
ABSTRACT
Given their recognized ability to kill acute myeloid leukemia (AML) blasts both in vitro and in vivo, Vγ9Vδ2 T cells are of growing interest in the design of new strategies of immunotherapy. We show that the Butyrophilin3A (BTN3A, CD277) subfamily is a critical determinant of Vγ9Vδ2 TCR-mediated recognition of human primary AML blasts ex vivo. Moreover, anti-BTN3A 20.1 agonist monoclonal antibodies (mAbs) can trigger BTN3A on AML blasts leading to further enhanced Vγ9Vδ2 T cell-mediated killing, but this mAb had no enhancing effect upon NK cell-mediated killing. We show that monocytic differentiation of primary AML blasts accounts for their AminoBisphosphonate (N-BP)-mediated sensitization to Vγ9Vδ2 T cells. In addition, anti-BTN3A 20.1 mAbs could specifically sensitize resistant blasts to Vγ9Vδ2 T cells lysis and overcome the poor effect of N-BP treatment on those blasts. We confirmed the enhancement of Vγ9Vδ2 T cells activity by anti-BTN3A 20.1 mAb using a human AML xenotransplantation mouse model. We showed that anti-BTN3A 20.1 mAb combined with Vγ9Vδ2 T cells immunotherapy could increase animal survival and decrease the leukemic burden in blood and bone marrow. These findings could be of great interest in the design of new immunotherapeutic strategies for treating AML.
Abbreviations
| Ab | = | Antibody |
| ADCC | = | Antibody-dependent cell cytotoxicity |
| AHSCT | = | Allogeneic hematopoietic stem cell transplantation |
| AML | = | Acute myeloid leukemia |
| BM | = | Bone marrow |
| BrHpp | = | Bromohydrine pyrophosphate |
| BTN | = | Butyrophilin |
| Chr | = | chromosome |
| DNAM-1 | = | DNAX accessory molecule-1 |
| GvHD | = | Graft-versus-host-disease |
| HV | = | Healthy volunteer |
| Ig | = | Immunoglobulin |
| IPP | = | Isopentenyl diPhosphate |
| mAb | = | Monoclonal antibody |
| MHC | = | Major histocompatibility complex |
| N-BP | = | Nitrogen-containing bisphosphonates |
| NKG2D | = | Natural killer group 2 member D |
| NSG | = | NOD-SCID-common-γ-chain-knockout mice |
| pAg | = | phosphoAntigen |
| RLI | = | Receptor-linked IL-15 |
| TCR | = | T cell receptor |
| UPN | = | Unique patient number |
| ZOL | = | Zoledronate |
Introduction
Allogeneic haematopoietic stem cell transplantation (AHSCT) has highlighted a major role for the immune system in the control of residual AML.Citation1 AHSCT still remains the most effective therapy for most AML, though its use is still restricted to selected cases and outcomes are limited by both relapse and graft-versus-host-disease (GvHD). New approaches including the development of immunotherapy strategies are being actively investigated that may improve outcomes in AHSCT. Innate immune cells, notably NK cells and γδ T cells, are of particular interest on account of their ability to kill hematologicalCitation2,3 or solid tumor cellsCitation4 without inducing GvHD.Citation5,6
The anti-leukemic role of γδ T cells was suggested by a correlation between Vδ1 T cells expansion and prognosis following AHSCT.Citation7,8 Aside from Vδ1 T cells, Vγ9Vδ2 T cells, the main circulating subset, account for 1–10% of human T lymphocytes. Their T cell receptors (TCR) sense non-peptidic-phosphorylated phosphoAntigens (pAgs), such as isopentenyl diphosphate (IPP), an intermediary metabolite of the human mevalonate pathway.Citation9 In malignant cells, the blockade of this pathway with pharmacological agents such as N-BPs leads to IPP accumulation that triggers Vγ9Vδ2 T cells MHC-unrestricted functions.Citation10,11
Furthermore, we have recently shown that Vγ9Vδ2 T cells can specifically recognize and kill AML blasts in vitro and in a xenograft model and that patients with active AML have lower counts of Vγ9Vδ2 T cells.Citation12 Those Vγ9Vδ2 T cells have reduced expansion potential and a differentiation profile biased toward an effector memory phenotype. This phenotype can be reversed in vitro after blasts removal or after patients achieve remission,Citation12 supporting a model of blast-induced impairment of Vγ9Vδ2 T cells differentiation and function. Attempts to bypass this mechanism of escape to Vγ9Vδ2 T cells rely on TCR direct agonists, including bromohydrin pyrophosphate (BrHpp), a synthetic pAg, or indirect agonists, such as zoledronate (ZOL), an N-BP.Citation10 The anti-leukemic effect of TCR agonists was supported by a trial showing partial remission in two AML patients receiving N-BP with IL-2.Citation13 As Vγ9Vδ2 T cells are altered in patients,Citation12,14 the adoptive transfer of these cells emerges as a promising alternative.Citation15 However, 50% of N-BP-treated blasts still failed to enhance allogeneic γδ T cells cytotoxicity in vitro, suggesting that novel-triggering agents are still warranted.Citation16
Among the B7-related family, the butyrophilin3A (BTN3A) subfamily comprises three isoforms (BTN3A1, BTN3A2 and BTN3A3)Citation17 that contain two extracellular immunoglobulin (Ig) domains (IgV and IgC) and a transmembrane domain. Only BTN3A1 and BTN3A3 contain a B30.2 cytoplasmic domain. BTN3A members are broadly expressed, notably by most immune cells and various tumors.Citation17,18 The anti-BTN3A 20.1 agonist monoclonal antibody (mAb) recognizes all three isoforms. Recently, the requirement of BTN3A molecules in pAg-sensing by Vγ9Vδ2 T cells was demonstrated.Citation19 Anti-BTN3A 20.1 mAb acts indirectly, through sensitization of target cells to Vγ9Vδ2 T cells recognition.Citation20 This mAb induced the immobilization of BTN3A on the target cell surface, suggesting an involvement of the extracellular domain in Vγ9Vδ2 T cells activation. However, only the B30.2 domain of the BTN3A1 isoform can bind pAgCitation21 through a positively charged pocket.Citation22,23 This intracellular binding induced extracellular conformational changes of BTN3A, similar to those observed with 20.1 mAb and resulting Vγ9Vδ2 T cell activation.Citation24
Together, these data open new perspectives in Vγ9Vδ2 T cells-based immunotherapies, with BTN3A appearing as an interesting target to regulate their functions. In this study, we decided to investigate how BTN3A molecules might contribute to AML cells clearance through Vγ9Vδ2 T cells-based immunotherapy.
Results
BTN3A specifically enhance Vg9Vd2 TCR-mediated lysis of primary AML blasts
BTN3A molecules are key players in Vγ9Vδ2 TCR activation.Citation19,20 Hence, we decided to assess whether anti-BTN3A 20.1 mAb impact on the recognition and killing of primary AML blasts by Vγ9Vδ2 T cells.
We first compared the effect of Vγ9Vδ2 TCR agonists such as BrHpp and ZOL to anti-BTN3A agonist 20.1 mAb. We observed that, for optimal stimulation conditions, ZOL and anti-BTN3A 20.1 mAb were significantly more efficient (p = 0.0170 and p < 0.0001, respectively) than BrHpp to enhance Vγ9Vδ2 T cells lysis of AML blasts ().
Figure 1. BTN3A triggering enhances TCR-mediated lysis of AML blasts by Vγ9Vδ2 T cells. AML blasts were pre-incubated for 1 h with Control Isotype, anti-BTN3A 20.1 agonist mAb (10 μg/mL) or O/N with Zoledronate (ZOL) (45 μM), before addition of effector cells. Each AML blast sample was tested with at least three different Vγ9Vδ2 T cells or NK cells donors. (A) Comparison of TCR agonists. Specific lysis of primary AML blasts (n = 25) by expanded Vγ9Vδ2 T cells from HV after treatment with BrHpp, ZOL, or anti-BTN3A 20.1 mAb. Data are shown for E:T 30:1. (B, C) Effect of TCR and BTN3A targeting. Primary AML blasts lysis (n = 11) by expanded Vγ9Vδ2 T cells from HV. (B) Vγ9Vδ2 T cells were pre-incubated for 30 min with anti-TCRγ9 mAb. The blasts were treated with ZOL or anti-BTN3A agonist 20.1 mAb. (C) The blasts were treated with ZOL, agonist (20.1) or antagonist (108.5) anti-BTN3A mAb or combination and then incubated with Vγ9Vδ2 T cells. Data are shown for E:T 30:1. The lysis is expressed as percentage of specific lysis. (D, E) Effect of anti-BTN3A mAb and ZOL on NK cells lysis compared to Vγ9Vδ2 T cells. Representative data from two independent experiments. Primary AML blasts lysis (n = 3) by (D) expanded Vγ9Vδ2 T cells from HV; (E) NK cells from HV isolated and treated O/N with IL2; DAUDI and K562 were used as positive controls respectively for Vγ9Vδ2 T cells and NK cells lysis. The lysis is expressed as percentage of specific lysis. Results were expressed as mean ± SEM and statistical significance was established using the non-parametric paired Wilcoxon U-test. *p < 0.05; **0.001 < p < 0.01; ***p < 0.001.
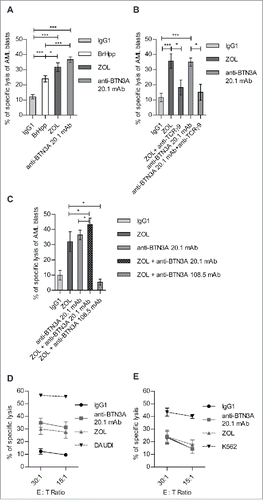
Secondly, to dissect the role of BTN3A in Vγ9Vδ2 T cells lysis of AML blasts, we performed cytotoxicity assays involving different combinations of TCR Vγ9 blocking mAb, ZOL and anti-BTN3A mAbs with agonist (20.1 mAb) or antagonist (108.5 mAb) functions. The TCR blockade resulted in significant decreases of ZOL (p = 0.0156) and anti-BTN3A 20.1-induced lysis of AML blasts ( p = 0.0156) (). This demonstrated that Vγ9Vδ2 TCR-induced lysis of AML blasts could be enhanced using agonists such as anti-BTN3A 20.1 mAb.
In this line, as both ZOL and BTN3A exerted strong agonist functions, we combined them in cytotoxic assays against AML blasts. The addition of anti-BTN3A 20.1 mAb to ZOL resulted in a poor but significant ( p = 0.0420) enhancement of AML blasts lysis by Vγ9Vδ2 T cells (). Conversely, the antagonist anti-BTN3A 108.5 mAb could significantly and drastically abrogate ZOL-induced lysis ( p = 0.0156) ().The lack of additivity or synergic effect of ZOL and agonist 20.1 mAb-combined stimulation suggested that they had overlapping functions through BTN3A molecules to mediate the enhancement of lysis. In addition, we confirmed BTN3A key function in the recognition of N-BP-treated blasts by Vγ9Vδ2 T cells with anti-BTN3A antagonist 108.5 mAb that totally abrogated the ZOL-induced activation of Vγ9Vδ2 T cells.
In order to test whether anti-BTN3A 20.1 mAb and ZOL effects were specifically related to Vγ9Vδ2 TCR activation, we assessed their effects on NK cells-mediated AML non-MHC-restricted blasts lysis. Unlike Vγ9Vδ2 T cells (), anti-BTN3A 20.1 mAb and ZOL had no effect upon NK cells-mediated AML blasts lysis (). Collectively, these observations show that TCR agonist-mediated enhancement of Vγ9Vδ2 T cells lysis of AML blasts specifically involves BTN3A molecules.
Triggering BTN3A with agonist 20.1 mAb enhances Vγ9Vδ2 T cells antitumor functions
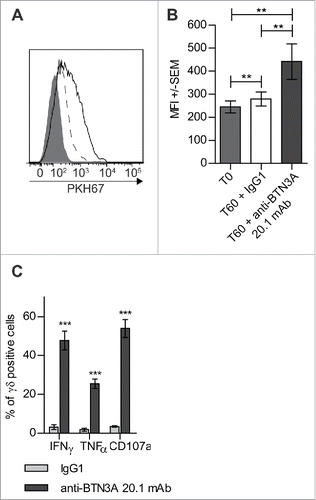
Treatment with BrHpp has been shown to enhance the contact-dependent capture of AML cell membrane via trogocytosisCitation12 leading us to hypothesize that the anti-BTN3A 20.1 agonist mAb might induce a similar effect. As shown in , treatment with anti-BTN3A 20.1 mAb significantly enhanced the trogocytosis of AML blasts by Vγ9Vδ2 T cells (p = 0.0039). Finally, we observed that when Vγ9Vδ2 T cells were co-cultured with AML blasts in the presence of anti-BTN3A 20.1 agonist mAb, they showed greater cytokine production of TNFα (p = 0.0313) and IFNγ (p = 0.0313) and a greater ability to degranulate as assessed by the expression of CD107a (p = 0.0313) (). Collectively, these data demonstrated a potent role of anti-BTN3A 20.1 mAb to enhance the recognition of resistant AML by Vγ9Vδ2 T cells.
Figure 2. Triggering of BTN3A with 20.1 mAb enhances Vγ9Vδ2 T cells antitumor functions toward AML blasts. (A) Representative experiment showing trogocytosis of AML blasts from patient UPN01 by Vγ9Vδ2 T cells from HV (full gray histogram) and after 1 h in co-culture with IgG1 (dashed line) or anti BTN3A 20.1 mAb (full line). (B) Vγ9Vδ2 T cells trogocytosis (PKH67 MFI ± SEM) of AML blasts (n = 10). Cumulative data at 0 and 60 min from three different experiments are provided. (C) CD107a expression, TNFα and IFNγ production by Vγ9Vδ2 T cells from HV after a 4-h co-culture with AML blasts (n = 6).
Agonist BTN3A 20.1 mAb enhances Vγ9Vδ2 T cells killing through fixation on AML blasts
Up to now, we have demonstrated that BTN3A agonist mAb enhanced Vγ9Vδ2 T cells antitumor functions. However, as BTN3A molecules are expressed both on Vγ9Vδ2 T cells and blasts cells, we do not know if the agonist molecules precisely exert their effects on the target, the effector cells or both.
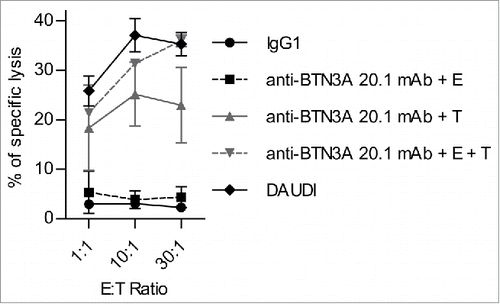
We thus examined whether sensitization to killing occurred through the interaction of anti-BTN3A 20.1 mAb with AML blasts themselves, or rather as a consequence of anti-BTN3A 20.1 mAb exerting its effects upon Vγ9Vδ2 T cells. We compared the lysis of AML blasts either (i) after blasts pre-incubation with anti-BTN3A 20.1 mAb (washed before co-incubation with Vγ9Vδ2 T cells), or (ii) after simultaneous incubation of blasts, anti-BTN3A 20.1 mAb and Vγ9Vδ2 T cells, or (iii) after Vγ9Vδ2 T cells were previously incubated with anti-BTN3A 20.1 mAb (then washed before co-incubation with AML blasts). The lysis achieved with Vγ9Vδ2 T cells pre-incubated with anti-BTN3A 20.1 mAb was comparable to basal lysis (4.36 ± 2.11%). The lysis was enhanced when blasts were pre-incubated with anti-BTN3A 20.1 mAb (22.92 ± 7.64%) and further enhanced by simultaneous incubation of effector cells, target cells and mAb (35.95 ± 1.16%) (). Those results show that anti-BTN3A 20.1 mAb biological effect is upon AML blasts rather than directly upon BTN3A molecules expressed by Vγ9Vδ2 T cells. The anti-BTN3A 20.1 mAb agonist effect was even more pronounced when effectors, targets and mAb were simultaneously present.
Figure 3. The 20.1 mAb triggers BTN3A on the surface of AML blasts. Primary AML blasts, target (T) lysis (n = 2) by Effector (E) Vγ9Vδ2 T cells from HV (n = 2) with IgG1 (full black line) or (i) after T pre-incubation with anti-BTN3A 20.1 mAb (anti-BTN3A + T) (full gray line) or (ii) after simultaneous incubation of E, T and anti-BTN3A 20.1 mAb (anti-BTN3A + E + T) (dashed gray line), or (iii) after E pre-incubation with anti-BTN3A 20.1 mAb (anti-BTN3A + E) (dashed black line). T or E cells were preincubated for 90 min with anti-BTN3A 20.1 mAb (10 μg/mL), and then extensively washed before E + T co-culture. Representative data from two independent experiments are depicted. Data are shown for E:T 1:1, 10:1, 30:1. The lysis is expressed as a mean percentage of specific lysis ± SEM and compared to DAUDI lysis.
Among BTN3A molecules, primary AML blasts mainly express BTN3A2
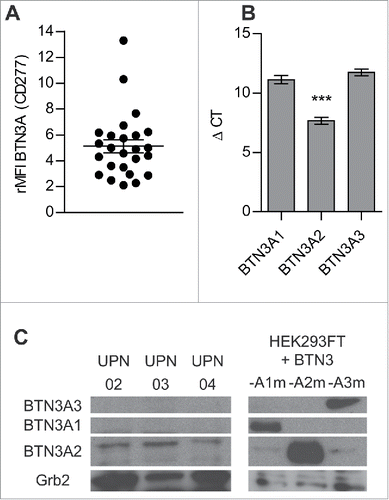
As we know that BTN3A subfamily gathers three different isoforms, we decided to describe the expression of each one of them on primary AML blasts. First of all, BTN3A was expressed at the surface of all the primary AML blasts tested (n = 25), all states of differentiation confounded (, Table S1). We assessed the level of expression of each BTN3A isoform at the transcriptional level by qRT-PCR and protein level by Western Blot. Compared to BTN3A1 (Mean ΔCT = 11.12 ± 0.34) and BTN3A3 (Mean ΔCT = 11.73 ± 0.29), BTN3A2 was the most abundant isoform both at transcriptional (Mean ΔCT = 7.67 ± 0.38; p <0.0001) () and protein levels in all primary AML blasts (). BTN3A1 and BTN3A3 were expressed at the transcriptional level () but were not detected at the expected size at the protein level (). We demonstrated for the first time that blasts mainly express the BTN3A2 isoform, i.e. the isoform without the B30.2 cytoplasmic domain.
Figure 4. BTN3A molecules expression on primary AML blasts. (A) BTN3A molecules surface expression on primary AML blasts (n = 25). The global BTN3A surface expression was assessed by flow cytometry analysis. The results are expressed as Median Fluorescence Intensity ratio (rMFI) compared to control isotype. (B, C) BTN3A isoform expression on primary AML blasts. (B) qRT PCR analysis of total RNA isolated from primary AML blasts (n = 24). Data were normalized using GAPDH as endogenous control (ΔCt = Cttarget gene – CtGAPDH). Statistical significance was established using the non-parametric paired Wilcoxon U-test. ***p < 0.001. (C) Western-blot analysis of total protein extracts from primary AML blasts (n = 3). BTN3A knock-down HEK293FT (sh#284; clone#30) transiently transfected with BTN3A1m, BTN3A2m, BTN3A3m are used as size controls. Extracts were loaded in 10% SDS PAGE gel and membranes were hybridized with anti-BTN3A 20.1 mAb and anti-Grb2 as a loading control.
The sensitization of AML blasts by N-BP depends on monocytic differentiation of blasts
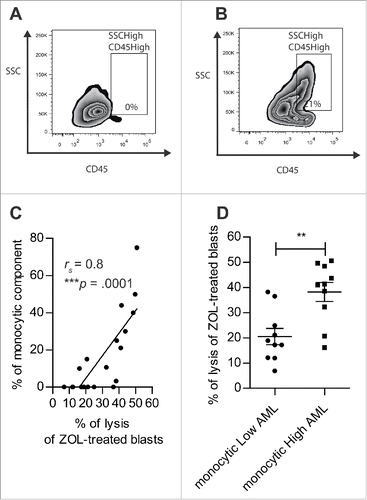
Finally, Gunderman et al. have demonstrated that AML samples with poor monocytic differentiation were poorly responsive to Vγ9Vδ2 T cells lysis despite ZOL treatment.Citation16 We thus assessed the level of monocytic differentiation in our samples by determining the percentage of monocytic component defined based on the SSC parameter and the level of expression of CD45 (). First, we confirmed that monocytic differentiation positively correlated with N-BP sensitization of AML blasts (rS = 0.8; p < 0.0001) (). Then, based on the percentage of monocytic component, we segregated two groups of AML blasts corresponding to monocyticLow AML with a monocytic component strictly below 10% () and monocyticHigh AML with a monocytic component greater than 10% (). The potentiation of Vγ9Vδ2 T cells lysis by N-BP-treated blasts was significantly higher in monocyticHigh AML (38.25 ± 3.76%) than in monocyticLow AML (20.53 ± 3.24%; p = 0.0029) (). Altogether, these data show that monocytic component accounts for N-BP sensitivity of primary AML blasts.
Figure 5. The sensitization of AML blasts by N-BP depends on monocytic component of blasts. (A, B) The percentage of monocytic component among AML blasts (SSCHigh CD45High) (n = 20) was assessed by flow cytometry. Representative dot plot of: (A) monocyticLow AML (UPN02) and (B) monocyticHigh AML (UPN10). (C) Linear regression of Vγ9Vδ2 T cells lysis of N-BP-treated AML blasts with respect to monocytic component (n = 20). The correlation was established using the non-parametric Spearman correlation coefficient (rs). ***p < 0.001. (D) Lysis of N-BP-treated blasts with low monocytic component (monocyticLow AML, % of SSCHigh CD45High<10) (n = 10) compared with the lysis of N-BP-treated blasts with high monocytic component (monocyticHigh AML, % of SSCHigh CD45High≥10) (n = 10) by Vγ9Vδ2 T cells. Results were expressed as mean ± SEM. Statistical significance was established using the Mann–Whitney test. 0.001 < **p < 0.01.
Anti-BTN3A 20.1 mAb can overcome the N-BP poor sensitization of monocyticLow AML
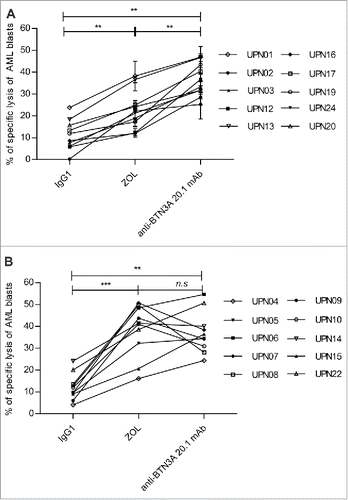
We next determined the extent to which monocyticLow AML blasts that were either resistant or poorly responsive to N-BP sensitization could be sensitized by anti-BTN3A 20.1 mAb. We thus decided to compare the effect of ZOL and anti-BTN3A 20.1 mAb upon monocyticLow and monocyticHigh AML samples. As shown in , anti-BTN3A 20.1 mAb were able to enhance Vγ9Vδ2 T cell lysis of both monocyticLow and monocyticHigh AML blasts (p = 0.002). Anti-BTN3A 20.1 mAb enhancing effect was significantly higher than ZOL regarding Vγ9Vδ2 T cell lysis of ZOL-resistant or ZOL-poorly responsive monocyticLow AML blasts (p = 0.002) (). Anti-BTN3A 20.1 mAb and ZOL had a comparable enhancing effect (37.23 ± 3% and 38.25 ± 3.8%, respectively) upon monocyticHigh AML blasts ().
Figure 6. Anti-BTN3A 20.1 mAb can overcome N-BP poor sensitization of monocyticLow AML. (A) Specific lysis of monocyticLow AML blasts (n = 10) and (B) monocyticHigh AML blasts (n = 10) by Vγ9Vδ2 T cells from HV after treatment with IgG1, ZOL (O/N, 45 μM), or anti-BTN3A 20.1 mAb. Data are shown for E:T 30:1. Results were expressed as mean ± SEM and statistical significance was established using the non-parametric paired Wilcoxon U-test. *p < 0.05; 0.001 <**p < 0.01; ***p < 0.001.
Altogether, these data converge to show that BTN3A triggering with the agonist 20.1 mAb can significantly enhance the lysis of AML blasts by Vγ9Vδ2 T cells, thus overcoming their resistance to N-BP treatment.
Anti-BTN3A mAb combined with γδ T cells infusion decreases leukemic burden in AML xenografted mice models
We next asked whether the in vitro potentiating effects of anti-BTN3A 20.1 mAb against leukemia cells could be reproduced in vivo.
We first used a validated AML xenograft model based on the inoculation of luciferase-transduced-U937 cell line, to monitor the engraftment of malignant cells by bioluminescent imaging.Citation12
We used RLI as an antitumor adjuvant tool combined with Vγ9Vδ2 T cells and anti-BTN3A 20.1 mAb infusions. RLI is an IL-15/IL15Rα fusion protein that has been shown to expand antitumor lymphocyte subsets including CD8+ and NK cells and to be an adjuvant to immune responses against murine and human cancers in vivo.Citation25,26 In this line, we first assessed whether RLI could boost Vγ9Vδ2 T cells proliferation and demonstrated for the first time that RLI could drive Vγ9Vδ2 T cells' expansion in vitro (Figs. S1A and B) and in vivo (Figs. S1C and D).
We confirmed that Vγ9Vδ2 T cells plus-RLI infusions decrease the tumor burden. This effect was even more striking and was associated with a significant increase of survival when anti-BTN3A 20.1 mAb was added along with Vγ9Vδ2 T cells (p = 0.0224) ().
Figure 7. Evaluation of anti-BTN3A agonist mAb anti-leukemic activity in vivo in AML xenograft models. (A) (B) NSG mice (n = 15) were inoculated with luciferase-transfected U937 cells on Day 0. They were intravenously injected with PBS, anti-BTN3A mAb alone, RLI alone or 30 × 106 Vγ9Vδ2 T cells-plus-RLI or 30 × 106 Vγ9Vδ2 T cells-plus-RLI and anti-BTN3A 20.1 mAb. (A) Representative and comparative bioluminescent imaging of five groups of mice on Day 12 post-graft. Day 12 corresponds to the last bioluminescent measurement before first mice deceased. Medians of survival of each group of mice are notified below. (B) Median survival time was 21 d for the PBS control group (n = 3), 25 d for the group treated with Vγ9Vδ2 T cells and RLI (n = 3) and 29 d for the group treated with Vγ9Vδ2 T cells, RLI and anti-BTN3A 20.1 mAb (n = 3). Median survival times were compared using Log-Rank (Mantel-Cox) Test. *p < 0.05. (C, D) NSG mice (three groups of n = 6) were intravenously injected with primary AML blasts. Since the Week 8 post-graft, they were treated twice a week during two weeks with injections of 15 × 106 Vγ9Vδ2 T cells-plus RLI and either anti-BTN3A mAb or IgG1. Control mice were injected with PBS. Mice were sacrificed at the Week 11 post-graft. (C) Percentage of variation of the absolute number of blasts in blood quantified by Flow Cytometry before and 4 d after the end of the treatment. (D) Absolute number of blastic cells quantified by Flow Cytometry in harvested BM. Representative and cumulative data from two independent experiments. Mann–Whitney test was used to compare differences between treatments. *p < 0.05; 0.001 < **p < 0.01; ***p < 0.001.
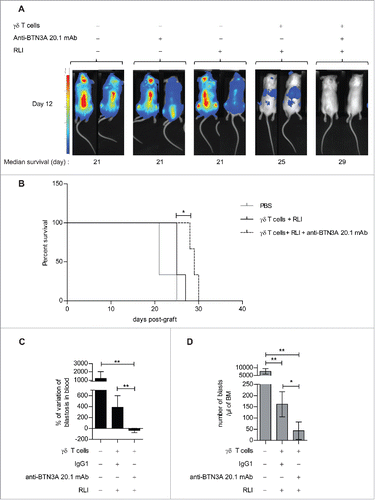
We next confirmed our data in a xenotransplantation model using primary AML blasts.Citation27 Blasts from UPN09 were highly sensitized to Vγ9Vδ2 T cells lysis after anti-BTN3A 20.1 mAb treatment in vitro (Fig. S2A). Blasts from UPN09 highly engrafted in BM and blood of NSG mice, thus recapitulating human disease (Fig. S2B). We asked whether anti-BTN3A 20.1 mAb and Vγ9Vδ2 T cells-immunotherapy could decrease the disease progression in this model. We showed a significant decrease of circulating blasts in the anti-BTN3A 20.1 mAb treated group (37.38 ± 39.89) compared to IgG1(388.1 ± 212.3; p = 0.0379) and untreated group (1208 ± 817; p = 0.0022) (). Similarly, anti-BTN3A 20.1 mAb treated group had a significantly lower absolute number of blasts in the BM (43.4 ± 39.28) than the IgG1 (161.5 ± 55.72; p = 0.0411) and untreated groups (7715 ± 1819; p = 0.022) ().
These data highlight the potent anti-leukemic effect exerted by anti-BTN3A 20.1 mAb combined to Vγ9Vδ2 T cells immunotherapy in vivo.
Discussion
In this study, we show that TCR activation by agonists such as N-BP involves BTN3A molecules and that agonist anti-BTN3A 20.1 mAb triggers BTN3A on blasts to specifically enhance Vγ9Vδ2 T cell-mediated lysis. Finally, we demonstrate the enhancement of Vγ9Vδ2 T cell anti-leukemic effects, using anti-BTN3A 20.1 mAb in vivo, in xenotransplantation models.
We demonstrated in this study the critical role of BTN3A subfamily in N-BP-mediated sensitization of blasts to Vγ9Vδ2 T cells lysis, using primary AML samples from patients. Indeed, treatment with the newly described antagonist anti-BTN3A 108.5 mAb totally abrogated Vγ9Vδ2 T cells lysis of N-BP treated blasts.
Conversely, triggering of BTN3A with the agonist 20.1 mAb induced the TCR-mediated activation of Vγ9Vδ2 T cells. This suggests that BTN3A molecules could interact with Vγ9Vδ2 TCR and the literature rather favors an indirect engagement of the TCR mediated by BTN3ACitation22. BTN3A 20.1 mAb and N-BP-combined treatment of AML blasts did not provide a synergic or an additive enhancing effect upon Vγ9Vδ2 T cells lysis of blasts. This is consistent with the hypothesis of overlapping effects between N-BP and anti-BTN3A 20.1 mAb. In this line, biochemical, biophysical and FRAP (fluorescence recovery after photobleaching) experiments have shown that pAg and BTN3A 20.1 mAb induced the same conformational activating changes of BTN3A molecules.Citation24,20
We confirmed anti-BTN3A 20.1 mAb's indirect effect, mainly on primary AML blasts.Citation20 But, interestingly, the simultaneous incubation of Vγ9Vδ2 T cells with anti-BTN3A 20.1 mAb and blasts resulted in even more enhanced cytotoxicity. This suggests a concomitant triggering of BTN3A by anti-BTN3A 20.1 mAb on both effector and target cells, which could be of interest in the perspective of a novel immunotherapy. In addition, as previously demonstrated,Citation20 this observed effect is probably not ADCC-dependent as anti-BTN3A 20.1 mAb did not have any biological effect on NK cells lysis of AML blasts.
Although N-BP could be a promising therapy in hematologic malignancies,Citation13,28 our data confirm that some primary AML blasts with poor monocytic differentiation are poorly sensitized by N-BP to Vγ9Vδ2 T cells lysis.Citation16 These monocyticLow AML were shown to have poorly active mevalonate pathway.Citation16 This could account for a poor pAg accumulation in these blasts upon N-BP treatment that would fail to activate Vγ9Vδ2 T cells.Citation29 We show that anti-BTN3A 20.1 mAb can sensitize N-BP-poorly sensitized primary AML blasts to Vγ9Vδ2T cells lysis, notably monocyticLow AML. The sensitization of these resistant blasts to Vγ9Vδ2 T cells lysis with anti-BTN3A 20.1 mAb points out a potential major role of this mAb to circumvent intrinsic resistance mechanisms in blasts. In addition, this observation underlies that different ways are involved in anti-BTN3A 20.1 mAb and N-BP-mediated activation. Indeed, whereas N-BP-mediated activation of Vγ9Vδ2 T cells requires the expression of the three BTN3A isoformsCitation19 and putative additional partners,Citation30 anti-BTN3A 20.1 mAb can trigger each one of the three BTN3A isoforms.Citation20
BTN3A2 was the most expressed isoform in primary AML blasts. As BTN3A2 lacks the intracellular B30.2 domain, it has been hypothesized to be a decoy receptor via the binding of a putative ligand.Citation31 Its predominant expression by AML blasts could constitute an immune escape mechanism to Vγ9Vδ2 T cells recognition. However, we assume that anti-BTN3A 20.1 mAb could by-pass N-BP resistance via the triggering of BTN3A2, through mechanisms involving decreased mobilityCitation20 and a multimerization of BTN3A molecules.Citation24 These 20.1 mAb-induced extracellular conformational changes of BTN3A molecules might require transmembrane or intracellular partners such as cytolinker proteins.Citation19 In addition, as BTN3A molecules might form heterodimeric complexes,Citation24 the 20.1 mAb binding to BTN3A2 might recruit BTN3A1 and BTN3A3 and transduce an intracellular signaling through their intracellular domain.
In the context of developing novel immunotherapies, we have adopted a preclinical xenograft mouse model employing human leukemia cells. We show that the addition of anti-BTN3A 20.1 mAb to allogeneic Vγ9Vδ2 T cells and cytokine infusions could decrease leukemic burden and increase the survival probability of U937-xenografted NSG mice. Hence, we confirmed our data in a more clinically applicable model consisting in the xenotransplantation of primary AML blast in NSG mice.Citation27 In this model, anti-BTN3A 20.1 mAb combined to Vγ9Vδ2 T cells-plus cytokines could decrease the leukemic progression in BM and blood. Accordingly, these models highlight the potential utility of anti-BTN3A 20.1 mAb in Vγ9Vδ2 T cell-based immunotherapy of AML.
In conclusion, our data demonstrate that Vγ9Vδ2 T cells lysis of primary AML blasts involves TCR and BTN3A-mediated activation. We showed that agonist anti-BTN3A 20.1 mAb can circumvent primary AML blasts resistance to allogeneic Vγ9Vδ2 T cells lysis. Anti-BTN3A 20.1 mAb effect is confirmed in vivo, using a NSG human AML-xenografted mice model. This study highlights the potential therapeutic utility of anti-BTN3A 20.1 mAb, combined with Vγ9Vδ2 T cells infusions, cytokine or chemotherapy, for the treatment of relapsed or refractory AML.
Material and methods
Patients
Written informed consent was obtained from the 25 AML patients included in this study, in accordance with the Declaration of Helsinki. Clinical data are detailed in Table S1. The study was approved by the local institutional review boards of the Institut-Paoli-Calmettes. Peripheral blood mononuclear cells (PBMCs) from 20 healthy volunteers (HV) were provided by the local Blood Bank (EFS). PBMCs from AML patients and HV were isolated by density gradient (Lymphoprep, Abcys) and frozen until use. Samples with more than 80% of blasts were selected for the study.
Reagents and antibodies
BrHpp was from Innate Pharma. ZOL was from Novartis. Recombinant human (Rh) IL-2 and rhIL-15 were from BD Biosciences. Receptor Linked IL-15 (RLI, CYP0150)Citation26 was provided by Cytune Pharma. The mAbs used are listed in Table S2.
Generation of anti-human BTN3A mAbs
Anti-BTN3A mAbs (clone 20.1 and 108.5) targeting the three BTN3A isoforms (Fig. S3) were generated as previously describedCitation17 and labeled using Alexa Fluor® 647 Protein Labeling Kit (Life Technologies).
Cell culture
Effector γδ -T cells were established as previously described.Citation32 PBMC from HV were stimulated with ZOL (1 μM) and rhIL-2 (200 IU/mL) at Day 0. From Day 5, rhIL-2 was renewed every two days and cells were kept at 1.5 × 106/mL for 15 d. NK cells were isolated using EasySep Human NK cell enrichment kit (StemCell Technologies). The purity of γδ -T cells and NK cells, assessed by flow cytometry, was greater than 80%. The Burkitt lymphoma cell line, DAUDI, the human AML cell line, U937, and the erythroleukemic cell line, K562, were obtained from the American Type Culture Collection and cultured (0.5 × 106/mL) in RPMI 1640 medium with 10% FCS. BTN3A Knock-down HEK293FT cells (sh#284; clone#30) were provided by E.Scotet (Inserm U892, Nantes), cultured and transfected with BTN3A1, BTN3A2, BTN3A3 mutated cDNA-containing plasmids, as described.Citation20
Flow cytometry
2 × 105 PBMC were incubated at 4°C for 20 min with specified mAb before analysis on LSRFortessa (Becton Dickinson) using DIVA software (BD bioscience). For analysis of CD107 expression, γδ-T cells were incubated at 37°C with anti-CD107a and Golgi stop, with or without anti-BTN3A 20.1 mAb. After 4 h, cells were collected and analyzed by flow cytometry. To study cytokine production, cells were permeabilized with Permwash (BD bioscience) to allow intracellular staining. Trogocytosis of PKH67 (Sigma Aldrich) labeled blasts by γδ-T cells was quantified as previously described.Citation33
Chromium release assay
0.5 × 106 target cells were incubated with 50 μCi of 51Cr (Perkin-Elmer) for AML blasts and 10 μCi for DAUDI and K562 cells for 90 min and mixed with effector cells in a Effector:Target (E:T) ratio of 30:1 ± 15:1 ± 10:1 ± 1:1. After 4 h of-incubation at 37°C, 50 μL supernatant of each sample was transferred in LUMA plates. Radioactivity was determined by a gamma counter. The percentage of specific lysis was expressed as the mean of triplicate, using the formula [(experimental – spontaneous release/total –spontaneous release) × 100]. DAUDI and K562 cell lines were used as respective positive controls for NK cells and γδ-T cells cytotoxicity.
Determination of BTN3A isoforms expression by quantitative RT-PCR (qRT-PCR)
cDNA was obtained after TRIzol extraction (Life Technologies) and M-MLV mediated reverse transcription(Life Technologies). qRT-PCR was performed with the Applied Bio-systems 7900HT Fast Real-Time PCR system. The Ct (threshold cycle) was calculated for each assay (Sequence Detection System Software 2.3, Applied Biosystems). Data were normalized using GAPDH as endogenous control (ΔCt = Cttarget gene – CtGAPDH). Higher ΔCt means lower expressions of analyzed genes. Probes were BTN3A1 (Hs01063368_m1), BTN3A2 (Hs00389328_m1), BTN3A3 (Hs00757230_m1) and GAPDH (Hs99999905_m1) Taqman gene expression Assays (Applied Biosystems).
Western blot
AML blasts (5–20 × 106) were washed in cold PBS, and lysed in 110 μL of ice-cold TIT buffer (50 mM Tris pH 7.4, 200 µM NaF, 2.5 mM EDTA pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% Nonidet-P40, 5% glycerol) containing protease inhibitors (Roche Applied Science) and 1 mM Na3VO4. Proteins in cell lysates were quantified according to the manufacturer's instructions (Pierce™ BCA Protein Assay Kit). Proteins were resolved by SDS-PAGE 10%, followed by western blotting with primary anti-BTN3A 20.1 and anti-GrB2 Ab (SantaCruz technology) and detection with peroxidase-conjugated anti-mouse IgG1 and anti-rabbit IgG Ab (Jackson Laboratory). Immuno-reactive bands were detected using enhanced chemo-luminescent reagents (Pierce).
NSG mouse/human AML model
NOD-SCID-common-γ-chain-knockout mice (NSG) were purchased from Charles River France or home-bred and maintained under pathogen-free conditions. All animal procedures were in accordance with protocols approved by the local Committee for Animal Experiments. Healthy 6-8-weeks-old female mice (n = 15) received 0.2 × 106 luciferase transduced-U937 cells on Day 0, as described,Citation12 and were randomly assigned in five groups to receive intravenous injections of post-expansion-purified Vγ9Vδ2 T cells on Day 1 (30 × 106, 98.8% CD3 + TCRVδ2+; Unité de Thérapie Cellulaire et Génique de Nantes) alone or combined to anti-BTN3A 20.1 mAb (250 μg) on Day 2 and 6. As RLI allows the proliferation of Vγ9Vδ2 T cells (Fig. S1), RLI (2 μg) was administered on Day 1, 2, 8 and 9 post-graft. Control mice were infused with PBS, RLI or anti-BTN3A 20.1 mAb. Mice were imaged with a Photon Imager (Biospace Lab, Paris, France) at different days. For the Primary AML blasts model, NSG mice received sublethal irradiation (2 Gy). On Day 1, they received 1 × 106 of CD3-depleted blasts (CD3 depletion Kit, Miltenyi Biotec) isolated from UPN09. Treatments comprising intravenous infusions of expanded Vγ9Vδ2 T cells (15 × 106, 90% CD3 + TCRVδ2+), RLI (2 μg) and either anti-BTN3A 20.1 mAb (250 μg) or IgG1 (250 μg), were administered twice a week, from 8 to 10 weeks post-graft. The peripheral blood number of blasts was quantified by Flow Cytometry (Fig. S2B) and expressed as percentage of variation: [(Absolute number of blasts/μL of blood after treatment (W10) – Absolute number of blasts/μL of blood before treatment (W8))/Absolute number of blasts/μL of blood before treatment (W8)] × 100. Mice were sacrificed at week 11th post-graft and the number of blasts was quantified in their bone marrow (BM).
Statistics
Results are expressed as median ± SEM. Statistical analysis was performed using Spearman correlation, Wilcoxon test and Mann–Whitney t test. p values <0.05 were considered significant. Analyses were performed using GraphPad Prism program.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
AB designed research, performed experimental work, analyzed and interpreted data and drafted the paper; ALR, FO, CC, AG, LP performed experimental work and analyzed data; EM contributed to draft the paper; JGD designed research, performed experimental work and analyzed data. NV, YC and ES contributed to the design of the project research. RC designed research, performed experimental work, analyzed and interpreted data and contributed to draft the paper. DO designed research, contributed to the analysis and interpretation of data and to draft the paper.
KONI_A_1146843_s02.pdf
Download PDF (2.1 MB)Acknowledgments
We thank the CRCM animal core, cytometry and immunomonitoring facilities for their technical assistance. We acknowledge P.Livrati, S.Just-Landi and S.Pastor for technical support.
Funding
This study was supported by research grant from Fondation ARC to AB and CC, ANR GDStress, Fondation pour la Recherche Médicale (FRMDEQ20140329534), and INCa Translationnel.
References
- Butturini A, Bortin MM, Gale RP. Graft-versus-leukemia following bone marrow transplantation. Bone Marrow Transplant 1987; 2(3):233–42; PMID:3332173
- Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood 2000; 96(2):384–92; PMID:10887096; http://dx.doi.org.gate2.inist.fr/
- Castella B, Vitale C, Coscia M, Massaia M. Vγ9Vδ2 T cell-based immunotherapy in hematological malignancies: from bench to bedside. Cell Mol Life Sci CMLS 2011; 68(14):2419–32; PMID:21584812; http://dx.doi.org/10.1007/s00018-011-0704-8
- Kabelitz D, Wesch D, Pitters E, Zöller M. Characterization of tumor reactivity of human V gamma 9V delta 2 gamma delta T cells in vitro and in SCID mice in vivo. J Immunol 2004; 173(11):6767–76; PMID:15557170; http://dx.doi.org/10.4049/jimmunol.173.11.6767
- Norell H, Moretta A, Silva-Santos B, Moretta L. At the Bench: Preclinical rationale for exploiting NK cells and γδ T lymphocytes for the treatment of high-risk leukemias. J Leukoc Biol 2013; 94(6):1123–39; PMID:24108703; http://dx.doi.org/10.1189/jlb.0613312
- Locatelli F, Merli P, Rutella S. At the Bedside: Innate immunity as an immunotherapy tool for hematological malignancies. J Leukoc Biol 2013; 94(6):1141–57; PMID:24096380; http://dx.doi.org/10.1189/jlb.0613343
- Godder KT, Henslee-Downey PJ, Mehta J, Park BS, Chiang KY, Abhyankar S, Lamb LS. Long term disease-free survival in acute leukemia patients recovering with increased gammadelta T cells after partially mismatched related donor bone marrow transplantation. Bone Marrow Transplant 2007; 39(12):751–7; PMID:17450185; http://dx.doi.org/10.1038/sj.bmt.1705650
- Meeh PF, King M, O'Brien RL, Muga S, Buckhalts P, Neuberg R, Lamb LS, Jr. Characterization of the gammadelta T cell response to acute leukemia. Cancer Immunol Immunother 2006; 55(9):1072–80; PMID:16328383; http://dx.doi.org/10.1007/s00262-005-0094-6
- Gober HJ, Kistowska M, Angman L, Jenö P, Mori L, De Libero G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med 2003; 197(2):163–8; PMID:12538656; http://dx.doi.org/10.1084/jem.20021500
- Li J, Herold MJ, Kimmel B, Müller I, Rincon-Orozco B, Kunzmann V, Herrmann T. Reduced expression of the mevalonate pathway enzyme farnesyl pyrophosphate synthase unveils recognition of tumor cells by Vgamma9Vdelta2 T cells. J Immunol 2009; 182(12):8118–24; PMID:19494338; http://dx.doi.org/10.4049/jimmunol.0900101
- Benzaïd I, Mönkkönen H, Stresing V, Bonnelye E, Green J, Mönkkönen J, Touraine JL, Clézardin P. High phosphoantigen levels in bisphosphonate-treated human breast tumors promote Vgamma9Vdelta2 T-cell chemotaxis and cytotoxicity in vivo. Cancer Res 2011; 71(13):4562–72; PMID:21646473; http://dx.doi.org/10.1158/0008-5472.CAN-10-3862
- Gertner-Dardenne J, Castellano R, Mamessier E, Garbit S, Kochbati E, Etienne A, Charbonnier A, Collette Y, Vey N, Olive D. Human Vγ9Vδ2 T cells specifically recognize and kill acute myeloid leukemic blasts. J Immunol 2012; 188(9):4701–8; PMID:22467661; http://dx.doi.org/10.4049/jimmunol.1103710
- Kunzmann V, Smetak M, Kimmel B, Weigang-Koehler K, Goebeler M, Birkmann J, Becker J, Schmidt-Wolf IG, Einsele H, Wilhelm M. Tumor-promoting versus tumor-antagonizing roles of γδ T cells in cancer immunotherapy: results from a prospective phase I/II trial. J Immunother 2012; 35(2):205–13; PMID:22306909; http://dx.doi.org/10.1097/CJI.0b013e318245bb1e
- Coscia M, Vitale C, Peola S, Foglietta M, Rigoni M, Griggio V, Castella B, Angelini D, Chiaretti S, Riganti C et al. Dysfunctional Vγ9Vδ2 T cells are negative prognosticators and markers of dysregulated mevalonate pathway activity in chronic lymphocytic leukemia cells. Blood 2012; 120(16):3271–9; PMID:22932792; http://dx.doi.org/10.1182/blood-2012-03-417519
- Wilhelm M, Smetak M, Schaefer-Eckart K, Kimmel B, Birkmann J, Einsele H, Kunzmann V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J Transl Med 2014; 12:45; PMID:24528541; http://dx.doi.org/10.1186/1479-5876-12-45
- Gundermann S, Klinker E, Kimmel B, Flierl U, Wilhelm M, Einsele H, Kunzmann V. A comprehensive analysis of primary acute myeloid leukemia identifies biomarkers predicting susceptibility to human allogeneic Vγ9Vδ2 T cells. J Immunother 2014; 37(6):321–30; PMID:24911793; http://dx.doi.org/10.1097/CJI.0000000000000043
- Compte E, Pontarotti P, Collette Y, Lopez M, Olive D. Frontline: Characterization of BT3 molecules belonging to the B7 family expressed on immune cells. Eur J Immunol 2004; 34(8):2089–99; PMID:15259006; http://dx.doi.org/10.1002/eji.200425227
- Le Page C, Marineau A, Bonza PK, Rahimi K, Cyr L, Labouba I, Madore J, Delvoye N, Mes-Masson AM, Provencher DM et al. BTN3A2 expression in epithelial ovarian cancer is associated with higher tumor infiltrating T cells and a better prognosis. PloS One 2012; 7(6):e38541; PMID:22685580; http://dx.doi.org/10.1371/journal.pone.0038541
- Rhodes DA, Chen HC, Price AJ, Keeble AH, Davey MS, James LC, Eberl M, Trowsdale J. Activation of Human γδ T Cells by Cytosolic Interactions of BTN3A1 with Soluble Phosphoantigens and the Cytoskeletal Adaptor Periplakin. J Immunol 2015; 194(5):2390–8; PMID:25637025; http://dx.doi.org/10.4049/jimmunol.1401064
- Harly C, Guillaume Y, Nedellec S, Peigné CM, Mönkkönen H, Mönkkönen J, Li J, Kuball J, Adams EJ, Netzer S et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood 2012; 120(11):2269–79; PMID:22767497; http://dx.doi.org/10.1182/blood-2012-05-430470
- Wang H, Henry O, Distefano MD, Wang YC, Räikkönen J, Mönkkönen J, Tanaka Y, Morita CT. Butyrophilin 3A1 plays an essential role in prenyl pyrophosphate stimulation of human Vγ2Vδ2 T cells. J Immunol 2013; 191(3):1029–42; PMID:23833237; http://dx.doi.org/10.4049/jimmunol.1300658
- Sandstrom A, Peigné CM, Léger A, Crooks JE, Konczak F, Gesnel MC, Breathnach R, Bonneville M, Scotet E, Adams EJ. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity 2014; 40(4):490–500; PMID:24703779; http://dx.doi.org/10.1016/j.immuni.2014.03.003
- Wang H, Morita CT. Sensor Function for Butyrophilin 3A1 in Prenyl Pyrophosphate Stimulation of Human Vγ2Vδ2 T Cells. J Immunol 2015; 195(10):4583–94; PMID:26475929; http://dx.doi.org/10.4049/jimmunol.1500314
- Palakodeti A, Sandstrom A, Sundaresan L, Harly C, Nedellec S, Olive D, Scotet E, Bonneville M, Adams EJ. The molecular basis for modulation of human Vγ9Vδ2 T cell responses by CD277/butyrophilin-3 (BTN3A)-specific antibodies. J Biol Chem 2012; 287(39):32780–90; PMID:22846996; http://dx.doi.org/10.1074/jbc.M112.384354
- Bessard A, Sole V, Bouchaud G, Quemener A, Jacques Y. High antitumor activity of RLI, an interleukin-15 (IL-15)-IL-15 receptor fusion protein, in metastatic melanoma and colorectal cancer. Mol Cancer Ther 2009; 8(9):2736–45; PMID:19723883; http://dx.doi.org/10.1158/1535-7163.MCT-09-0275
- Mortier E, Quéméner A, Vusio P, Lorenzen I, Boublik Y, Grötzinger J, Plet A, Jacques Y. Soluble interleukin-15 receptor α (IL-15R α)-sushi as a selective and potent agonist of IL-15 action through IL-15R β/gamma. Hyperagonist IL-15 x IL-15R α fusion proteins. J Biol Chem 2006; 281(3):1612–9; PMID:16284400; http://dx.doi.org/10.1074/jbc.M508624200
- Sanchez PV, Perry RL, Sarry JE, Perl AE, Murphy K, Swider CR, Bagg A, Choi JK, Biegel JA, Danet-Desnoyers G et al. A robust xenotransplantation model for acute myeloid leukemia. Leukemia 2009; 23(11):2109–17; PMID:19626050; http://dx.doi.org/10.1038/leu.2009.143
- Wilhelm M, Kunzmann V, Eckstein S, Reimer P, Weissinger F, Ruediger T, Tony HP. Gammadelta T cells for immune therapy of patients with lymphoid malignancies. Blood 2003; 102(1):200–6; PMID:12623838; http://dx.doi.org/10.1182/blood-2002-12-3665
- Idrees ASM, Sugie T, Inoue C, Murata-Hirai K, Okamura H, Morita CT, Minato N, Toi M, Tanaka Y. Comparison of γδ T cell responses and farnesyl diphosphate synthase inhibition in tumor cells pretreated with zoledronic acid. Cancer Sci 2013; 104(5):536–42; PMID:23387443; http://dx.doi.org/10.1111/cas.12124
- Riaño F, Karunakaran MM, Starick L, Li J, Scholz CJ, Kunzmann V, Olive D, Amslinger S, Herrmann T. Vγ9Vδ2 TCR-activation by phosphorylated antigens requires butyrophilin 3 A1 (BTN3A1) and additional genes on human chromosome 6. Eur J Immunol 2014; 44(9):2571–6; http://dx.doi.org/10.1002/eji.201444712
- Messal N, Mamessier E, Sylvain A, Celis-Gutierrez J, Thibult ML, Chetaille B, Firaguay G, Pastor S, Guillaume Y, Wang Q et al. Differential role for CD277 as a co-regulator of the immune signal in T and NK cells. Eur J Immunol 2011; 41(12):3443–54; PMID:21918970; http://dx.doi.org/10.1002/eji.201141404
- Gertner J, Wiedemann A, Poupot M, Fournié J-J. Human gammadelta T lymphocytes strip and kill tumor cells simultaneously. Immunol Lett 2007; 110(1):42–53; PMID:17451812; http://dx.doi.org/10.1016/j.imlet.2007.03.002
- Gertner-Dardenne J, Poupot M, Gray B, Fournié JJ. Lipophilic fluorochrome trackers of membrane transfers between immune cells. Immunol Invest 2007; 36(5–6):665–85; PMID:18161524; http://dx.doi.org/10.1080/08820130701674646