
ABSTRACT
The presence of tumor immune infiltrating cells (TILs), particularly CD8+ T-cells, is a robust predictor of outcome in patients with colorectal cancer (CRC). We revisited TIL abundance specifically in patients with microsatellite stable (MSS) CRC without evidence of lymph node or metastatic spread. Examination of the density of CD8+ T-cells in primary tumors in the context of other pro-oncogenic markers was performed to investigate potential regulators of TILs. Two independent cohorts of patients with MSS T2-4N0M0 CRC, enriched for cases with atypical relapse, were investigated. We quantified CD8+ and CD45RO+ -TILs, inflammatory markers, NFkBp65, pStat3, Cyclo-oxygenase-2 (COX2) and GRP78 as well as transcription factors (TF), β-catenin and MYB. High CD8+ TILs correlated with a better relapse-free survival in both cohorts (p = 0.002) with MYB and its target gene, GRP78 being higher in the relapse group (p = 0.001); no difference in pSTAT3 and p65 was observed. A mouse CRC (CT26) model was employed to evaluate the effect of MYB on GRP78 expression as well as T-cell infiltration. MYB over-expressing in CT26 cells increased GRP78 expression and the analysis of tumor-draining lymph nodes adjacent to tumors showed reduced T-cell activation. Furthermore, MYB over-expression reduced the efficacy of anti-PD-1 to modulate CT26 tumor growth. This high MYB and GRP78 show a reciprocal relationship with CD8+ TILs which may be useful refining the prediction of patient outcome. These data reveal a new immunomodulatory function for MYB suggesting a basis for further development of anti-GRP78 and/or anti-MYB therapies.
Introduction
Early stage CRC is managed by surgery, followed with adjuvant chemotherapy in higher risk cases. There is now compelling evidence that a poor immune response in CRC, as defined by the absence of lymphocytes within the primary tumor, is a robust predictor of relapse.Citation1,2 Initiating a specific, or reawakening a latent, immune response in patients might improve CRC control but how this might be achieved remains unclear. The mechanism for tumor infiltrating lymphocytes (TILs) in CRC is intriguing and remains to be fully determined. Perhaps, there is a role for T-regulatory cells (T-regs), myeloid derived suppressor cells (MDSC), as well as immune check point control in suppressing the patient's immune response as measured by the absence of TILs.Citation3 Thus, it is paramount to understand the central mediators of both positive and negative host responses as this will inform future CRC therapeutic strategies.
Inflammatory pathways mediated by the TF NFκB and phosphoStat3 (pStat3) are considered to be central players in the gastro-intestinal tract immune system.Citation4 However, it is unclear whether these TFs are responsible for the attraction of TILs or suppression of antitumor immune responses. It also remains unclear if the actions of these TFs are tumor-driven or stroma-mediated. Thus, defining prognostic markers that are independent of the traditional indicators such as T and N stage are likely to influence patient management. To this effect, the demonstrated interplay between TILs and patient outcome has initiated a paradigm shift in thinking.
To help determine further markers of patient outcome independent of T and N staging, we have defined the TILs status in patients, who as a group, would normally be considered to be at low risk of relapse with markers measuring inflammatory pathway activation. Patients with microsatellite unstable (MSI-H) tumors were deliberately excluded because on the whole they have superior survival Citation5 and typically higher TILs. Patients with MSI-H CRC also appear to respond to anti-immune check point therapy likely due to an increased mutational load.Citation6,7,8 In addition to TILs, we examined the role of the TF MYB, as it has also been found to predict outcome in CRC patients,Citation9 but had not yet been explored in early stage CRC. As MYB drives the expression of a large number of pro-oncogenic genes, including cyclooxygenase 2 (COX2; PTGS2), BCL2, BCLXL (BCL2L1) and MYC,Citation10 we investigated its expression in these cohorts. To confirm that MYB was actively operating as a TF, we determined whether target gene, GRP78 (glucose regulatory protein-78 (HSPA5, BIP) Citation11 was also elevated in concordance with nuclear MYB.
The endoplasmic reticulum (ER) protein GRP78 is a sentinel for the induction of the unfolded protein response (UPR).Citation12 GRP78 has also been found to modulate both immune and therapy responses in cancer.Citation13 As the development of cancer accompanies a spectrum of elevated cellular stress responses,Citation14 we decided to use GRP78 as measure of the UPR and elevated cellular stress in tumors which are responding to glucose deprivation and hypoxia.Citation13 GRP78 has also been implicated in modulating cytotoxic T-cell-mediated tumor killing Citation15-17 and here we asked if there was any association with reduced CD8+ TILs in patients with CRC relapse and GRP78.
Using IHC, RNA expression and a relevant animal model, we found that high MYB and high GRP78 were robust predictors of poor patient outcome. Furthermore, the high expression of these proteins tracked with low CD8+ TILs in patients who relapsed with MSS CRC. Using the CT26 mouse model, we directly showed that increased MYB expression led to a downregulation of immune activation and a reduction in anti-PD-1 immunotherapy efficacy. Collectively, we have identified a new role for MYB in modulating immune responses, which is shown by the linked relationship between high TILs, low MYB and good outcome on the one side and low TILs, high MYB and poor outcome in early CRC on the other.
Results
Lymphocyte status
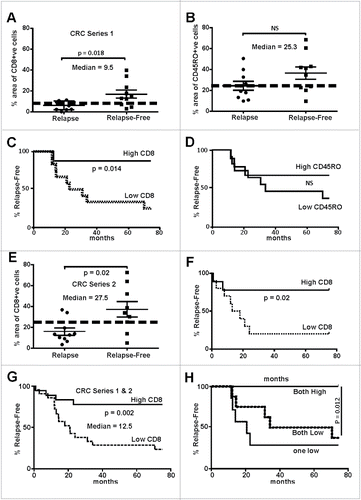
Whole tumor sections were evaluated for the presence of CD8+ cytotoxic or CD45RO+ memory T-cells in five randomly selected tumor regions determined using histology and/or β-catenin staining to demark tumor boundaries. The tumor core, tumor margin, as well as proximal stroma and stroma distal to the tumor were analyzed in each patient. In silico analysis was used to measure stained T-cells, with the tumor and stroma averaged separately. We observed no significant difference with regards to T-cell abundance in the tumor stroma in either cohort. However, in CRC Series 1, the presence of CD8+ cells was significantly greater in the tumor core and margin in sections from relapse-free patients. This trend was not observed with CD45RO+ staining (). Median scores were calculated and used to generate Kaplan–Meir plots. Using the median cut-off for CD8+ density, a significant separation between relapse-free patients from those that relapsed can be observed (). This again was not consistent with CD45RO staining in tumors (). Similar discrimination was evident in Series 2, although the area of CD8+ cells was on the whole higher in this series compared to the first series (). Analysis of Series 2 using Kaplan–Meir plots also demonstrated a clear difference in relapse between the CD8+-high and low groups (). Combining the two cohorts and using the collective median cut-off, the presence of high CD8+ cells remained highly significantly different between patients who relapsed and those that did not (). Although the presence of CD45RO+ TILs was unable to discriminate between the two patient groups alone in terms of relapse, low CD45RO+ or CD8+ cells was indicative of poor patient outcome ().
Figure 1. Abundance of tumor infiltrating lymphocytes in early stage MSS colorectal cancers track with relapse-free survival. (A) CD8+ cells were evaluated in two cohorts of patients (Series 1- RMH), one that had tumor relapse (n = 10) that other without (n = 10). The area of positive CD8+ IHC signal was found to the significantly higher in relapse-free patients (two-way t-test). (B) CD45RO+ cells IHC showed no significant difference between the two cohorts. (C) Data from (A) were used to calculate the statistical median which was used to partition with an area above 9.5% and those 9.5% and below. Relapse-free patients were found to have significantly higher CD8+ cells that tracked with relapse-free survival (Log-rank, Mantel–Cox test). (D) Data from (B) were used to calculate the median percentage of CD45RO+ cells whereby tumors with a median above 25.3% and those below were indistinguishable in terms of outcome. (E) CD8+ cells were evaluated in two cohorts of patients from a separate hospital (Series 2- SJOG), one that had tumor relapse (n = 11) that other without (n = 9). The area of positive CD8+ IHC signal was found to the significantly higher in relapse-free patients (two-way t-test, n = 10). (F) Data from (E) were used to calculate the statistical median which was used to partition with an area above 27.5% and those below whereby the relapse-free patients were found to have significantly higher CD8+ cells tracked with relapse-free survival (Log-rank, Mantel–Cox test). (G) Data from Series 1 and 2 (Panels A and E) were pooled and the median recalculated as above and whereby tumors with a median above 12.5% and those below found to have significantly higher CD8+ cells tracked with relapse-free survival (Log-rank, Mantel–Cox test). (H) When the percentage of CD8+ plus CD45RO+ cells were evaluated those tumors where high expression was calculated patients were found to be essentially relapse-free.
The unfolded protein stress response, stromal inflammation and MYB
We next evaluated a range of potential biomarkers in CRC specifically related to TILs. The inflammatory marker COX-2 and CD8+ TILs in general co-localized, however, they were not significantly correlated with pSTAT3 or nuclear NFkBp65 within the tumor or adjacent stroma. COX-2 expression was found predominantly within the tumor epithelia. NFkBp65 showed intense epithelial cytosolic as well as nuclear expression (data not shown). Neither, COX-2, pSTAT3 or p65 expression or localization provided any insights as to the basis of high or low TILs. Therefore, we explored other potential biomarkers that might correspond to the differences in TIL density in patients with different cancer outcomes.
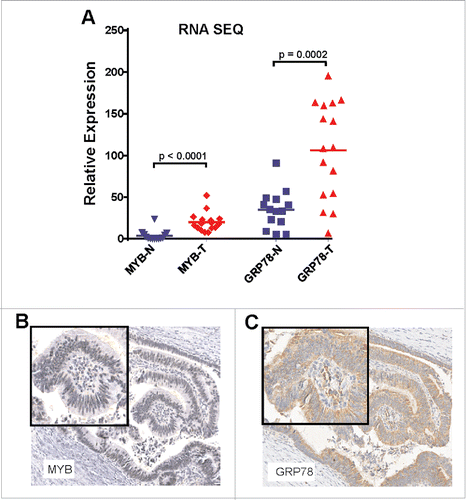
Increasingly, there has been recognition that cellular stress in the form of the unfolded stress response (UPR) pathway might be important in carcinogenesis.Citation18 Here, we focused on GRP78/BIP which is a robust marker of the UPR. We have previously reported that (i) GRP78 expression is modulated by the TF MYB,Citation11 (ii) that GRP78 is elevated when additional exogenous MYB is expressed in CRC and breast cancer cell lines and (iii) that it serves as a direct MYB-target gene.Citation19 The relevance of GRP78/BIP was of further interest in the context of TILs because it has been shown to modulate cytotoxic T-cell-mediated tumor killing.Citation15-17 We therefore evaluated both MYB and GRP78 expression in a third series of CRC and matched normal mucosa by RNAseq. This confirmed our previous finding that both MYB and GRP78 mRNA are significantly elevated in CRC ().Citation11 Analysis of tissue blocks from CRC Series 1 and 2, examining the same tumor regions as consecutive sections 4 μm apart, showed that MYB protein expression was predominantly nuclear, while GRP78 expression was cytoplasmic and that these signals were co-localized to the same cells ().
Figure 2. MYB and GRP78 expression is generally higher in colorectal tumors than in normal adjacent mucosa. (A) RNAseq was used to determine significantly higher GRP78 and MYB expression in CRC (T) compared the matched normal mucosa (N). (Two-way, t-test, n = 14 plus 2 addition CRC samples). (B) IHC for MYB and GRP78 antigens in the same region of an early stage MSS CRC.
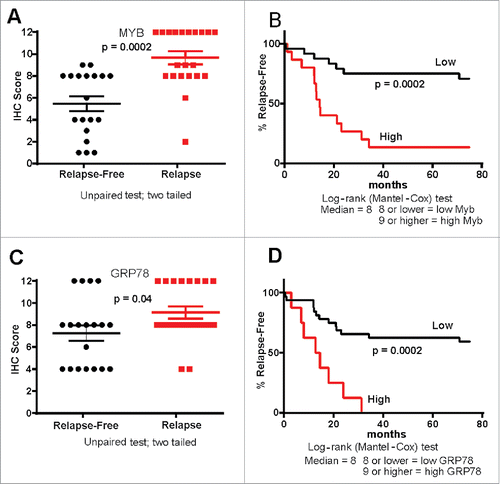
When MYB protein expression was evaluated in the combined CRC cohorts its expression (0–12 Histoscore) was significantly higher in tumors of patients who had relapsed, with this also corresponding to patient outcome (). Similarly, as might be predicated of a MYB target gene, GRP78 expression was also found to be higher in relapsed patients, with those having the highest GRP78 relapsing more rapidly ().
Figure 3. MYB and GRP78 expression tracks with CRC patient outcome. (A, B) IHC Histoscore was used to determine high and low MYB expression in relapse (n = 21) and relapse-free (n = 19) MSS CRC showing that high MYB tracks with poor outcome as well as tumor relapse (C, D) IHC Histoscore was used to determine high and low GRP78 expression in relapse and relapse-free CRC showing that high GRP78 tracks with poor outcome as well as relapse-free survival.
Absence of CD8+ TILS where MYB target GRP78 expression is strongest
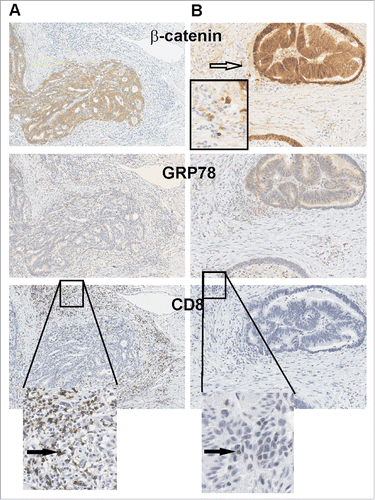
Given that both MYB and GRP78 independently predicted early relapse, we next examined their spatial expression in consecutive tumor sections to determine their relationship with CD8+ cells. To distinguish tumor from normal tissue, β-catenin staining was employed to define the boundaries of the tumor foci (). β-catenin also allowed analysis of the relapsed cases providing evidence of local tumor invasion beyond the tumor margin as has been reported by others.Citation20 We noted that in the relapse-free CRC cases CD8+ cells were most abundant at the tumor margin and in the immediate tumor stroma region (). A reciprocal relationship was found in CRC relapse cases, with a marked reduction in CD8+ cells observed where there was strong GRP78 staining. These observations were consistent across both Series 1 and 2 CRC cohorts, further establishing the reciprocal relationship between MYB, its target gene GRP78, and CD8+ cells TILs.
Figure 4. GRP78 expression is higher in CRC where CD8+ TILs cells are absent. (A) Two consecutive series of IHC representative of a relapse-free and (B) a relapse patient CRC are shown. β-catenin demarks tumor epithelium as well as invading tumor cells (open arrow) along with low GRP78 which is associated with greater CD8+ TILs. Black arrows identify CD8+ TILs.
MYB upregulation directly reduces T-cell activation in CT26 CRC mouse model
To directly explore the relationship between high GRP78/MYB expression and low CD8+ TILs observed in human CRC, we employed the BALB/c mouse colon tumor model, CT26. The use of the CT26 tumor cell line allowed us to investigate the effect of over-expressing MYB in a syngeneic primary tumor setting.Citation21 CT26 cells were transduced with a retrovirus expressing full length MYB linked with an IRES GFP reporter gene (data not shown). Successful transduction was confirmed by FACS sorting for GFP-expression and analysis of Gfp mRNA (), as well as upregulation of the MYB target gene, Grp78 ().
Figure 5. MYB overexpression increases GRP78. (A) Green fluorescent protein (Gfp) gene reporter expression in mouse CT26 CRC cells transduced with a MYB-expressing retrovirus is shown. (B) Induction of endogenous Grp78 mRNA expression in MYB-GFP-CT26 cells compared to parental cells. (C) Tumor formation kinetics (volume) in parental verses MYB-GFP-CT26 cells was determined. (D) IHC for GRP78 in parental verses MYB-GFP-CT26 including secondary antibody control (2nd Ab). (E) Percentage area of tumor expressing GR78 quantified by MetamorphR analyses is shown.
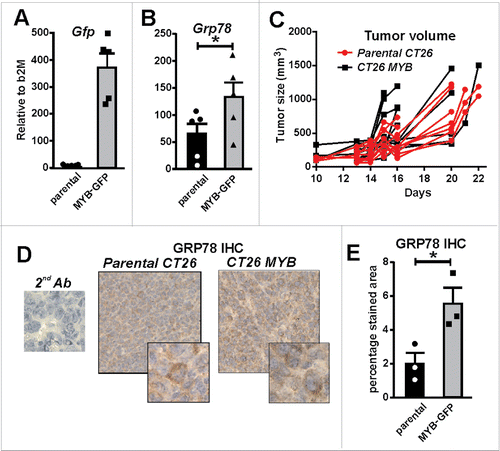
As parental CT26 cells have a high rate of proliferation in vitro, no increase was observed with upregulation of MYB (data not shown). When the MYB overexpressing CT26 cells were injected in vivo, no overall difference in tumor growth was observed when compared to control cells (). However, we did note that tumors became palpable earlier in the mice with MYB-over-expressing cells. Concordant with our in vitro observations, an increase in the GRP78 was also observed in vivo when tumors were examined by IHC ().
When we examined established CT26 tumors very few CD8+ cells were observed by IHC in either parental or MYB-transduced CT26 tumors (data not shown). A low number of CD8+ cells confined to the stroma surrounding the tumor, mirroring our findings in relapse CRC patients. As the TILs could not be quantified numerically using IHC, flow cytometry of primary tumors was employed. While flow cytometry does not allow for accurate quantification of total CD8+ cells, it does facilitate an evaluation of the quality of the TILs from both CT26 parental and CT26-MYB tumors to be determined using cell surface markers CD44 and CD62L. Analysis of CT26 parental and CT26-MYB tumors showed a modest but statistically significant reduction in the activation state of MYB over-expressing tumors compared to parental ().
Figure 6. Increased MYB is associated with suppressed immune activation. (A) CD8+ TILs were isolated from tumors generated by parental and MYB-GFP-CT26 cells and assessed for activation marker CD44. (B) Tumor draining lymph nodes were isolated on the basis of CD4+ or CD8+ expression and then evaluated for T-cell activation markers CD62L and CD44. Significantly reduced activation status was observed in mice bearing MYB-GFP-CT26 tumors; (*p < 0.05, t-test). (C) Tumor growth was indistinguishable when CT26 cells with either MYB-GFP or GFP were compared following doses of isotype control antibodies (200 mg IP) at day 5, 10 and 15 post tumor cell inoculation (arrows). (D) When mice bearing CT26 MYB-GFP cells were treated with anti-PD-1 antibodies (200 mg IP) at day 5, 10 and 15 (arrows) 3/6 mice escaped growth inhibition compared to 0/6 mice in the CT26-GFP control group.
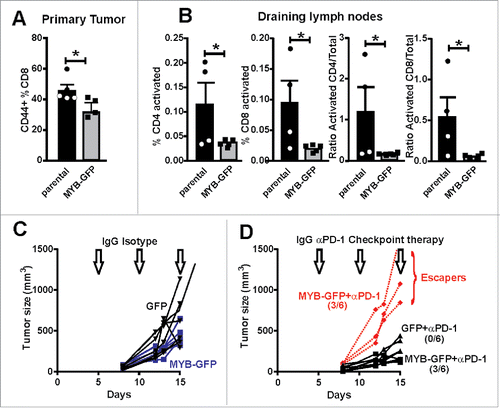
Given MYB appeared to be suppressing the infiltration of activated T cells at the primary tumor site, we then investigated whether a similarly reduced activation state could also be found at the tumor draining lymph node. Analysis of tumor-draining lymph nodes from CT26 and CT26-MYB tumor bearing mice showed that the percentage of activated (CD44+; CD62L−) CD4+ and CD8+ T cells were significantly lower in MYB over-expressing tumors (). Similarly, we found a significant reduction in the activation status of CD4+ and CD8+ T cells when we looked at the ratio of activated CD44+ CD62L− cells to naïve CD44− CD62L+ cells (). These analyses were performed on relatively small tumors (∼0.2 g), suggesting that MYB directly influences (early on) the ability of the host animal to mount a robust immune response against tumors. These observations are thus in accord with what we found in human CRC and suggest that the reciprocal relationship between high MYB, its target GRP78 and low CD8+ TILs is a direct result of the expression levels of MYB.
As MYB appeared to be directly influencing the activation state of the immune system in CT26 tumor bearing mice, we next determined whether this would impact upon immunotherapy. Other studies,Citation22 including our own,Citation23 have shown the CT26 model to be responsive to anti-PD-1 check-point blockade. Employing the same therapeutic strategy as our previous study, we demonstrated that overexpression of MYB eliminated any therapeutic benefit in half of the treated mice compared to uniform tumor control in the GFP-expressing CT26 parental tumors (). These data directly highlight the immunomodulatory capabilities of MYB in CRC and how it might affect the implementation of immunotherapy in CRC.
Discussion
In this study, we have shown that that high CD8+ TILs are associated with relapse-free survival in patients with early stage, node-negative MSS CRC, similar to previous findings when all stages are considered.Citation1 This analysis of early stage MSS CRC shows that the immune system remains an important prognostic indicator of relapse, even when neo-epitopes are likely to be very low. Given the importance of the immune microenvironment in cancer, the lack of association with either recurrence or relapse free survival for COX-2, pSTAT3 and NFkBp65 when analyzed within the tumor or in surrounding stroma, was unexpected (data not shown). In view of the lack of obvious associations with transcriptional drivers of inflammation, pSTAT3 and NFκBp65, we investigated another TF, MYB, which we had shown to predict CRC outcome in more advanced disease.Citation9
We were additionally interested in GRP78 expression because we had previously found it to be a MYB regulated gene, with high expression observed in CRC cell lines, particularly those with amplified MYB.Citation11 Beyond GRP78 being a sentinel of MYB activity, it also serves to show induction of the immune-modulatory UPR pathway. Others have also noted the potential importance of the UPR in immune modulation.Citation24,25 When GRP78 is over-expressed, it is mis-localized to the cell surface and has also been shown to be secreted.Citation26 With specific relevance to immune-modulation, GRP78 has been found to reduce cytotoxic T-cell activity Citation15 and induce T-regs.Citation27 When we analyzed MYB expression, we found that it was concordant with GRP78 and combined, these proteins were inversely correlated with CD8+ TILs.
Although the role of the UPR in cancer immunity is emerging, it is important to emphasize that MYB is a master regulator of a plethora of pro-oncogenic genes. There are more than 80 pro-oncogenic genes that are directly regulated by MYB, including GRP78 and GRP94Citation11,10 Accordingly, the concept of targeting transcriptional regulation directly is building currency.Citation28 With specific reference to MYB, it is the case that it directly regulates genes including VEGFA, COX2 and BCL2. VEGFA for instance is also an immune modulator while BCL2 protects cells from apoptosis. Therefore, focusing on any single MYB target gene is unlikely to explain MYB's association with immune evasion as the multifaceted orchestration of oncogenesis by MYB is certain to involve many of its targets.
To directly address whether high MYB expression in CRC was driving low CD8+ TILs, we used a mouse CRC model. We expressed additional MYB in this mouse CRC line that generates highly aggressive tumors in an immunocompetent host. We found no significant differences in cell line growth in vitro, and there was at best, only a trend to more rapid induction of tumors in vivo. However, we were able to demonstrate more uniform and extensive cell-surface expression of GRP78 in the MYB-transduced cells that formed tumors.
As we found very few CD8+ TILs in the parental CT26 tumors, it was not surprising that they were numerically too few to evaluate robustly in tumors generated from MYB-over-expressing cells. Nevertheless, the activation status of TILs was statistically significantly decreased by the expression of elevated MYB. Perhaps more interestingly, when we evaluated CD4+ and CD8+ cells in the tumor-draining lymph nodes, we were also able to establish that these were also substantially reduced in terms of activation. When we then challenged mice with established tumors with anti-PD-1 immune check-point blockade, we found that half of the mice with CT26-MYB were able to escape the characteristic tumor control normally observed in this model. These observations suggest that MYB has strong immunomodulatory effects, perhaps through induced GRP78 expression or another undefined mechanism, that have the potential to operate at a distance from the primary tumor site. Thus, these data go some of the way to explain why high MYB is associated with a reduced antitumor immunological response and poor outcome.
In conclusion, these studies have shown the prognostic significance of a robust CD8+ TIL response in patients with early stage, presumably mutation-low, CRC. The recognition that MYB and its downstream transcriptional target GRP78 track with CRC development provides opportunities to advance new therapeutic strategies in treating CRC and to better predict treatment outcome. This study is the first direct demonstration that MYB expressed in tumor cells can modulate the host immune response, which has the potential to influence the use of immunotherapy in CRC patients. Emerging strategies to exploit cell surface expression of GRP78 in cancer are being reported,Citation29-31 and our results suggest they may synergize with immunotherapy in CRC. While MYB has numerous gene targets, many with a pro-tumorigenic action, we have considered approaches that directly target MYB over-expression in CRC using immunological strategies with some success in pre-clinical animal models.Citation23,32,33 The data presented here further elevate the importance of MYB in cancer, highlighting a new pro-tumorigenic role in negatively modulating the immune response to tumors.
Methods
Patients' characteristics
Characteristics of tumors from atypical relapse (n = 21) and relapse-free (n = 19) patients are tabulated (). Tumor blocks were used with consent according to the institutional ethics approval at Royal Melbourne Hospital (RMH), Peter MacCallum Cancer Center and St John of God Subiaco Hospital (SJGSH), Western Australia. All patients were diagnosed with Stage B (T2-4N0M0) CRC, gave informed consent. This study was approved by the ethics committees at all sites.
Table 1. Patient and tumor characteristics.
Patients with Stage B CRC (T2-4N0M0) were identified and their FFPE blocks were retrieved with associated outcome data. In addition to overall staging, every attempt was made to match patients regardless of outcome. Cases with deficient mismatch repair (MSI-H) were excluded due to the recognized association with increased TILs and increased stage specific survival.Citation6,7,34 Accordingly in our study, only MSS tumors were evaluated. The two cohorts examined were from geographically distant sites and managed by separate health systems. Patients who relapsed did so at a median of 24 (Series 1, RMH) and 12 (Series 2, SJGSH) months, respectively. The RMH series had CRC blocks from 10 relapse and 10 closely matched relapse-free patients, while the SJGSH series had 11 relapse and nine closely matched relapse-free patients. Tumor sites were evenly distributed from the right to the left side of the colon, as was the site of relapse.
Human CRC sections
Tumor blocks from patients with early stage CRC were sourced from the RMH and SJGSH. These were enriched for cases with atypical recurrence. These were processed into ten sections and subjected to immunohistochemistry (IHC) to assess TILs and biomarkers expression.
IHC
Antibodies and antigen retrieval protocols are described in Tables S1 and S2. Briefly, antigen retrieval was achieved by placing the slides in 500 mL of EDTA (1 mM pH 6.0) or citrate buffer (10 mM, pH 6.0) in a pressure cooker. To detect primary antibodies the appropriate antibody was diluted in TBS and 150 μL was added to dewaxed slides at either RT ˚C for 1 h or 4˚C O/N. Corresponding secondary HRP-conjugated antibody (ImmPRESS, Vector Laboratories) was added for either 30 min or 1 h and for slides that did not use the ImmPRESS secondary antibody an additional step was included with the ABC detection kit (Vector Laboratories). Following incubation slides were washed twice in TBS-Tween for 10 min and developed using the DAB with chromagen substrate (DAKO) system.
Slide vizualization and quantitative analysis
Slides were coded and then scanned using the AperioR ScanScope scanner (Aperio in the Tissue Bank Facility at PMCC) and visualized on a computer using the ImageScope software (Aperio). For CD8+ and CD45RO IHC images were taken of each slide and quantitation analysis was carried out using the MetaMorph (Molecular Devices) microscopy image analysis to determine the number of brown stained cells. The MetaMorph software quantitates the number of brown cells/mm2 by setting a color threshold to first demark the brown cells to be counted. A second threshold is then set based on pixel size and any cell that is greater or less than the set threshold will be omitted from the cell count. The number of cells counted was then divided by the total area selected and multiplied by one hundred to obtain a percentage/mm2.
IHC assessment for other antigens involved inspection of AperioR generated images and scoring on the basis of a 0–12 Histoscore which was the product of staining intensity (0 = none, 1 = weak, 2 = moderate, 3 = strong) and extent (0 = none, 1 = 5% or less, 2 = 6–15 %, 3 = 51–75%, 4 => 76%). Two investigators in a blinded fashion scored de-identified slides.
Generation of CT26 CRC cells expressing exogenous MYB
The mouse colon adenocarcinoma cell line CT26 Citation21 has been authenticated by the authors based upon gene expression, tumor morphology and capacity to form adenocarcinoma in immune competent BALB/c mice. The retroviral packaging cell line PT67 were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS. PT67 cell line was used to produce MSCV-MYB retroviral particles as previously described.Citation19 The MSCV vector contains GFP allowing sorting the MSCV-MYB transduced cells by FACS. Transduction was carried out by replacing the culture media with retrovirus supernatant supplemented with 8 μg/mL polybrene (Sigma) every 24 h until cells were confluent. As a control, CT26 cells were mock transduced under similar conditions or expressed GFP alone. GFP+ cells were sorted after two weeks using a FACS DIVA sorter (BD).
Human CRC
Total RNA from 14 match matched normal mucosa and different stages of CRC including three additional CRC specimens were processed for RNA sequencing using methods described elsewhere.Citation35
Mouse CRC group assays
CT26 tumor cells (0.5 × 106) were subcutaneously injected into syngeneic BALB/c mice in the right lower flank. Tumors were measured using an electronic caliper. Experiments were conducted according to the Peter MacCallum Cancer Center's AECC guidelines. qRT-PCR for Grp78 has been described previously.Citation19
Fluorescent cell sorting of tumor and draining lymph node TILs
Tumor or Lymph Nodes were crushed in cold PBS using a 10 mL syringe plunger and gently pushed through a 70 μm filter into 50 mL falcon tube. The cells were spin down for 5 min at 1200 rpm at 4°C. Supernatant was decanted and discarded. Resuspended cells were transferred to round bottom 96 well plates in 200 μL of PBS, 2 mM EDTA (FACS Buffer) and the cells were spun down for 5 min at 1200 rpm at 4°C. These were then resuspended in antibody cocktail, 40–50 uL/sample and incubated on ice in the dark for 20–30 min. Cells were stained with a cocktail of antibodies CD44 (BV785, Biolegend, Karrinyup, Western, Australia), CD62L (BV510, Biolegend), CD8α (BV711, Biolegend), CD4+ (APC-Cy7 BD Biosciences), TCR (BV421, BD Biosciences, Australia) and PI. TCR, Samples were analyzed by flow cytometry (Fortessa; BD Biosciences). The data was analyzed using FlowJo software v8.5.3 (Stanford University, Palo Alto, California), and the mean percentage of viable cells for each cell type +/− SEM was determined for each group as previously reported.Citation36
Anti-PD-1 therapy
Mice were inoculated with CT26 cells and treated with isotype control IgG or anti-PD-1antibody at 200 μg IP on days 5, 10 and 15 as described.Citation23
Statistical analyses
The difference in tumor growth between the treatment groups was analyzed by one-way ANOVA with p < 0.05 considered significant. The difference in mean percentage +/− SEM of immune cell types in the primary tumor or in the draining lymph nodes and the IHC staining for GRP78 and COX-2 in the tumors plus qRT-PCR data were analyzed by student t-test or Mann and Whitney test.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1149667_s02.docx
Download MS Word (20.5 KB)Acknowledgment
We would like to thank Lisa Spalding who was responsible for coordinating the SJGSH cases and all associated clinical information.
Funding
RGR is supported by a Fellowship from the National Health and Medical Research Council, Australia.
References
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960-4; PMID:17008531; http://dx.doi.org/10.1126/science.1129139
- Galon J, Fridman WH, Pages F. The adaptive immunologic microenvironment in colorectal cancer: a novel perspective. Cancer Res 2007; 67:1883-6; PMID:17332313; http://dx.doi.org/10.1158/0008-5472.CAN-06-4806
- Baxevanis CN, Papamichail M, Perez SA. Immune classification of colorectal cancer patients: impressive but how complete? Expert Opin Biol Ther 2013; 13:517-26; PMID:23289642; http://dx.doi.org/10.1517/14712598.2013.751971
- Ernst M, Ramsay RG. Colorectal cancer mouse models: Integrating inflammation and the stroma. J Gastroenterol Hepatol 2012; 27:39-50; PMID:22188027; http://dx.doi.org/10.1111/j.1440-1746.2011.06883.x
- Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Eng J Med 2000; 342:69-77; PMID:10631274; http://dx.doi.org/10.1056/NEJM200001133420201
- Lal N, Beggs AD, Willcox BE, Middleton GW. An immunogenomic stratification of colorectal cancer: Implications for development of targeted immunotherapy. Onco Immunol 2015; 4:e976052; PMID:25949894; http://dx.doi.org/10.4161/2162402X.2014.976052
- Michael-Robinson JM, Biemer-Huttmann A, Purdie DM, Walsh MD, Simms LA, Biden KG, Young JP, Leggett BA, Jass JR, Radford-Smith GL. Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status. Gut 2001; 48:360-6; PMID:11171826; http://dx.doi.org/10.1136/gut.48.3.360
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Eng J Med 2015; 372:2509-20; PMID:26028255; http://dx.doi.org/10.1056/NEJMoa1500596
- Biroccio A, Benassi B, D'Agnano I, D'Angelo C, Buglioni S, Mottolese M, Ricciotti A, Citro G, Cosimelli M, Ramsay RG et al. c-Myb and Bcl-x overexpression predicts poor prognosis in colorectal cancer: clinical and experimental findings. Am J Pathol 2001; 158:1289-99; PMID:11290547; http://dx.doi.org/10.1016/S0002-9440(10)64080-1
- Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nature Rev Cancer 2008; 8:523-34; PMID:18574464; http://dx.doi.org/10.1038/nrc2439
- Ramsay RG, Ciznadija D, Mantamadiotis T, Anderson R, Pearson R. Expression of stress response protein glucose regulated protein-78 mediated by c-Myb. Int J Biochem Cell Biol 2005; 37:1254-68; PMID:15778089; http://dx.doi.org/10.1016/j.biocel.2004.12.011
- Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol 2004; 16:343-9; PMID:15261665; http://dx.doi.org/10.1016/j.ceb.2004.06.012
- Li Z, Li Z. Glucose regulated protein 78: a critical link between tumor microenvironment and cancer hallmarks. Biochim Biophys Acta 2012; 1826:13-22; PMID:22426159; http://dx.doi.org/10.1016/j.bbcan.2012.02.001
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Wang M, Zhao XR, Wang P, Li L, Dai Y, Huang H, Lei P, Zhu HF, Shen GX. Glucose regulated proteins 78 protects insulinoma cells (NIT-1) from death induced by streptozotocin, cytokines or cytotoxic T lymphocytes. Int J Biochem Cell Biol 2007; 39:2076-82; PMID:17689130; http://dx.doi.org/10.1016/j.biocel.2007.05.022
- Jamora C, Dennert G, Lee AS. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc Natl Acad Sci U S A 1996; 93:7690-4; PMID:8755537; http://dx.doi.org/10.1073/pnas.93.15.7690
- Sugawara S, Takeda K, Lee A, Dennert G. Suppression of stress protein GRP78 induction in tumor B/C10ME eliminates resistance to cell mediated cytotoxicity. Cancer Res 1993; 53:6001-5; PMID:8261413
- Sato M, Yao VJ, Arap W, Pasqualini R. GRP78 signaling hub a receptor for targeted tumor therapy. Adv Genet 2010; 69:97-114; PMID:20807604; http://dx.doi.org/10.1016/S0065-2660(10)69006-2
- Miao RY, Drabsch Y, Cross RS, Cheasley D, Carpinteri S, Pereira L, Malaterre J, Gonda TJ, Anderson RL, Ramsay RG. MYB is essential for mammary tumorigenesis. Cancer Res 2011; 71:7029-37; PMID:21948968; http://dx.doi.org/10.1158/0008-5472.CAN-11-1015
- Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 2001; 98:10356-61; PMID:11526241; http://dx.doi.org/10.1073/pnas.171610498
- Corbett TH, Griswold DP, Jr, Roberts BJ, Peckham JC, Schabel FM, Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res 1975; 35:2434-9; PMID:1149045
- Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol 2005; 17:133-44; PMID:15611321; http://dx.doi.org/10.1093/intimm/dxh194
- Cross RS, Malaterre J, Davenport AJ, Carpinteri S, Anderson RL, Darcy PK, Ramsay RG. Therapeutic DNA vaccination against colorectal cancer by targeting the MYB oncoprotein. Clin Translational Immunol 2015 4, e30; PMID:25671128; http://dx.doi.org/10.1038/cti.2014.29
- Mahadevan NR, Rodvold JJ, Zanetti M. A Janus-faced role of the unfolded protein response in antitumor immunity. Oncoimmunol 2013; 2:e23901; PMID:23762789; http://dx.doi.org/10.4161/onci.23901
- Zanetti M, Rodvold JJ, Mahadevan NR. The evolving paradigm of cell-nonautonomous UPR-based regulation of immunity by cancer cells. Oncogene 2016; 35:269-78; PMID:25893303; http://dx.doi.org/10.1038/onc.2015.108
- Panayi GS, Corrigall VM. BiP, an anti-inflammatory ER protein, is a potential new therapy for the treatment of rheumatoid arthritis. Novartis Found Symp 2008; 291:212-6; discussion 6–24; PMID:18575276; http://dx.doi.org/10.1002/9780470754030.ch16
- Corrigall VM, Vittecoq O, Panayi GS. Binding immunoglobulin protein-treated peripheral blood monocyte-derived dendritic cells are refractory to maturation and induce regulatory T-cell development. Immunol 2009; 128:218-26; PMID:19740378; http://dx.doi.org/10.1111/j.1365-2567.2009.03103.x
- Gonda TJ, Ramsay RG. Directly targeting transcriptional dysregulation in cancer. Nature Rev Cancer 2015; 15:686-94; PMID:26493648; http://dx.doi.org/10.1038/nrc4018
- Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013; 32:805-18; PMID:22508478; http://dx.doi.org/10.1038/onc.2012.130
- Park HR, Tomida A, Sato S, Tsukumo Y, Yun J, Yamori T, Hayakawa Y, Tsuruo T, Shin-ya K. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst 2004; 96:1300-10; PMID:15339968; http://dx.doi.org/10.1093/jnci/djh243
- Miao YR, Eckhardt BL, Cao Y, Pasqualini R, Argani P, Arap W, Ramsay RG, Anderson RL. Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin Cancer Res 2013; 19:2107-16; PMID:23470966; http://dx.doi.org/10.1158/1078-0432.CCR-12-2991
- Williams BB, Wall M, Miao RY, Williams B, Bertoncello I, Kershaw MH, Mantamadiotis T, Haber M, Norris MD, Gautam A et al. Induction of T cell-mediated immunity using a c-Myb DNA vaccine in a mouse model of colon cancer. Cancer Immunol Immunother 2008; 57:1635-45; PMID:18386000; http://dx.doi.org/10.1007/s00262-008-0497-2
- Carpinteri S, Williams BB, Miao Yu R, Cullinane C, Malaterre J, Norris MD, Haber M, Anderson RL, Darcy PK, Ramsay RG. Optimizing DNA Vaccines Against Nuclear Oncogenes. Immuno-Gastroenterol 2012; 1:108-18; http://dx.doi.org/10.7178/ig.19
- Prall F, Duhrkop T, Weirich V, Ostwald C, Lenz P, Nizze H, Barten M. Prognostic role of CD8+ tumor-infiltrating lymphocytes in stage III colorectal cancer with and without microsatellite instability. Hum Pathol 2004; 35:808-16; PMID:15257543; http://dx.doi.org/10.1016/j.humpath.2004.01.022
- Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, Arango D, Strausberg RL, Buchanan D, Wormald S et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res 2014; 74:3238-47; PMID:24755471; http://dx.doi.org/10.1158/0008-5472.CAN-14-0013
- John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CP, Stewart TJ, Westwood JA, Guo ZS, Bartlett DL et al. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res 2012; 72:1651-60; PMID:22315352; http://dx.doi.org/10.1158/0008-5472.CAN-11-2788