTregs,Citation23 which may provide advantages for physiologic iTregs on GVHD attenuation. Second, the difference between the third polarizing cytokine (rapamycin vs. RA) has a significant effect on the iTreg product. Interestingly, the use of rapamycin resulted in an unstable iTreg therapy,Citation20 which could only be effective if rapamycin was continuously administered post-transplant, whereas RA has been shown to yield a stable iTreg product.Citation12,13,16 The mechanism of action of these two compounds is quite different, with rapamycin indirectly suppressing expansion of contaminating Teffs through mTOR inhibitionCitation24, whereas RA directly enhances the accessibility of the conserved non-coding sequence within the Foxp3 promoter, thus specifically enhancing Foxp3 stability. In this study, we established the optimal generation conditions for iTreg therapy on GVHD, physiological activation (allogeneic DCs) and RA polarization.
Of biological importance, we uncovered unique roles for CD4+ and CD8+ iTregs following allo-HCT. To our knowledge, this is the first study to show preferential suppression of CD4+ Teff by CD4+ iTregs and CD8+ Teff by CD8+ iTregs. It is possible that CD4+ iTregs may engage with alloantigens with high affinity presented by MHC II+ DCs. Furthermore, high levels of Neurophilin-1 on CD4+ iTregs () may potentiate a sustained interaction between Tregs and DCs, resulting in decreased DC maturation. Maturation statute of DCs would in turn affect activation of naïve CD4+ Teffs more than CD8+ Teffs. Our cellular and molecular analysis uncovered increased expression of multiple cytolytic molecules on CD8+, but not on CD4+ iTregs, which likely contributes to the ability of CD8+ iTregs to delay tumor relapse on their own. Of clinical importance, human alloreactive CD8+ iTregs were also found to possess direct cytotoxicity against LCL tumor cell lines,Citation14 providing a correlation between our findings and human specimens. Given their innate cytotoxic nature, these findings could be further expanded to use CD8+ iTregs as a novel cell source for donor lymphocyte infusion (DLI) given to patients suffering from primary disease relapse.Citation25-27 The stability of Foxp3 expression was another unique difference harbored by CD4+ and CD8+ iTregs, with CD4+ iTregs displaying increased expression of multiple Treg stabilizing molecules, like the “Treg quintet,” which was lacking in CD8+ iTregs. These molecular findings mirror our cellular time course study of alloreactive iTregs and other's observationsCitation20 that CD4+ iTregs represent a stable cell subset following allo-HCT, whereas CD8+ iTregs are less stable, especially in GVHD target organs. Importantly, we observed that non-enriched CD4+, CD8+, or combinational therapy, even with loss of Foxp3 expression, was not pathogenic following allo-HCT, suggesting those iTregs are safe in cellular therapy. Together, our study proves that CD4+ and CD8+ iTregs, even when generated under similar conditions, possess distinctly different characteristics. More work is needed to optimize the dose ratio of CD4+ and CD8+ iTregs and to manipulate either CD4+ or CD8+ iTreg characteristics to create a superior singular iTreg for GVH prevention and GVL maintenance. Similarly, multiple infusions of either CD4+ or CD8+ iTregs may be necessary to achieve sufficient efficacy and superior outcome in clinical settings.
Primary disease relapse is the leading cause of mortality, occurring in 37% of recipients,Citation28 following allo-HCT. Therefore, when establishing a new therapeutic option for GVHD attenuation, maintaining GVL function is essential. A consensus on whether Tregs may preserve or impair the GVL effect has not been reached, due to the limited number of tumor models tested. Notably, our studies show CD4+ iTregs abrogate GVL activity against evasive P815 mastocytoma, which correlates with impaired CML clearance found by others.Citation20 Mirroring our observations, alloreactive nTregCitation29 therapy resulted in maintained GVL activity against A20 but not against P815. The authors hypothesize that clearance of P815 is strongly dependent on alloreactive T cells, which are sharply reduced by infused Tregs, which we also observed in our CD4+ iTreg therapy. In the current study, we demonstrate a striking reduction in cytolytic molecules, such as granzyme B and fas-L on Teffs by CD4+ iTregs. This finding also provides a rationale to combine CD4+ iTregs with cytolytic CD8+ iTregs for GVL preservation.
We consider the most exciting finding in this work to be the combinational therapy's ability to overcome the impaired GVL effect mediated by CD4+ singular therapy. Two main factors contribute to the unhindered GVL effect observed within the combinational therapy: (1) CD8+ iTregs express cytolytic molecules (Fig. S4B), directly lyse tumor cells in vitro () and delay tumor mortality in vivo (Fig. S5A–C), which likely significantly accounted for the ability to overcome relapse in the combinational therapy; (2) although the expansion of Teffs is dramatically inhibited, the residual cells are polyfunctional, with sustained expression of IFNγ, granzyme B, and fas-L. While underlying mechanisms of GVL maintenance may still contribute, we identify these two factors, which provided a significant therapeutic effect. An effective therapy following allo-HCT requires conflicting immune responses, both suppression and cytolytic potential, and rarely can one immune cell type simultaneously possess each of these functions without complicated cellular manipulation. Uniquely, our combinational therapy utilizes the innate strengths of two immune cell types to achieve both suppression (CD4+) and cytolytic potential (CD8+) following allo-HCT. In summary, these findings provide the rationale for a targeted dual cellular therapy that reduces GVHD propensity and primary disease relapse, the two major hindrances obstructing allo-HCT success.
Materials and methods
Mice
C57BL/6 (B6; H-2b), BALB/c (H-2d), B6-Ly5.2 (H-2b), B6DF1 (B6 × DBA2) F1 (H-2b/d) were purchased from NCI. B6-Foxp3-GFP (H-2b) was kindly provided by A. Rudensky. All animals were housed in specific pathogen-free conditions in the America Association for Laboratory Animal Care-accredited Animal Resource Center at the Medical University of South Carolina. The Institutional Animal Care and Use Committee of the Medical University of South Carolina approved all work.
iTreg generation and enrichment
CD4+CD25− and CD8+CD25− resting T cells were purified from C57BL6 spleen and lymph nodes as previously described.Citation13 CD11c+ dendritic cells were purified from BALB/c mice using CD11c microbeads (Miltenyi). CD4+ or CD8+ T cells were stimulated with CD11c DCs at a 1:10 (DC: T cell) ratio with IL-2 (5 ng/mL), TGFβ (5 μg/mL), and retinoic acid (RA) (40 nM) for 5 or 4 d, respectively. iTregs were enriched from bulk culture using positive selection with CD25 microbeads and LS columns (Miltenyi). Purity of iTregs was routinely 85–95% as in our previous publication.Citation13
GVHD/GVL models
MHC-mismatched (B6→BALB/c)Citation30 and haploidentical (B6→BDF1)Citation31 BMT models were used as previously established. In all experiments, Teffs indicate CD25 depleted CD4+ and CD8+ T cells freshly isolated from naive donor mice. Recipients were monitored for survival and body weight loss twice/week. In GVL models, tumor doses ranged from 5,000 to 10,000 per mouse. Tumor mortality and GVHD mortality were distinguished as previously described.Citation30 Representative samples of GVHD target organs were excised from recipient's 14 d post-BMT and subjected to pathology scoring as previously described.Citation32
Flow cytometry and intracellular cytokine staining
Mononuclear cells were isolated from recipient spleen and liver as previously describedCitation33 and stained for surface molecules using standard flow cytometry protocols. Foxp3 was detected using established manufacturer's instructions Foxp3 fixation/permeablization kit (ebiosciences). Intracellular cytokines and granzyme expression were detected from spleen and liver lymphocytes following in vitro phorbol-myristate-acetate/ionomycin stimulation as previously described.Citation31 Cells were analyzed using the LSRII Diva software (BD biosciences) and FlowJo (Treestar).
Microarray
CD8+Foxp3+ and CD4++Foxp3+ cells were generated three independent times from Foxp3-GFP reporter mice and sorted purified (BD FACSAria II cell sorter), CD8+Foxp3− and CD4+Foxp3− were used as controls. RNA was extracted using Trizol and isolated using Qiagen RNAeasy Mini Kit and quality assessed using 2100 Bioanalyzer (Aglient). Total RNA (250 ng) was converted using the WT Plus Kit (Affymetrix) and hybridized to Mouse 430 2.0T Genechips (Affymetrix) according to manufacturer's instructions. Raw data (CEL files) were normalized by Robust Multi-array average (RMA) 70 using the gene-level summarization method in Expression Console software (Affymetrix). Comparisons were performed using dChip software 71 using strict criteria of absolute fold change> 2 and p <0.05. Calculations based on 50 iterations estimated false discovery rate (FDR) of < 1 % for all comparisons.
Statistics
For comparison of recipient survival among groups in GVHD experiments and tumor mortality, the log-rank test was used to determine statistical significance. To compare pathology scores, cytokine levels, and effector T cells, a Student t test was performed.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1146842_s02.pdf
Download PDF (1.5 MB)Acknowledgments
The authors would like to thank the technical assistance of the Moffitt Cancer Center and Medical University of South Carolina Hollings Cancer Center Flow Cytometry Core Facilities (Zachary Mcpherson). Further Alfred Moore of the small animal imaging core at Medical University of South Carolina for his assistance with animal imaging. We also extend great thanks to our vivarium staffs at both Moffitt Cancer Center and the Drug Discovery Building at Medical University of South Carolina.
Funding
Microarray studies were performed by the MUSC Proteogenomics Facility, which is supported by NIH grants P30GM103342 and P20GM103499 and through institutional support from MUSC's Office of the Provost. This work was supported in part by grants from the National Institutes of Health, National Cancer Institute (R01s CA118116 and CA169116) (X.-Z.Y).
References
- Fu J, Heinrichs J, Yu XZ. Helper T-cell differentiation in graft-vs.-host disease after allogeneic hematopoietic stem cell transplantation. Archivum Immunologiae Et Therapiae Experimentalis 2014; 62:277-301; PMID:24699629; http://dx.doi.org/10.1007/s00005-014-0284-z
- Pasquini MC, Zhu X. Current uses and outcomes of hematopoietic stem cell transplantation: CIBMTR Summary Slides, 2015. Available at: http://www.cibmtr.org.
- Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol 2003; 15:430-5; PMID:12819471; http://dx.doi.org/10.1097/00002281-200307000-00010
- Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med 2005; 201:723-35; PMID:15753206; http://dx.doi.org/10.1084/jem.20041982
- Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol 2001; 2:301-6; PMID:11276200; http://dx.doi.org/10.1038/86302
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med 2003; 198:1875-86; PMID:14676299; http://dx.doi.org/10.1084/jem.20030152
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299:1057-61; PMID:12522256; http://dx.doi.org/10.1126/science.1079490
- Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, Mysliwska J, Hellmann A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol 2009; 133:22-6; PMID:19559653; http://dx.doi.org/10.1016/j.clim.2009.06.001
- Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 2011; 117:1061-70; PMID:20952687; http://dx.doi.org/10.1182/blood-2010-07-293795
- Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, Del Papa B, Zei T, Ostini RI, Cecchini D et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011; 117:3921-8; PMID:21292771; http://dx.doi.org/10.1182/blood-2010-10-311894
- Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL, June CH, Miller JS, Wagner JE, Blazar BR. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplantation 2011; 11:1148-57; PMID:21564534; http://dx.doi.org/10.1111/j.1600-6143.2011.03558.x
- Semple K, Yu Y, Wang D, Anasetti C, Yu XZ. Efficient and selective prevention of GVHD by antigen-specific induced Tregs via linked-suppression in mice. Biol Blood Marrow Transplantation 2011; 17:309-18; PMID:21224010; http://dx.doi.org/10.1016/j.bbmt.2010.12.710
- Li J, Heinrichs J, Haarberg K, Semple K, Veerapathran A, Liu C, Anasetti C, Yu XZ. HY-Specific Induced Regulatory T Cells Display High Specificity and Efficacy in the Prevention of Acute Graft-versus-Host Disease. J Immunol 2015; 195:717-25; PMID:26048147; http://dx.doi.org/10.4049/jimmunol.1401250
- Zheng J, Liu Y, Liu Y, Liu M, Xiang Z, Lam KT, Lewis DB, Lau YL, Tu W. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Sci Translational Med 2013; 5:168ra9; PMID:23325802; http://dx.doi.org/10.1186/1479-5876-11-168
- Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, McKenna DH, Bromberg JS, Levine BL, Riley JL et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Translational Med 2011; 3:83ra41; PMID:21593401; http://dx.doi.org/10.1126/scitranslmed.3001809
- Sela U, Olds P, Park A, Schlesinger SJ, Steinman RM. Dendritic cells induce antigen-specific regulatory T cells that prevent graft versus host disease and persist in mice. J Exp Med 2011; 208:2489-96; PMID:22084406; http://dx.doi.org/10.1084/jem.20110466
- Beres AJ, Haribhai D, Chadwick AC, Gonyo PJ, Williams CB, Drobyski WR. CD8+ Foxp3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol 2012; 189:464-74; PMID:22649199; http://dx.doi.org/10.4049/jimmunol.1200886
- Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, Huehn J, Ganser A, Forster R, Prinz I. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur J Immunol 2009; 39:3091-6; PMID:19750478; http://dx.doi.org/10.1002/eji.200939432
- Beres A, Komorowski R, Mihara M, Drobyski WR. Instability of Foxp3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clin Cancer Res 2011; 17:3969-83; PMID:21558402; http://dx.doi.org/10.1158/1078-0432.CCR-10-3347
- Zhang P, Tey SK, Koyama M, Kuns RD, Olver SD, Lineburg KE, Lor M, Teal BE, Raffelt NC, Raju J et al. Induced regulatory T cells promote tolerance when stabilized by rapamycin and IL-2 in vivo. J Immunol 2013; 191:5291-303; PMID:24123683; http://dx.doi.org/10.4049/jimmunol.1301181
- Robb RJ, Lineburg KE, Kuns RD, Wilson YA, Raffelt NC, Olver SD, Varelias A, Alexander KA, Teal BE, Sparwasser T et al. Identification and expansion of highly suppressive CD8(+)FoxP3(+) regulatory T cells after experimental allogeneic bone marrow transplantation. Blood 2012; 119:5898-908; PMID:22538855; http://dx.doi.org/10.1182/blood-2011-12-396119
- Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, Gazit R, Adoro S, Glimcher L, Chan S et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol 2012; 13:972-80; PMID:22961053; http://dx.doi.org/10.1038/ni.2420
- Zhao C, Shi G, Vistica BP, Hinshaw SJ, Wandu WS, Tan C, Zhang M, Gery I. Induced regulatory T-cells (iTregs) generated by activation with anti-CD3/CD28 antibodies differ from those generated by the physiological-like activation with antigen/APC. Cell Immunol 2014; 290:179-84; PMID:25038545; http://dx.doi.org/10.1016/j.cellimm.2014.06.004
- Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 2009; 9:324-37; PMID:19390566; http://dx.doi.org/10.1038/nri2546
- Luznik L, Fuchs EJ. Donor lymphocyte infusions to treat hematologic malignancies in relapse after allogeneic blood or marrow transplantation. Cancer Control 2002; 9:123-37; PMID:11965233
- Loren AW, Porter DL. Donor leukocyte infusions after unrelated donor hematopoietic stem cell transplantation. Curr Opin Oncol 2006; 18:107-14; PMID:16462177; http://dx.doi.org/10.1097/01.cco.0000208781.61452.d3
- Porter D, Levine JE. Graft-versus-host disease and graft-versus-leukemia after donor leukocyte infusion. Seminars Hematol 2006; 43:53-61; http://dx.doi.org/10.1053/j.seminhematol.2005.09.005
- Pasquini MC ZX. Current uses and outcomes of hematopoietic stem cell transplantation: 2014 CIBMTR Summary Slides. 2014
- Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, Cohen JL. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest 2003; 112:1688-96; PMID:14660744; http://dx.doi.org/10.1172/JCI17702
- Haarberg KM, Li J, Heinrichs J, Wang D, Liu C, Bronk CC, Kaosaard K, Owyang AM, Holland S, Masuda E et al. Pharmacologic inhibition of PKCalpha and PKCtheta prevents GVHD while preserving GVL activity in mice. Blood 2013; 122:2500-11; PMID:23908466; http://dx.doi.org/10.1182/blood-2012-12-471938
- Wu Y, Heinrichs J, Bastian D, Fu J, Nguyen H, Schutt S, Liu Y, Jin J, Liu C, Li QJ et al. MicroRNA-17-92 controls T-cell responses in graft-versus-host disease and leukemia relapse in mice. Blood 2015; 126(11):1314-23; PMID:26138686; http://dx.doi.org/10.1182/blood-2015-02-0627356.
- Liang Y, Liu C, Djeu JY, Zhong B, Peters T, Scharffetter-Kochanek K, Anasetti C, Yu XZ. Beta2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood 2008; 111:954-62; PMID:17928532; http://dx.doi.org/10.1182/blood-2007-05-089573
- Yu Y, Wang D, Liu C, Kaosaard K, Semple K, Anasetti C, Yu XZ. Prevention of GVHD while sparing GVL effect by targeting Th1 and Th17 transcription factor T-bet and RORgammat in mice. Blood 2011; 118:5011-20; PMID:21856864; http://dx.doi.org/10.1182/blood-2011-03-340315