
ABSTRACT
Cancers can escape immunesurveillance by diminishing the expression of MHC class-I molecules (MHC-I) and components of the antigen-processing machinery (APM). Developing new approaches to reverse these defects could boost the efforts to restore antitumor immunity. Recent studies have shown that the expression of MHC-I and antigen-processing molecules is transcriptionally regulated by NOD-like receptor CARD domain containing 5 (NLRC5). To investigate whether NLRC5 could be used to improve tumor immunogenicity, we established stable lines of B16-F10 melanoma cells expressing NLRC5 (B16-5), the T cell co-stimulatory molecule CD80 (B16-CD80) or both (B16-5/80). Cells harboring NLRC5 constitutively expressed MHC-I and LMP2, LMP7 and TAP1 genes of the APM. The B16-5 cells efficiently presented the melanoma antigenic peptide gp10025–33 to Pmel-1 TCR transgenic CD8+ T cells and induced their proliferation. In the presence of CD80, B16-5 cells stimulated Pmel-1 cells even without the addition of gp100 peptide, indicating that NLRC5 facilitated the processing and presentation of endogenous tumor antigen. Upon subcutaneous implantation, B16-5 cells showed markedly reduced tumor growth in C57BL/6 hosts but not in immunodeficient hosts, indicating that the NLRC5-expressing tumor cells elicited antitumor immunity. Following intravenous injection, B16-5 and B16-5/80 cells formed fewer lung tumor foci compared to control cells. In mice depleted of CD8+ T cells, B16-5 cells formed large subcutaneous and lung tumors. Finally, immunization with irradiated B16-5 cells conferred protection against challenge by parental B16 cells. Collectively, our findings indicate that NLRC5 could be exploited to restore tumor immunogenicity and to stimulate protective antitumor immunity.
Abbreviations
| APM | = | antigen processing machinery |
| B16-5 | = | B16-F10 cells expressing NLRC5 |
| B16-80 | = | B16-F10 cells expressing CD80 |
| B16-5/80 | = | B16-F10 cells expressing NLRC5 and CD80 |
| β2M | = | β2 microglobulin |
| CITA | = | MHC-I trans-activator |
| CIITA | = | MHC-II trans-activator |
| CTL | = | cytotoxic T lymphocytes |
| DC | = | Dendritic cells |
| ER | = | endoplasmic reticulum |
| MHC-I | = | major histocompatibility class-I |
| NLRC5 | = | NOD-like receptor CARD domain containing 5 |
| TAP | = | transporters associated with antigen processing. |
Introduction
An important immune evasion mechanism of tumor cells is to reduce their immunogenic potential and thereby escape destruction by cytotoxic T lymphocytes (CTL). For this purpose, cancer cells use several strategies such as downregulation of dominant tumor antigens, blockade of the APM and repression of the MHC-I molecules.Citation1 Expression of MHC-I requires the assembly of MHC-I heavy chain, β2 microglobulin (β2M) and an antigenic peptide within the endoplasmic reticulum (ER).Citation2,3 The antigenic peptide is generated by proteasomes, transported across the ER by ‘transporters associated with antigen processing’ TAP1 and TAP2, loaded onto MHC-I:β2M complex and delivered to the cell surface. By sampling these peptides, CD8+ T cells carry out their immunesurveillance functions and eliminate neoplastic cells expressing tumor-specific and tumor-associated antigens.Citation4-6 Many cancers show defects in the MHC-I antigen presentation pathway that would allow cancer cells to escape destruction by CTLs.Citation1,7,8 In some cancers, the MHC-I defects correlate with high tumor grading, disease progression, reduced survival and failure of CTL-based immunotherapies.Citation1,8 Notably, in melanoma patients undergoing immunotherapy, all regressing metastatic lesions expressed residual MHC-I while progressing metastases did not.Citation9,10 The defects in the expression of MHC-I and APM in cancer cells are classified into reversible “soft” and irreversible “hard” lesions.Citation7,11 While hard lesions arise from gene loss or structural mutations, soft lesions could arise from epigenetic, transcriptional and post-translational alterations.Citation12-14 The soft lesions are exemplified by restoration of MHC-I expression by DNA demethylating agents or IFNγ.Citation15,16 Even though defects in MHC-I expression and antigen presentation in cancer cells have been extensively documented, the transcriptional regulation of MHC-I and antigen-processing pathway genes, and their deregulation in cancer cells are not yet well understood.
NLRC5 is a member of the NLR family proteins that contain a nucleotide-binding domain and leucine-rich repeats, which are conserved in pattern recognition receptors that regulate inflammatory responses and cell death.Citation17 NLRC5 is conserved in vertebrates and is expressed in various tissues, although high expression occurs in immune tissues and mucosal epithelia.Citation18-22 The NLRC5 promoter is inducible by IFNγ. Citation18,21,23 NLRC5 is structurally related to NLRA, also known as MHC-II trans-activator (CIITA), which is essential for the transcription of MHC-II genes.Citation24 Similar to CIITA that induces MHC-II genes, NLRC5 promotes MHC-I gene expression and thus called MHC-I trans-activator (CITA).Citation23,24 Several groups studying the role of NLRC5 in innate immune functions have generated Nlrc5−/− mice, which have confirmed the essential role of NLRC5 in MHC-I expression.Citation18-23,25-29 The promoters of MHC-I genes contain cis-regulatory elements (SXY module) that bind distinct transcription factors, with which NLRC5 interacts and recruits transcriptional modifiers to assemble the MHC-I enhanceosome transcriptional complex.Citation24,30-33 NLRC5 also induces genes coding for B2M, LMP2 (large multifunctional proteasome 2, a proteasome component) and TAP1 involved in antigen processing and presentation to CD8+ T cells.Citation23,26,27 In agreement, Nlrc5-/- mice show impaired CTL responses, and NLRC5-null target cells are not efficiently cleared by CTLs.Citation26,27
Given the role of NLRC5 in the transcription of MHC-I and APM genes, we postulated that NLRC5 may play important roles in antitumor immunity and its loss may promote tumor immune evasion. In this study, we investigated the ability of NLRC5 to elicit antitumor immunity using the B16-F10 (referred hereafter as B16) mouse melanoma model. The B16 melanoma is a poorly immunogenic tumor that grows aggressively in syngeneic C57Bl/6 mice.Citation34 B16 cells express several melanoma antigens such as gp100 (also called Pmel-1), tyrosinase, tyrosinase-related protein 1 and dopachrome tautomerase.Citation34 The poor immunogenicity of B16 cells has been linked to low expression of MHC-I and APM.Citation35 To determine whether NLRC5 could be used to improve tumor immunogenicity, we generated stable lines of B16 cells expressing NLRC5 either alone or along with the T cell co-stimulatory ligand CD80. We evaluated the ability of these cells to present exogenously added or endogenous gp100 antigenic peptide in vitro and in vivo, their growth as tumors in C57BL/6 mice and their ability to activate gp100-specific CD8+ T cells. We also investigated the ability of NLRC5-expressing B16 cells to confer immune resistance to challenge by parental B16 cells. Our findings show that NLRC5 expression renders B16 cells immunogenic and raise the possibility that NLRC5 could be exploited to elicit antitumor immunity.
Results
Stable expression of NLRC5 enhances MHC-I expression in B16 cells and increases their ability to activate CD8+ T cells
B16 cells express low levels of MHC-I, β2M and several genes of the APM such as LMP2, LMP7, TAP1, TAP2, PA28α and PA28β.Citation35 As NLRC5 is a master regulator of MHC-I and some of the APM,Citation24 we evaluated Nlrc5 gene expression in B16 cells. Wild type B16 cells (B16-Wt) showed negligible level of Nlrc5 gene expression at steady state that was increased >1500-fold following IFNγ stimulation (). On the other hand, some of the mouse cancer cell line that we examined did not upregulate Nlrc5 upon IFNγ stimulation and showed defective MHC-I gene expression (Fig. S1). These results indicate that B16 cells are not inherently defective in Nlrc5 gene expression. To test whether NLRC5 would enable B16 cells to activate tumor antigen-specific CD8+ T cells, we derived stable lines expressing human NLRC5 (B16-5), which has been previously shown to induce MHC-I expression in murine B16 cells.Citation31 Human and mouse NLRC5 show 62.3% amino acid sequence identity and 80% similarity (Fig. S2).Citation20 Moreover, human and mouse MHC-I gene promoters harbor similar cis-regulatory elements that are occupied by transcriptional enhanceosomes containing NLRC5.Citation33 To increase the ability of B16-5 cells to activate tumor antigen-specific CD8+ T cells, we generated B16-5 cells expressing the costimulatory ligand CD80 (B16-5/80).Citation36 As controls, cells expressing only CD80 (B16-80) or the empty vectors (B16-v), and B16-Wt cells were used. When compared with B16-Wt cells, the transfected cells showed slightly increased level of endogenous murine Nlrc5 expression that was significant only in B16-v cells ().
Figure 1. Stable expression of NLRC5 induces MHC-I and a subset of antigen processing pathway genes in B16-F10 melanoma cells. B16-F10 melanoma cells (B16-Wt) were transfected with expression constructs of human NLRC5 (EBSB-PL-EGFP-NLRC5) and mouse CD80 (pcDNA3.0-CD80), either alone or together. Transfected cells were selected with blasticidin, G418 or both to generate the stable lines B16-5, B16-80 and B16-5/80 expressing NLRC5, CD80 or both, respectively. Control cells were transfected with both vectors (B16-v) and selected by antibiotics. (A) B16-derived cell lines were evaluated by qPCR for the expression of endogenous Nlrc5 and genes coding for MHC-I (H-2D, H-2K), β2 micoglobulin, and the antigen-processing machinery: proteasome components LMP2 and LMP7, proteasome activators PA28α and PA28β, transporter associated with antigen processing Tap1, and the Tap1-associated protein tapasin. B16-Wt cells treated with 500 pg/mL of IFNγ were used as control, along with the induction of the Stat1 gene. Gene expression was normalized to the housekeeping gene Rplp0 (36B4) and then compared to B16-Wt cells to measure fold change. Mean ± SEM from three experiments are shown. Statistical comparison of the indicated groups was done by Mann–Whitney test: ****p < 0.0001. (B) Relative expression of human NLRC5 transgene in B16-5 and B16-5/80 cells. (C) Cell surface expression of MHC class-I (H-2Db) and CD80 were measured in parental and B16-derived stable cell lines by flow cytometry. Expression level in B16-Wt cells (dark gray histograms) was overlapped with that of transfected cells (white histograms). GFP fluorescence served as marker for the NLRC5 expression construct. (D) Mean fluorescence intensity (MFI) values of H-2D, H-2K and CD80 expression in B16-derived cell lines. Data shown are mean + SEM from five experiments, normalized to the expression level in B16-Wt cells. Mann–Whitney test: **p < 0.01, ***p < 0.001, ****p < 0.0001.
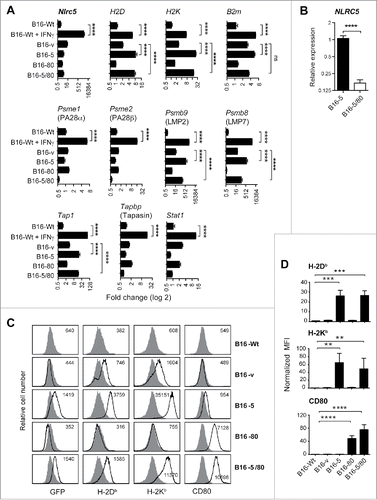
As expected, the transfected human NLRC5 transcripts were detected only in B16-5 and B16-5/80 cells. As there is no expression of human NLRC5 in the control groups (B16-Wt, B16-v), it was not possible to calculate fold increase of NLRC5 expression relative to controls. Nonetheless, the Ct values for human NLRC5 in B16-5 (22.3) and B16-5/80 (24.8) indicated that the transfected gene was well expressed in these cells. Ct value for the housekeeping gene Rplp0 was 14.9 in both B16-5 and B16-5/80 cells. Notably, B16-5/80 cells showed a 6-fold less NLRC5 transcript level with respect to B16-5 cells (), suggesting the possibility of competition for the transcription machinery in cells harboring both NLRC5 and CD80 expression constructs.
Next we examined the expression of genes coding for MHC-I and molecules involved in antigen processing (). Consistent with previous reports,Citation23,26 B16-5 and B16-5/80 cells showed constitutive expression of MHC-I genes H2D and H2K, although B2M was significantly upregulated only in B16-5 cells. The expression levels of H2D and H2K genes in NLRC5 expressing cells were comparable to the levels induced by IFNγ in B16-Wt cells. Both B16-5 and B16-5/80 cells showed significant upregulation of Psmb9 and Psmb8 coding for the proteasomal components LMP2 and LMP7, respectively, and Tap1 compared to B16-v controls (). In general, the expression of the above NLRC5-induced antigen-processing genes was slightly lower in B16-5/80 cells than in B16-5 cells, presumably due to weaker expression of NLRC5 in the latter (). Strikingly, B16-5 and B16-5/80 cells did not show expression of several other MHC-I pathway genes that were induced by IFNγ such as Psme1, Psme2 and Tapbp coding for the proteasomal activators PA28α and PA28β and TAP binding protein, respectively. These results indicate that NLRC5 induces only a subset of IFNγ-inducible MHC-I antigen-processing pathway genes.
B16-5 cells showed elevated cell surface expression of MHC-I molecules H-2Db and H-2Kb (), consistent with the expression of H2D, H2K and B2M genes. B16-5/80 cells showed reduced expression of MHC-I when compared with B16-5 cells, corresponding to the reduced level of NLRC5 transcripts and B2M gene expression in B16-5/80 cells (). B16-v cells expressing the control vector showed slight augmentation of MHC-I surface expression, presumably arising from the induction of endogenous Nlrc5 gene (). B16 cells do not express CD80, which was markedly elevated in B16-80 and B16-5/80 cells harboring the murine CD80 expression construct ().
Next we tested the ability of the B16-derived cell lines to present the mgp100 tumor antigenic peptide to Pmel-1 TCR transgenic CD8+ T cells and stimulate cell proliferation. While parental (B16-Wt) and control (B16-v) cells did not induce proliferation of naive Pmel-1 cells in response to exogenously added cognate peptide mgp10025-33, B16-5 and B16-5/80 cells stimulated robust cell proliferation (). B16-80 cells also induced strong proliferation of Pmel-1 cells in the presence of mgp100, presumably by delivering co-stimulatory signals that supplement the basal TCR signaling.Citation37 However, B16-5/80 cells did not elicit a response superior to B16-5 cells. Together, the above data indicated that stable NLRC5 expression in cancer cells can upregulate the expression of functional MHC-I molecules and present tumor antigenic peptides to CD8+ T cells and stimulate their proliferation.
Figure 2. NLRC5 increases the antigen-presenting capacity of B16 cells and enables presentation of endogenous tumor antigenic peptide. (A) B16-derived cell lines (• B16-Wt; ○ B16-v; ♦ B16-5, ▪ B16-80; ▴ B16-5/80) were gamma irradiated (100Gy) and incubated with purified Pmel-1 TCR transgenic CD8+ T cells along with the indicated concentrations of mouse gp100 melanoma antigen-derived peptide mgp10025–33. Proliferation of T cells was evaluated by [3H]-thymidine incorporation in triplicates. Representative data from three experiments with comparable results are shown. Statistical comparison was done by Mann–Whitney test. B16-v versus B16-5, B16-80, B16-5/80: ***p <0.001. (B) Irradiated B16-derived cell lines were incubated with Pmel-1 cells that were pre-stimulated for 3 d with IL-15 and IL-21 (cytokine-primed) in the presence of the indicated concentrations of mgp100 peptide. T cell proliferation was evaluated by [3H]-thymidine incorporation. Representative data from three experiments are shown. Mann–Whitney test: B16-v versus B16-5, B16-80, B16-5/80: ***p < 0.001; B16-v versus B16-5/80: ### p < 0.005. (C) Freshly purified (naive: columns a, b) or cytokine-primed Pmel-1 cells (columns c, d) were labeled with CFSE and incubated with irradiated B16-derived cell lines in the presence (columns b, d) or absence (columns a, c) of mgp100 peptide. At the indicated days (1, 2, 4, 6) of culture, cell proliferation was evaluated by dilution of the CFSE fluorescence intensity. Representative data from three independent experiments with comparable results are shown.
![Figure 2. NLRC5 increases the antigen-presenting capacity of B16 cells and enables presentation of endogenous tumor antigenic peptide. (A) B16-derived cell lines (• B16-Wt; ○ B16-v; ♦ B16-5, ▪ B16-80; ▴ B16-5/80) were gamma irradiated (100Gy) and incubated with purified Pmel-1 TCR transgenic CD8+ T cells along with the indicated concentrations of mouse gp100 melanoma antigen-derived peptide mgp10025–33. Proliferation of T cells was evaluated by [3H]-thymidine incorporation in triplicates. Representative data from three experiments with comparable results are shown. Statistical comparison was done by Mann–Whitney test. B16-v versus B16-5, B16-80, B16-5/80: ***p <0.001. (B) Irradiated B16-derived cell lines were incubated with Pmel-1 cells that were pre-stimulated for 3 d with IL-15 and IL-21 (cytokine-primed) in the presence of the indicated concentrations of mgp100 peptide. T cell proliferation was evaluated by [3H]-thymidine incorporation. Representative data from three experiments are shown. Mann–Whitney test: B16-v versus B16-5, B16-80, B16-5/80: ***p < 0.001; B16-v versus B16-5/80: ### p < 0.005. (C) Freshly purified (naive: columns a, b) or cytokine-primed Pmel-1 cells (columns c, d) were labeled with CFSE and incubated with irradiated B16-derived cell lines in the presence (columns b, d) or absence (columns a, c) of mgp100 peptide. At the indicated days (1, 2, 4, 6) of culture, cell proliferation was evaluated by dilution of the CFSE fluorescence intensity. Representative data from three independent experiments with comparable results are shown.](/cms/asset/ab1c17d2-701a-4734-9bb7-3ae009ee08d3/koni_a_1151593_f0002_b.gif)
NLRC5 expression in B16 cells enables presentation of endogenous tumor antigenic peptide
B16-5 cells induced low but discernible proliferation of Pmel-1 cells in the absence of exogenous peptide (), suggesting that NLRC5 may also facilitate the processing and presentation of endogenous mgp100 peptide. This idea was supported by increased expression of the genes coding for LMP2, LMP7 and TAP1 in B16-5 cells (). Therefore, we examined the ability of NLRC5-expressing B16 cells to present endogenous tumor antigenic peptides. Theoretical estimates indicate that processing and presentation of a specific endogenous antigenic peptide via the MHC-I pathway is a very inefficient process.Citation3 This could limit the ability of B16 cells to activate naive T cells. We have previously shown that “priming” of naive CD8+ T cells with IL-15 and IL-21 increases their responsiveness to limiting concentrations of antigenic peptides.Citation38 Therefore, we pre-stimulated Pmel-1 cells with IL-15 and IL-21 and then tested their responsiveness to B16 cells in the presence or absence of mgp100 peptide. As expected, cytokine-primed Pmel-1 cells showed significant DNA synthesis in response to lower concentrations of mgp100 presented by B16-Wt or B16-v cells (; 0.1 μg/mL), while naive Pmel-1 cells showed negligible proliferation at this peptide concentration (). Compared to B16-Wt and B16-v cells, B16-5 and B16-5/80 cells induced strong DNA synthesis in cytokine-primed Pmel-1 cells at limiting concentrations of mgp100 (, 0.01 μg/mL). Importantly, B16-5/80 cells induced strong proliferation of cytokine-primed Pmel-1 cells in the absence of exogenous peptide (), confirming the processing of endogenous antigen and presentation of the mgp100 peptide by B16 cells expressing NLRC5.
Next, we evaluated the proliferation kinetics of naive and cytokine-primed Pmel-1 cells by CFSE assay (), which assesses cell division while thymidine incorporation reflects DNA synthesis. In the presence of mgp100, all B16-derived cell lines induced strong proliferation of cytokine-primed Pmel-1 cells that was clearly discernible within 2 d of stimulation (: all cells, column d). Consistent with the thymidine incorporation data (), only B16-5/80 cells induced robust proliferation of cytokine-primed cells in the absence of exogenously added mgp100 peptide (: B16-5/80 cells, column-c, days 2, 4, 6). This response was almost comparable in kinetics and magnitude to that of naïve Pmel-1 cells stimulated by exogenous mgp100 peptide (, B16-5/80 cells, column-b, days 2, 4, 6). Strikingly, B16-5/80 cells also induced proliferation of naive Pmel-1 cells in the absence of exogenous peptide, albeit with a delayed kinetics (: B16-5/80 cells, column a, day 6). These results indicate that melanoma cells can be rendered immunogenic that are capable of activating naive CD8+ T cells by expressing NLRC5 along with CD80.
Cytokine-primed Pmel-1 cells activated by B16-5/80 cells acquire memory-like phenotype with reduced differentiation toward effector cells
Next we examined the phenotype and functionality of Pmel-1 cells activated by B16-5 and B16-5/80 cells. Most of the naive or cytokine-primed Pmel-1 cells incubated with B16-v cells, in the presence or absence of mgp100 peptide, showed CD44loCD62Lhi phenotype reflecting inefficient T cell activation (, first row). While B16-5 cells did not cause appreciable change in this phenotype even with mgp100 peptide, B16-80 cells presenting the exogenous peptide caused an increase in CD44 expression in about 40% of cytokine-primed but not naive Pmel-1 cells (, second and third rows). Only B16-5/80 cells induced marked activation of naive Pmel-1 cells without the addition of exogenous peptide, as reflected by the increased frequency of CD44loCD62Llo and CD44hiCD62Llo cells (, bottom row, first two plots). Importantly, cytokine-primed Pmel-1 cells exposed to B16-5/80 cells displayed not only an increased proportion of cells with CD44hiCD62Llo activated phenotype but also a notable increase in CD44hiCD62Lhi memory-phenotype cells (, bottom row, last two plots).
Figure 3. B16 cells expressing both NLRC5 and CD80 efficiently activate Pmel-1 CD8+ T cells. Naive and cytokine-primed Pmel-1 cells were co-cultured with irradiated (100Gy) B16-derived cell lines in the presence or absence of mgp100 peptide. Activation of Pmel-1 cells were evaluated after 3 d by analyzing (A) modulation of the expression of CD44 and CD62L, (B) production of IL-2, (C) expression of the effector cytokine TNFα and (D) mobilization of CD107b to the cell surface. Numbers within the histograms represent the percentage of cells within the quadrant or the indicated marker boundaries. Representative data from at least two experiments with similar results are shown.
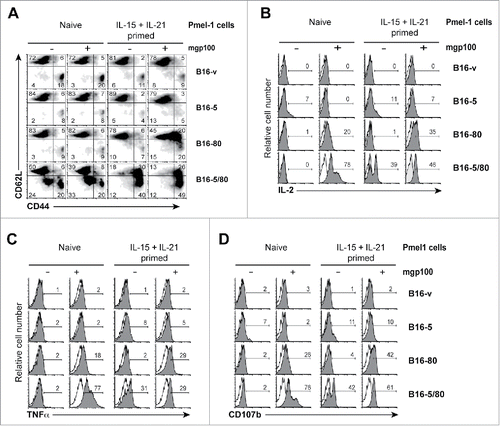
Evaluation of IL-2 and TNFα production by cytokine-primed Pmel-1 cells under the above conditions revealed that B16-5/80 cells strongly induced these cytokines, even in the absence of exogenous mgp100 peptide ( bottom rows). B16-5/80 cells also activated cytokine-primed Pmel-1 cells to display CD107b at the cell surface, which is associated with granule exocytosis in CTLsCitation39 (). While B16-5 and B16-v cells induced minimal IL-2 and TNFα production or CD107b expression in cytokine-primed Pmel-1 cells, B16-80 cells elicited a discernible response only in the presence of exogenous peptide (, third row). Even though B16-5/80 cells elicited similar phenotypic changes in “naive” Pmel-1 cells with or without peptide addition (), the responder cells acquired effector functions only in the presence of mgp100 peptide (). Together, the data shown in indicate that expression of both NLRC5 and CD80 in B16 cells is necessary to promote efficient activation of CD8+ T cell functions.
NLRC5 reduces tumor formation by B16 cells in a CD8+ T cell-dependent manner
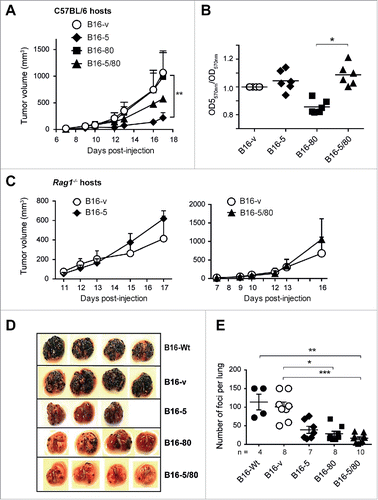
To evaluate the effect of NLRC5 on the tumor-forming potential of B16 cells in vivo, we implanted the B16-derived cells lines subcutaneously and monitored tumor growth in C57BL/6 mice. Whereas control and B16-80 cells rapidly established as tumors, B16-5 cells showed significantly reduced tumor growth (). Surprisingly, B16-5/80 cells, which were more efficient than B16-5 cells in activating Pmel-1 cells in vitro, displayed an intermediate rate of tumor growth. The reduced ability of B16-5 and B16-5/80 cells to form tumors is unlikely to result from cell-intrinsic difference in growth rates, as their in vitro expansion was comparable to that of B16-v cells (). In fact, B16-5/80 cells showed a stronger in vitro expansion than B16-80 cells, but showed reduced in vivo tumor growth. Moreover, B16-5 and B16-5/80 cells showed comparable tumor forming ability in Rag1−/− mice (), suggesting that their reduced growth in C57BL/6 mice was mediated by antitumor immune response elicited by NLRC5 expression. Next, we compared the ability of B16-derived cells to form tumor foci in the lungs following intravenous injection. As shown in , expression of NLRC5, CD80 or both significantly reduced the numbers of lung tumor foci compared to B16-Wt of B16-v cells. Interestingly, B16-5/80 cells developed the least number of lung tumor foci (), although they formed larger subcutaneous tumors than B16-5 cells (). These results indicated that NLRC5 expression retarded tumor growth and metastatic potential of B16 cells, which could be further enhanced in certain anatomic sites by simultaneous expression of CD80.
Figure 4. NLRC5 expression reduces the tumor forming ability of B16 cells. (A) The B16-derived cell lines (○ B16-v; ♦ B16-5, ▪ B16-80; ▴ B16-5/80) were injected subcutaneously into C57BL/6 mice (1 × 105 cells per injection site) and tumor growth was monitored. Error bars represent standard error. n = 4–6 mice/group from three experiments. Statistical comparisons were performed using two-way ANOVA with Tukey's multiple comparisons. **p < 0.01. (B) Growth of B16-derived cells in vitro. The B16-derived cell lines were seeded at different concentrations and in triplicates in 96-well plates and their growth was evaluated by MTT assay. Data were pooled from at least three independent experiments. Error bars represent SEM. Samples were normalized to B16-vector cells. (C) Subcutaneous tumor growth of B16-5 (♦), B16-5/80 (▴) and control B16-v (○) cells in Rag1−/− mice. Data represent four mice per group from two experiments. (D, E) The B16-derived cell lines were injected intravenously into C57BL/6 mice (2 × 105 cells/mouse) and formation of tumor foci in the lungs was evaluated 17 d later. Representative images of the lungs (D) and the number of tumor foci (E) are shown. Mean ± SE from the indicated number of mice per group from two–three independent experiments are shown. (B, D) Data were compared by one-way ANOVA with Kruskal–Wallis test adjusted for Dunn's multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.005.
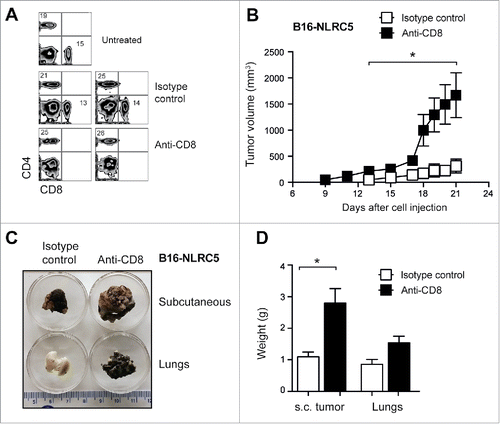
Next we examined whether the reduced tumor growth of B16-5 cells was mediated by CD8+ T cells. To this end, we depleted CD8+ T cells one day prior to tumor cell inoculation () and monitored tumor growth. Treatment with CD8+-depleting antibody, but not the isotype control, resulted in massive growth of the subcutaneously inoculated B16-5 cells (). CD8+ T cell depletion also enabled B16-5 cells to obliterate the lungs following intravenous inoculation, although this did not cause a significant increase in the lung mass (). Collectively, the above results indicate that CD8+ T-cell-mediated antitumor immunity underlies the reduced tumor forming ability of NLRC5 expressing B16 cells.
Figure 5. Rapid growth of NLRC5-expressing B16 cells in mice depleted of CD8+ T cells. (A) C57BL/6 mice were administered via intraperitoneal route anti-CD8+ or isotype control monoclonal antibody. Twenty-four hours later, depletion of CD8+ T cells was evaluated by flow cytometry. Representative data from five mice per group are shown. (B–D) CD8+-depleted mice were injected 24 h later with B16-5 cells via subcutaneous or intravenous route and tumor growth was monitored. Anti-CD8+ or control Ab was administered twice weekly throughout the evaluation period. (B) Subcutaneous tumor growth from two experiments is shown. Student's t test: *p < 0.05. (C) Representative images of subcutaneous tumors and lung tissues collected 20 d after challenge are shown in C. (D) Mass of s.c. tumors and lung tissues obtained from the challenged mice. Mann–Whittney test: *p < 0.05.
B16 cells expressing NLRC5 elicit potent antitumor immunity
To directly examine whether B16 cells expressing NLRC5 activated tumor-specific CD8+ T cells in vivo, irradiated B16-v, B16-5 or B16-5/80 cells were inoculated into the footpad of C57BL/6 mice followed by intravenous injection of naïve or cytokine-primed Pmel-1 cells (). Examination of the draining and non-draining lymph nodes and spleen showed that B16-5 cells and B16-5/80 cells induced robust proliferation of cytokine-primed Pmel-1 cells (). However, naive Pmel-1 cells underwent proliferation only in mice inoculated with B16-5/80 cells (). These results indicated that NLRC5 expression enhanced the cross-presentation of tumor cell-derived mgp100, however activation of naive CD8+ T cells required a costimulatory signal.
Figure 6. NLRC5 expression in B16 cells enables cross-presentation of endogenous mgp100 and elicits protective antitumor immunity. (A) Experimental plan for evaluating antigen cross-presentation in vivo. B16-v, B16-5 and B16-5/80 cells were irradiated and 2 × 105 cells were injected into the footpad of C57BL/6 mice. One day later, CFSE-labeled naive or cytokine-primed Pmel-1 cells were injected intravenously (1 × 106 cells/mouse). Four days later, lymph nodes (LN) draining the footpad (popliteal), non-draining (contralateral popliteal) LN and spleen were harvested to evaluate CFSE fluorescence on CD8+Vβ13+ gated of Pmel-1 cells. (B) Proliferation of Pmel-1 cells elicited by cross presentation of endogenous mgp100 by B16-derived cells. Representative data from three–four mice per group from two experiments are shown. (C) Protocol for evaluating the ability of B16-derived cells to elicit protective antitumor immunity. (D, E) Eight week-old C57BL/6 mice were immunized with irradiated B16-derived cell lines (○ PBS; • B16-Wt; ♦ B16-5, ▪ B16-80; ▴ B16-5/80) by intradermal route (2 × 105 cells/mouse). Four weeks later, the immunized mice were challenged with parental B16-F10 (B16-Wt) cells injected subcutaneously (1 × 105 cells/mouse), and tumor growth was monitored. (D) Tumor growth kinetics from two experiments (n=4–6 mice per group). Error bars = SE. Statistical comparisons were performed using two-way ANOVA with Tukey's multiple comparisons. **p < 0.01, ****p < 0.001. (E) Pooled end-point data of palpable tumor growth 21 d after challenge from the indicated number of mice from more than three experiments. (F) C57BL/6 mice immunized with irradiated B16-derived cell lines were challenged with parental B16-Wt cells via intravenous route. Twenty days later, the recipient mice were sacrificed and the lung tumor foci were evaluated. Representative images of the lungs (left) and number of lung tumor foci (right) are shown. Kruskal–Wallis test adjusted for Dunn's multiple comparisons: *p < 0.05.
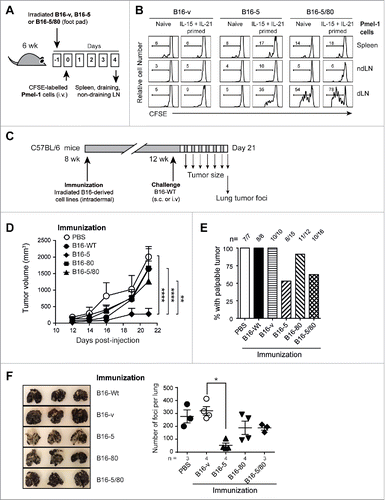
Next we investigated whether NLRC5 expression enabled B16 cells elicit protective antitumor immunity by endogenous CD8+ T cells. C57BL/6 mice were immunized with irradiated B16 cell lines and then challenged 4 weeks later with parental (B16-Wt) cells (). Measurement of tumor size in challenged mice showed significant reduction in tumor growth in mice vaccinated with B16-5 cells (). Notably, B16-5 cells conferred superior protection compared to B16-5/80 cells (). Moreover, immunization with B16-5 cells prevented the development of palpable tumors in nearly 50% of the challenged mice (), while the rest showed negligible tumor growth (). Although mice immunized with B16-5/80 cells also resisted tumor growth (), those that developed tumors showed considerably larger tumors than mice immunized with B16-5 cells (). Mice immunized with B16-5 also showed better resistance to lung tumor foci formation by B16-Wt cells than mice immunized with B16-5/80 cells (). These findings indicate that stable expression of NLRC5 alone is sufficient to render B16 cells immunogenic and elicit protective antitumor immunity.
Discussion
Several immunotherapeutic approaches are currently being evaluated to restore functional antitumor immunity against established tumors that challenge surgical resection and defy other treatment regimens.Citation40,41 Among these, inhibitors of immune checkpoint blockade have shown impressive success in prolonging patient survival. Despite these advances, reduced tumor immunogenicity remains an important obstacle for eliciting efficient antitumor CTL response.Citation42 Most tumors are initially immunogenic, but undergo T-cell-dependent selection toward less immunogenic outgrowth.Citation5,6 Tumors progressively become less immunogenic by downmodulating dominant tumor antigens, the APM or MHC-I.Citation1,7 Although restoration of MHC-I and improving the presentation of subdominant antigens are considered promising approaches to improve tumor immunogenicity, there has been only limited success in achieving this goal.Citation7 Our findings on the B16 melanoma model indicate that NLRC5 could be exploited to restore MHC-I expression and antigen presentation in cancer cells, and thereby elicit antitumor immunity.
Several groups have generated NLRC5-deficient mice independently.Citation26,27,29,43 Even though these mice show markedly reduced expression of MHC-I in lymphoid and myeloid cells, this defect does not impair CD8+ T cell development, maturation or homeostasis, all of which require MHC-I. Hence, NLRC5 is dispensable for the basal expression of MHC-I and its critical functions in immune cell homeostasis. Induction of MHC-I genes by IFNγ still occurs in NLRC5-deficient cells, albeit to a reduced level,Citation27,29 suggesting that NLRC5-independent transactivation of MHC-I genes can also occur. NLRC5-deficient B lymphocytes and macrophages stimulated with LPS efficiently present the OVA peptide to OT-I TCR transgenic CD8+ T cells,Citation26,27 suggesting that inducers of MHC-I expression may overcome NLRC5 deficiency to a certain extent. However, NLRC5-deficient T lymphocytes pulsed with the OVA-peptide were not efficiently killed by OT-I cells.Citation27 Moreover, NLRC5-deficient mice were inefficient in clearing the intracellular bacteria Listeria monocytogenes, although they efficiently cleared vesicular stomatitis virus.Citation26,43 Collectively, these reports suggest that NLRC5 may be dispensable to mount an efficient CTL response during acute inflammatory conditions but is critical when the inflammatory response is chronic and not robust, which is probably the case in cancers. It is noteworthy that experimental evidence for the role of NLRC5 in tumor immune surveillance using NLRC5-deficient mice has not been reported yet. Although the residual MHC-I expression and NLRC5-independent induction of MHC-I genes by inflammatory mediators may confound tumor immunesurveillance experimentsCitation44 in NLRC5-deficient mice, such studies are needed to understand whether loss of NLRC5 would compromise immunesurveillance and contribute to immune evasion.
Intriguingly, B16-5/80 cells, which were more efficient than B16-5 cells in activating Pmel-1 cells in vitro (), were less efficiently controlled than B16-5 cells in vivo when inoculated subcutaneously, although both were efficiently controlled from forming lung tumor foci (). When used for vaccination, B16-5 cells were again more efficient in preventing both subcutaneous tumor growth and lung tumor foci formation following challenge with B16-Wt cells (). These observations raise the possibility that CD80 expressed on B16 cells may actually contribute to tumor growth rather than enhancing the anti-immune response. For instance, although CD80 may initially promote T cell activation, it could engage CTLA-4 on activated T cells and attenuate their ability to control tumor growth. It has been recently shown that IFNγ promotes PD-L1 expression in tumor cells and that can limit the efficacy of immunotherapy-induced antitumor CTLs, which has been referred to as “adaptive immunosuppression”.Citation45 This raises the possibility that activated T cells producing IFNγ might induce PD-L1 that would engage PD-1 on activated T cells, delivering a negative signal. Clearly, further studies are needed to understand the complexity of immunoregulatory circuits elicited by B16-5 and B16-5/80 cells growing as tumors in different anatomical locations namely, subcutaneous versus the lung tissue.
Even though immune checkpoint inhibitors have limited success in improving overall survival in treating cancer patients (a few months), their impressive ability to restore antitumor immune response (22% complete response, 50% objective response) have raised hopes to achieve further improvements through combinatorial approaches.Citation40 We propose that NLRC5 can be exploited for this purpose, particularly in eliciting a productive immune response toward diverse tumor antigens. The success of immune checkpoint blockade inhibitors lies partly in restoring polyclonal antitumor immune response. Similarly, expansion of tumor-infiltrating lymphocytes for adoptive cell therapy outside the immunosuppressive tumor microenvironment would permit expansion of CTLs toward diverse tumor antigens.Citation46 Dendritic cells (DCs) pulsed with tumor cell lysates may also elicit polyclonal antitumor immune response, although this process would be inefficient because only 0.1% of endogenous peptides would survive intracellular antigen processing.Citation3 However, DCs can directly present MHC-I:peptide complexes acquired via a process called trogocytosis.Citation47 We propose that the ability of NLRC5 to improve tumor cell immunogenicity could be exploited, for example, (i) to generate tumor cell lysates with improved antigenic potential for DC-based vaccines, (ii) to expand tumor-reactive CTLs for adoptive cell therapy, and (iii) to identify and characterize protective CTL epitopes. These approaches, when combined with checkpoint inhibitors and with strategies to enhance the self-renewal potential of tumor-reactive CTLs,Citation48 could help to improve disease-free survival in cancer patients.
Materials and methods
Cells and animals
B16 cells were obtained from ATCC and were tested for their ability to form lung tumor foci upon intravenous injection into C57BL/6 mice. C57BL/6, Rag1−/− and Pmel-1 TCR transgenic miceCitation34 were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). For all experiments 6- to 8-week-old mice were used. Animal experiments were carried out with approval of the Université de Sherbrooke Ethics Committee for Animal Care and Use.
Reagents
The Pmel-1 melanoma antigen-derived peptide mgp10025-33 (EGSRNQDWL)Citation34 was custom synthesized by GenScript (Scotch Plains, NJ). RPMI-1640 cell culture medium and fetal bovine serum were from Sigma-Aldrich (Oakville, Canada). Recombinant mouse IL-2, human IL-15 and mouse IL-21 were from R&D Systems (Minneapolis, MN). Five-(6)carboxyfluoresceindiacetatesuccinimidyl ester (CFSE) was purchased from Molecular Probes, Life Technologies Inc. (Burlington, Canada).
Plasmids, transfection and stable cell lines
Plasmid encoding EGFP-tagged human NLRC5 in EBSB-PL vector has been previously described.Citation31 Mouse CD80 (NCBI NM_009855.2) cDNA was amplified from murine dendritic cells (Forward: CCCAAGCTTGGG-ATGGCTTGCAATTGTCAG; Reverse: CCGCTCGAGCGG-CTAAAGGAAGACGGTCTG) and cloned into pcDNA3.0 vector (Invitrogen) at HindIII and XhoI sites. B16-F10 cells were transfected with NLRC5, CD80 or both expression vectors using polyethylenimine-Max reagent (Polysciences Inc., PA). Stable B16-F10 cell populations expressing NLRC5 (B16-5) were selected using blasticidin (8 μg/mL) whereas CD80 expressing cells (B16-80) were selected using G418 (3 mg/mL). Both antibiotics were used to select B16-F10 cells expressing NLRC5 and CD80 (B16-5/80) or the control vectors (B16-v).
Lymphocyte proliferation assays
To assess the ability of B16 cells to present exogenous or endogenous gp100 tumor antigenic peptide, cells were irradiated (100 Gy) and 2.5 × 104 cells were plated in 96-well plates in presence or absence of the indicated concentrations of the gp100 peptide. After overnight incubation, cells were washed and freshly isolated CD8+ T cells from Pmel-1 TCR transgenic mice, enriched by negative selection using magnetic beads (Dynal, Invitrogen), were added (5 × 104 cells/well). In some experiments, Pmel-1 cells were pre-stimulated with cytokines hIL-15 and mIL-21 (10ng/mL) for 48–72 h (cytokine priming).Citation38 Irradiated C57BL/6 splenocytes (1 × 106 cells/mL) served as control antigen-presenting cells. One μCi of methyl-[3H]-thymidine (NEN Life Sciences, Boston, MA) was added during the last 8 h of culture period and the incorporated radioactivity measured.Citation38 For CFSE proliferation assay, Pmel-1 cells were labeled with CFSE before adding to irradiated B16 cells and CFSE fluorescence was evaluated on gated CD8+ T cells at different time points.Citation49
Tumor growth, CD8+ T cell depletion, immunization and injections
All B16-derived cell lines were washed twice in PBS, filtered using a 70 μM nylon filter to remove cell aggregates. To evaluate subcutaneous tumor growth, 1 × 105 cells were inoculated subcutaneously in the flank. Tumor growth was monitored every 2–3 d until the maximal diameter of 2 cm. Tumor volume was calculated using the modified ellipsoidal formula: ½(length × width2). To evaluate tumor formation in the lung, each mouse received 2 × 105 cells in 100 μL PBS via the tail vein. All injected mice were euthanized 17–20 d later. Lung tissues were excised, photographed and the tumor nodules counted. To deplete CD8+ T cells, anti-CD8+ (clone 53-6.72) or isotype control (Rat IgG2a, clone 2A3) monoclonal antibodies purchased from BioXCell (West Lebanon, NH, USA) was administered via intra-peritoneal route (25 μg/mouse). Depletion of CD8+ T cells was confirmed 24 h later by flow cytometry. Antibody-treated mice were inoculated with B16-5 cells subcutaneously or intravenously. For immunization, B16 cells were irradiated, washed, suspended in PBS and 2 × 105 cells in 40 μL volume were injected via intradermal route on the left flank of anesthetized, 6–8-weeks-old C57BL/6 mice. Immunized mice were challenged 4–5 weeks later with 1 × 105 parental B16 cells, inoculated subcutaneously on the right flank or 2 × 105 cells intravenously. Tumor growth was monitored as described above.
In vivo antigen cross-presentation assay
Irradiated B16 cells (2 × 105 cells in 40 μL PBS) were inoculated into the footpad of C57BL/6 mice. After 18 h, CFSE-labeled Pmel-1 cells were injected intravenously. Four days later, proliferation of Pmel-1 cells was assessed in the draining (popliteal) and non-draining (popliteal from the non-inoculated limb) lymph nodes and spleen by CFSE dilution on gated CD8+TCRVβ13+ cells.
Statistical analyses
The Prism software (GraphPad, La Jolla, CA, USA) was used to plot graphs and to calculate statistical significance.
Supplementary methods
Gene expression analysis, MTT assay and Flow cytometry are detailed in Supplementary Methods.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1151593_s02.pdf
Download PDF (439.5 KB)Acknowledgment
We thank Ms. Carmen R. Carrasquilla for help with PCR reactions.
Funding
This project is funded by Canadian Cancer Society Research Institute (CCSRI) Innovation grant to SI (Grant number 702798). TAK acknowledges support from the German Research Foundation (DFG, grant number KU 1945/2-1). GMR is a recipient of FRQS graduate scholarship. DB is a recipient of FRQS postdoctoral fellowship. CRCHUS is an FRQS-funded research center.
References
- Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst 2013; 105:1172-87; PMID:23852952; http://dx.doi.org/10.1093/jnci/djt184
- Gromme M, Neefjes J. Antigen degradation or presentation by MHC class I molecules via classical and non-classical pathways. Mol Immunol 2002; 39:181-202; PMID:12200050; http://dx.doi.org/10.1016/S0161-5890(02)00101-3
- Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol 2003; 3:952-61; PMID:14647477; http://dx.doi.org/10.1038/nri1250
- Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004; 21:137-48; PMID:15308095; http://dx.doi.org/10.1016/j.immuni.2004.07.017
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; 482:400-4; PMID:22318521; http://dx.doi.org/10.1038/nature10755
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012; 482:405-9; PMID:22318517; http://dx.doi.org/10.1038/nature10803
- Lampen MH, van Hall T. Strategies to counteract MHC-I defects in tumors. Curr Opin Immunol 2011; 23:293-8; PMID:21295956; http://dx.doi.org/10.1016/j.coi.2010.12.005
- Bukur J, Jasinski S, Seliger B. The role of classical and non-classical HLA class I antigens in human tumors. Semin Cancer Biol 2012; 22:350-8; PMID:22465194; http://dx.doi.org/10.1016/j.semcancer.2012.03.003
- Cabrera T, Lara E, Romero JM, Maleno I, Real LM, Ruiz-Cabello F, Valero P, Camacho FM, Garrido F. HLA class I expression in metastatic melanoma correlates with tumor development during autologous vaccination. Cancer Immunol Immunother 2007; 56:709-17; PMID:16960691; http://dx.doi.org/10.1007/s00262-006-0226-7
- Carretero R, Romero JM, Ruiz-Cabello F, Maleno I, Rodriguez F, Camacho FM, Real LM, Garrido F, Cabrera T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics 2008; 60:439-47; PMID:18545995; http://dx.doi.org/10.1007/s00251-008-0303-5
- Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer 2010; 127:249-56; PMID:20178101; http://dx.doi.org/10.1002/ijc.25270
- Seliger B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol Immunother 2008; 57:1719-26; PMID:18408926; http://dx.doi.org/10.1007/s00262-008-0515-4
- Respa A, Bukur J, Ferrone S, Pawelec G, Zhao Y, Wang E, Marincola FM, Seliger B. Association of IFN-gamma signal transduction defects with impaired HLA class I antigen processing in melanoma cell lines. Clin Cancer Res 2011; 17:2668-78; PMID:21248298; http://dx.doi.org/10.1158/1078-0432.CCR-10-2114
- Seliger B. Novel insights into the molecular mechanisms of HLA class I abnormalities. Cancer Immunol Immunother 2012; 61:249-54; PMID:22120755; http://dx.doi.org/10.1007/s00262-011-1153-9
- Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother 2008; 57:647-54; PMID:18046553; http://dx.doi.org/10.1007/s00262-007-0402-4
- Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene 2008; 27:5869-85; PMID:18836468; http://dx.doi.org/10.1038/onc.2008.273
- Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007; 27:549-59; PMID:17967410; http://dx.doi.org/10.1016/j.immuni.2007.10.002
- Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol 2010; 185:1681-91; PMID:20610642; http://dx.doi.org/10.4049/jimmunol.0903900
- Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, Büning H, Schwarzenbacher R, Kufer TA. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem 2010; 285:26223-32; PMID:20538593; http://dx.doi.org/10.1074/jbc.M110.109736
- Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010; 141:483-96; PMID:20434986; http://dx.doi.org/10.1016/j.cell.2010.03.040
- Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, Grötzinger J, Fickenscher H, Schreiber S, Rosenstiel P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol 2010; 184:1990-2000; PMID:20061403; http://dx.doi.org/10.4049/jimmunol.0900557
- Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol 2011; 186:1333-7; PMID:21191067; http://dx.doi.org/10.4049/jimmunol.1003111
- Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A 2010; 107:13794-9; PMID:20639463; http://dx.doi.org/10.1073/pnas.1008684107
- Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol 2012; 12:813-20; PMID:23175229; http://dx.doi.org/10.1038/nri3339
- Kumar H, Pandey S, Zou J, Kumagai Y, Takahashi K, Akira S, Kawai T. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J Immunol 2011; 186:994-1000; PMID:21148033; http://dx.doi.org/10.4049/jimmunol.1002094
- Biswas A, Meissner TB, Kawai T, Kobayashi KS. Cutting edge: impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J Immunol 2012; 189:516-20; PMID:22711889; http://dx.doi.org/10.4049/jimmunol.1200064
- Staehli F, Ludigs K, Heinz LX, Seguin-Estevez Q, Ferrero I, Braun M, Schroder K, Rebsamen M, Tardivel A, Mattmann C et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J Immunol 2012; 188:3820-8; PMID:22412192; http://dx.doi.org/10.4049/jimmunol.1102671
- Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, Wang H, Shi Y, Qian Y. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res 2012; 22:836-47; PMID:22491475; http://dx.doi.org/10.1038/cr.2012.56
- Robbins GR, Truax AD, Davis BK, Zhang L, Brickey WJ, Ting JP. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins. J Biol Chem 2012; 287:24294-303; PMID:22645137; http://dx.doi.org/10.1074/jbc.M112.364604
- Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, van den Elsen PJ, Kobayashi KS. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol 2012; 188:4951-8; PMID:22490869; http://dx.doi.org/10.4049/jimmunol.1103160
- Neerincx A, Rodriguez GM, Steimle V, Kufer TA. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J Immunol 2012; 188:4940-50; PMID:22490867; http://dx.doi.org/10.4049/jimmunol.1103136
- Meissner TB, Li A, Liu YJ, Gagnon E, Kobayashi KS. The nucleotide-binding domain of NLRC5 is critical for nuclear import and transactivation activity. Biochem Biophys Res Commun 2012; 418:786-91; PMID:22310711; http://dx.doi.org/10.1016/j.bbrc.2012.01.104
- Ludigs K, Seguin-Estevez Q, Lemeille S, Ferrero I, Rota G, Chelbi S, Mattmann C, MacDonald HR, Reith W, Guarda G. NLRC5 exclusively transactivates MHC class I and related genes through a distinctive SXY module. PLoS Genet 2015; 11:e1005088; PMID:25811463; http://dx.doi.org/10.1371/journal.pgen.1005088
- Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol 2001; Chapter 20:Unit 20 1; PMID:18432774
- Seliger B, Wollscheid U, Momburg F, Blankenstein T, Huber C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res 2001; 61:1095-9; PMID:11221838; http://dx.doi.org/10.1002/0471142735.im2001s39.
- Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science 1993; 259:368-70; PMID:7678351; http://dx.doi.org/10.1126/science.7678351
- Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol 2003; 3:939-51; PMID:14647476; http://dx.doi.org/10.1038/nri1248
- Ramanathan S, Dubois S, Chen XL, Leblanc C, Ohashi PS, Ilangumaran S. Exposure to IL-15 and IL-21 enables autoreactive CD8 T cells to respond to weak antigens and cause disease in a mouse model of autoimmune diabetes. J Immunol 2011; 186:5131-41; PMID:21430227; http://dx.doi.org/10.4049/jimmunol.1001221
- Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 2003; 281:65-78; PMID:14580882; http://dx.doi.org/10.1016/S0022-1759(03)00265-5
- Lizee G, Overwijk WW, Radvanyi L, Gao J, Sharma P, Hwu P. Harnessing the power of the immune system to target cancer. Annu Rev Med 2013; 64:71-90; PMID:23092383; http://dx.doi.org/10.1146/annurev-med-112311-083918
- Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 2015; 27:439-49; PMID:25858803; http://dx.doi.org/10.1016/j.ccell.2015.03.007
- Blankenstein T, Coulie PG, Gilboa E, Jaffee EM. The determinants of tumour immunogenicity. Nat Rev Cancer 2012; 12:307-13; PMID:22378190; http://dx.doi.org/10.1038/nrc3246
- Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF. Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res 2012; 22:822-35; PMID:22473004; http://dx.doi.org/10.1038/cr.2012.53
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001; 410:1107-11; PMID:11323675; http://dx.doi.org/10.1038/35074122
- McGray AJ, Hallett R, Bernard D, Swift SL, Zhu Z, Teoderascu F, Vanseggelen H, Hassell JA, Hurwitz AA, Wan Y et al. Immunotherapy-induced CD8+ T cells instigate immune suppression in the tumor. Mol Ther 2014; 22:206-18; PMID:24196579; http://dx.doi.org/10.1038/mt.2013.255
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008; 8:299-308; PMID:18354418; http://dx.doi.org/10.1038/nrc2355
- Yewdell JW, Dolan BP. Immunology: Cross-dressers turn on T cells. Nature 2011; 471:581-2; PMID:21455165; http://dx.doi.org/10.1038/471581a
- Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer 2012; 12:671-84; PMID:22996603; http://dx.doi.org/10.1038/nrc3322
- Gagnon J, Ramanathan S, Leblanc C, Ilangumaran S. Regulation of IL-21 signaling by suppressor of cytokine signaling-1 (SOCS1) in CD8(+) T lymphocytes. Cell Signal 2007; 19:806-16; PMID:17126530; http://dx.doi.org/10.1016/j.cellsig.2006.10.003