
ABSTRACT
Hodgkin lymphoma (HL) presents with a unique histologic pattern. Pathognomonic Hodgkin and Reed–Sternberg (HRS) cells usually account for less than 1% of the tumor and are embedded in a reactive infiltrate mainly comprised of CD4+ T cells. HRS cells induce an immunosuppressive microenvironment and thereby escape antitumor immunity. To investigate the impact of interactions between HRS cells and T cells, we performed long-term co-culture studies that were further translated into a xenograft model. Surprisingly, we revealed a strong antitumor potential of allogeneic CD4+ T cells against HL cell lines. HRS and CD4+ T cells interact by adhesion complexes similar to immunological synapses. Tumor-cell killing was likely based on the recognition of allogeneic major histocompatibility complex class II (MHC-II) receptor, while CD4+ T cells from MHC-II compatible donors did not develop any antitumor potential in case of HL cell line L428. However, gene expression profiling (GEP) of co-cultured HRS cells as well as tumor infiltration of matched CD4+ T cells indicated cellular interactions. Moreover, matched CD4+ T cells could be activated to kill CD30+ HRS cells when redirected with a CD30-specific chimeric antigen receptor. Our work gives novel insights into the crosstalk between HRS and CD4+ T cells, suggesting the latter as potent effector cells in the adoptive cell therapy of HL.
Introduction
The striking differences between HL and B-cell derived non-HL (NHL) have engaged researchers for decades.Citation1 Whereas tumor cells dominate biopsy specimens derived from NHL patients, HL-affected tissue features a high number of activated immune cells surrounding the HRS cells. Although HRS cells usually account for less than 1% of the cellular infiltrate, diagnosis of HL is primarily based on the giant and multinuclear HRS-cell morphology.Citation2-5
HRS cells produce large amounts of cytokines and chemokines resulting in the infiltration of various types of immune cells. Thereby, HRS cells shape a HL-specific cellular and cytokine microenvironment.Citation6,7 CD4+ T cells display the largest cell population of the infiltrate and predominantly present with a Th2 or regulatory T-cell (Treg) phenotype.Citation8-10 Indeed, HRS cells secrete chemokines attracting CCR4-expressing CD4+ T cells,Citation11-13 thus orchestrating the so-called T-cell rosette, another histological hallmark of HL.Citation14 But HRS cells not only attract CD4+ T cells to avoid cytotoxic CD8+ T cells in their close proximity, they also seem to induce Treg-cell differentiation directly.Citation15,16 This in consequence improves Treg-cell-mediated suppression of cytotoxic CD8+ T cells and natural killer (NK) cells by secretion of immunosuppressive cytokines. Besides the fact that HRS cells produce some immunosuppressive cytokines themselves, they can also directly drive CD8+ T cells into anergy or apoptosis, as they often express programmed cell death ligand 1 (PD-L1) and CD95L (Fas ligand).Citation17,18 Moreover, major histocompatibility complex class I (MHC-I) expression is often downregulated on HRS cells, reducing their recognition by CD8+ T cells. These and other mechanisms are likely crucial key players in preventing tumor cell clearance in HL.Citation19,20
Research to elucidate the functional co-operation between tumor cells and the cells of the immune system is so far hampered in HL because of the unavailability of primary HRS-cell cultures and a paucity in the reconstruction of HL-like infiltrates in vitro. HL cell lines currently represent the only model system to study HRS-cell physiology.Citation14,21-23 While only few HL cell lines could be established to date,Citation24,25 their common potential to activate T cells was unraveled.Citation26,27 Several studies showed that co-cultured T cells spontaneously bind to HRS cells, a process defined as rosetting due to its similarity to T-cell rosettes in primary tumor sections.Citation15,28,29 Moreover, the ability of antigen presentation of HRS cells was described.Citation30 In this regard, it is commonly accepted that HRS cells act like antigen presenting cells (APCs) and stimulate T-cell proliferation in co-culture, but an anti-tumor reaction has not been described so far, although T cells of allogeneic donors have been used frequently in previous studies.Citation14,15,27,29,31,32 Moreover, as T-cell stimulation by HRS cells only takes few days, the necessity of long-term co-culture observation has not been considered so far. Hence, a profound characterization of the underlying interaction network with respect to T-cell function is still missing; indeed, the impact of T cells on HRS cells is neglected so far.
The aim of the presented study was to develop a new model system for HL, to illuminate the interaction between HRS cells and CD4+ T cells and thereby shed light on potential novel therapeutic strategies. Therefore, we analyzed the interaction between HRS cells and CD4+ T cells in vitro as well as in a xenograft mouse model using unmatched and MHC-II compatible CD4+ T cells.
Results
Allogeneic CD4+ T cells, but not CD8+ T cells efficiently eradicate HRS cells in vitro and HL tumors in vivo
To gain deeper insight into cellular interactions between primary human T cells and HRS cells, these cells were co-incubated in long-term assays and characterized in detail by flow cytometry (, Figs. S1A–C). Co-incubation of bulk allogeneic T cells abrogated expansion of HRS cells (, Fig. S1A). Although allogeneic CD8+ T cells were expected to mediate a cytotoxic effect, co-cultures of the HL cell line L428 either with CD4+ or CD8+ T cells revealed that only CD4+ T cells abolished proliferation of L428 cells, whereas CD8+ cells did not (). In addition, L428 cells activated only CD4+ T cells, since CD4+ T cells but not CD8+ T cells amplified in the long-term co-incubation assays (). This might be due to the lack of expression of MHC-I by L428 cells.Citation20 Therefore, experiments were repeated with MHC-I expressing HL cell line KMH2 showing that CD4+ as well as CD8+ T cells abrogate proliferation of KMH2 cells (Fig. S1A). Interestingly, T-cell response to KMH2 cells was induced faster compared to co-culture experiments with L428 cells and did not result in a remarkable T-cell proliferation (Fig. S1B).
Figure 1. CD4+ T cells show cytotoxic activity against HRS cells. (A–E) Bulk (CD3), CD4+ or CD8+ T cells were isolated from PBMC of healthy donors and subsequently co-cultured with HRS cells (HL cell line L428) in a ratio of 20:1 for 14 d. Every 2–3 d T cells and L428 cells were counted by FACS analysis after staining for CD3 and CD30. The experiment was performed in triplicates and repeated with two different donors showing similar results. (A) Growth curves of HRS cells co-cultured with (w) bulk, CD4+ or CD8+ T cells in comparison to a corresponding monoculture. (B) Growth curves of bulk, CD4+ or CD8+ T cells co-cultured with HRS cells in comparison to a corresponding monoculture of unstimulated T cells. (C) Frequency of CD3-positive HRS cells (rosettes) over time. (D) Representative phase contrast images of cluster formation in 7 d-old co-cultures of HRS cells and CD4+ T cells. (E) Cluster formation was recorded by assessment of cluster diameters. The experiment was performed twice with n > 50 and in addition, HL cell lines KMH2 and L1236 were included in the cluster formation assay. (F–H) T cells and L428 cells were transplanted into NSG mice. NSG mice were first inoculated with HRS cells i.p. (F) or s.c. (G+H) and 7 d later additionally transplanted with bulk, CD4+ or CD8+ T cells. The experiment was performed with n = 5 mice per group and repeated once with similar results. (F) Mice were sacrificed after 6 weeks and analyzed for tumor burden. Representative images show i.p. tumor growth in absence of T cells. (G) Volume of s.c. tumors was measured every 3–4 d. (H) Relative (rel.) weight (% of initial weight) was assessed every 3–4 d for 6 weeks.
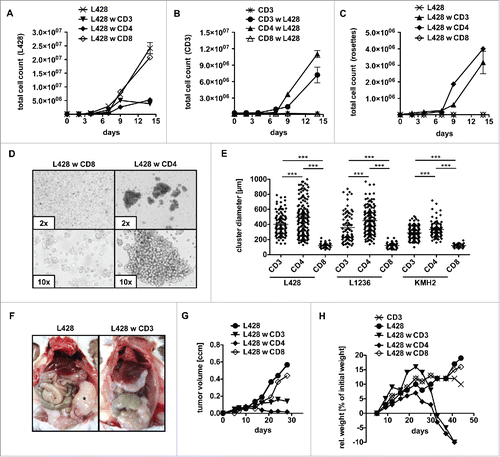
In accordance with these observations, only CD4+ T cells formed rosettes with L428 cells (, Fig. S1D), while CD8+ T cells were unable to attach to L428 cells and to form clusters (). In case of KMH2 cells, earlier onset of the T-cell response hindered rosette detection by flow cytometry (Fig. S1C). Prolonged rosette formation produced cell clusters of about 300–500 µm in diameter (). The analyses were performed with three different allogeneic donors per cell line and reproduced with the HL cell lines KMH2 and L1236 showing that the rosetting by CD4+ T cells is a general phenomenon of established HL cell lines in co-culture with T cells (). Notably, cluster formation assay revealed that T cells also attach to KMH2 cells; however, with clusters of smaller size compared to co-cultures of L428 or L1236 cells, again in accordance to the earlier onset of the T-cell response.
To address whether co-incubated T cells exhibit antitumor cell activity in vivo we used a xenograft mouse model based on subsequent injection of HRS cells and T cells. Immunodeficient NOD SCID γc−/− (NSG) mice were intraperitoneally (i.p.) inoculated with L428 cells. Seven days later, T cells were injected intravenously (i.v.) into the mice. Interestingly, necropsy revealed that control mice developed solid i.p. tumors 6 weeks after transplantation of L428 cells, while i.p. tumors were absent in mice that received additional transfer of allogeneic T cells (). Furthermore, established subcutaneous (s.c.) HL tumors were completely rejected by CD4+ T cells but not by CD8+ cells (). Experiments were repeated with KMH2 cells, but without distinguishing between T-cell subtypes, as both CD4+ and CD8+ T cells showed an antitumor potential in vitro. While tumors of KMH2 cells were rejected likewise to tumors of L428 cells, s.c. growth of the Burkitt lymphoma (BL) cell line Raji was not influenced by adoptive transfer of T cells (Fig. S1E). Of note, L1236 cells showed no in vivo growth potential, neither after i.p. nor s.c. transplantation and were therefore not assessable in the xenograft model.
Transplantation of human T cells into immunodeficient mice frequently induces xenograft-versus-host disease (xeno-GvHD) with weight loss.Citation33 Comparison of naive to tumor-bearing mice after administration of bulk T cells showed an accelerated onset of xeno-GvHD in the presence of the HL tumor (). This is due to CD4+ T cells, since adoptive transfer of CD4+ T cells but not CD8+ T cells induced weight loss after 3–4 weeks (). This finding was not restricted to HL cell line L428 but confirmed using the HL cell line KMH2 (Fig. S1F). In accordance, histological analyses of animals succumbed to disease revealed massive lymphocyte infiltrations of multiple organs including the lung and liver (Fig. S1G). Infiltrates mainly consisted of CD4+ T cells in the case of bulk T-cell administration and CD8+ T cells represented a minority (Fig. S1H, upper panel). Furthermore, CD8+ T cells did not infiltrate organs in absence of CD4+ T cells (Fig. S1I). Noteworthy, tumors of control mice showed a strong CD30 signal by HRS cells with few cells expressing CD15 (Fig. S1H, lower panel). T-cell infiltrates also partly expressed CD30, a well-known feature of activated T cells (Fig. S1J).
Interaction between HRS cells and CD4+ T cells is mediated by adhesion molecules similar to the immunological synapse
To reveal the interaction between L428 cells and CD4+ T cells on the molecular level, we blocked the interaction of CD2/CD58 (LFA-2/LFA-3) and CD11a/CD54 (LFA-1/ICAM-1) together with an inhibition of CD40 by addition of blocking antibodies, as previously described.Citation34 The co-cultures were recorded one week after single-shot antibody addition. The frequency of CD3-positive HRS cells, representing T-cell rosetted HRS cells, was determined and recorded as a value of T cell–HRS cell interaction. Administration of all antibodies substantially reduced rosette formation, but also blocking of CD58 alone was sufficient to abrogate rosette formation to some extent (). Moreover, our cluster formation assay revealed that blocking of the adhesion molecules CD2, CD11a, CD40 and CD58 but not blocking of CD54 showed a significant reduction of T-cell adhesion to HRS cells, while the anti-CD40 antibody did not influence rosette persistence (). Both investigated adhesion molecule pairs are components of the immunological synapse formed between T cells and APCs upon T-cell stimulation. In co-culture these molecules are distributed in clusters formed between HRS cells and CD4+ T cells ().
Figure 2. CD4+ T cells and HRS cells interact via immunological synapses. CD4+ T cells were isolated from PBMC of healthy donors and subsequently co-cultured with HRS cells (HL cell line L428) in a ratio of 20:1 for up to 14 d. Co-cultures were incubated either with antibodies against CD2, CD11a, CD40, CD54 and CD58 (A–B), left untreated (C) or initially treated with 75 µM dexamethasone (Dex) (D–I). (A) After 1 week, the frequency of CD3-positive HRS cells (rosettes) was assessed by flow cytometry. The experiment was performed in triplicates and repeated 3 times with similar results. (B+F) Cluster formation was recorded in parallel by phase contrast imaging to determine cluster diameter. Experiments were performed 3 times with n > 50. C, smears of co-cultures were stained for the adhesion molecule pairs CD2/CD58 (LFA-2/LFA-3) or CD11a/CD54 (LFA-1/ICAM-1) and analyzed by fluorescence microscopy. (D–E) Expression of CD2 (D) or CD11a (E) on CD4+ T cells within Dex-treated co-cultures was determined on day 7. (G-I) Every 2–3 d T cells and HRS cells were counted by flow cytometry after staining for CD3 and CD30. Experiments were performed in triplicates and repeated with two different donors showing similar results. (G) Growth curves of HRS cells co-cultured with (w) CD4+ T cells in dependence of Dex administration and in comparison to corresponding monocultures. (H) Growth curves of CD4+ T cells co-cultured with HRS cells in dependence of Dex administration and in comparison to untreated control. (I) Frequency of CD3-positive HRS cells (rosettes) over time. Experiments were performed in triplicates and repeated 3 times.
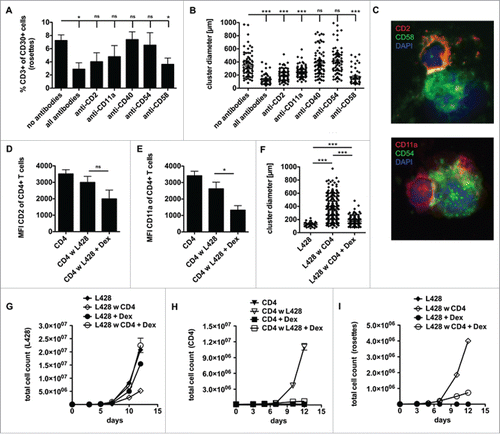
Glucocorticoid reduces expression of LFA-1 (CD11a/CD18) and LFA-2 (CD2) on T cells.Citation35 For this reason and because glucocorticoids are implemented in chemotherapy regimens for the treatment of HL,Citation36 we asked whether steroids impact cluster formation between HRS cells and CD4+ T cells. First, we confirmed that dexamethasone downregulates CD2 and CD11a on T cells, as a slight reduction in CD2 expression but a significantly reduced level of CD11a was observed on co-cultured CD4+ T cells upon dexamethasone treatment (). Moreover, dexamethasone strongly impaired cluster formation between HRS cells and CD4+ T cells (). As the cluster formation of CD4+ T cells with HRS cells seemed to be based on strong binding of these adhesion molecules, we tested if dexamethasone leads to increased survival of HRS cells in the presence of CD4+ T cells. In fact, dexamethasone-treated co-cultures showed restored HRS-cell proliferation accompanied by inhibited binding of T cells to HRS cells which occurs simultaneously to an abrogated formation of T-cell rosettes (). Using KMH2 cells instead of L428 cells, cluster formation assay revealed a reduction of cluster size due to dexamethasone treatment (Fig. S2A), but KMH2 cells were more sensitive than L428 cells to dexamethasone showing strongly impaired proliferation, which might account for the observed effect (Fig. S2B+C). However, blocking of CD2 and CD11a to disrupt T-cell adhesion showed comparable results to dexamethasone treatment of co-cultures with L428 cells (Fig. S2D+E). In conclusion, these data show that CD4+ T cells can be reduced in their capacity to form rosettes with HRS cells by treatment with steroids abrogating their antitumor potential.
HLA-matched CD4+ T cells show reduced reactivity against HL cell lines
To corroborate the hypothesis that TCR/MHC engagement is involved in rosette formation, we wanted to block MHC-II on HRS cells during co-culture studies. However, antibody binding to MHC-II strongly induced tumor-cell death in monocultures of L428 as well as KMH2 cells (Fig. S2F+G). The toxicity of the antibody might be due to cross-linking of HRS cells, clearly visible after treatment and resembling cluster formation of T cells in co-cultures (Fig. S2H).
Therefore, as a next step, we co-incubated L428 cells with MHC-II matched and unmatched CD4+ T cells in order to reveal the impact of an allogeneic response in the assay (Tables S1+S2). Intriguingly, HLA-matched CD4+ T cells exhibited a reduced antitumor potential compared to CD4+ T cells isolated from non-matched donors (). L428 cells showed unaltered growth kinetics in the presence of matched CD4+ T cells (). L428 cells did not stimulate matched CD4+ T cells and no T-cell rosettes were detected (). We speculated that a strong TCR/MHC interaction is inevitable for extended T-cell binding to HRS cells with rosette formation and subsequent tumor-cell killing.
Figure 3. Anti-HRS cell activity of CD4+ T cells displays an allogeneic reaction. HLA genotype of HL cell line L428 was determined. Thereafter, CD4+ T cells were isolated from MHC-II compatible donors and subsequently co-cultured with HRS cells (A–D) or co-injected into tumor-bearing NSG mice (E, F). (A–D) HLA-matched (mCD4+) or unmatched (uCD4+) CD4+ T cells were co-cultured with HRS cells in a ratio of 20:1 for 14 d. Every 2–3 d T cells and HRS cells were counted using flow cytometry after staining for CD3 and CD30. Experiment was performed in triplicates and repeated with two different MHC-II compatible donors showing similar results. (A) Growth curves of HRS cells co-cultured with (w) mCD4+ or uCD4+ T cells in comparison to a corresponding monoculture. (B) Growth curves of mCD4+ or uCD4+ T cells co-cultured with HRS cells in comparison to untreated control. (C) Frequency of CD3-positive HRS cells (rosettes) over time. (D) Cluster formation was recorded by assessment of cluster diameters. Experiment was performed twice with n > 50. (E–G) Co-culture experiments were translated into a murine xenograft model. NSG mice were first inoculated with HRS cells s.c. and 7 d later co-injected with mCD4+ or uCD4+ T cells. Experiment was performed twice with n = 5. (E) Volume of s.c. tumors was measured every 3–4 d. (F) Relative (rel.) weight (% of initial weight) was assed every 3–4 d for 6 weeks. (G) Mice were sacrificed after 6 weeks and tumors (*) were processed by histological analysis. Representative images of s.c. tumors stained for CD3 and CD30 showing tumor-infiltration by mCD4+ T cells.
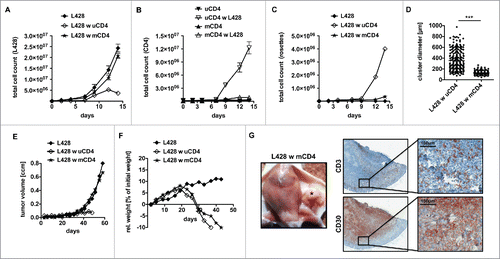
The same experiment was performed using KMH2 cells and CD4+ T cells from two different MHC-II compatible donors. However, in both cases growth kinetics showed that expansion of KMH2 cells was abrogated in the presence of matched CD4+ T cells. For the more reactive donor, the T-cell response was even comparable to the afore investigated allogeneic setting (Fig. S3). Although the reason for the antitumor response is so far unclear, this finding highlights the unique opportunity to study L428 cells with mCD4+ T cells as a HL model system. Therefore, further experiments were focused on this cell line.
The impact of matched CD4+ T cells on HL-tumor formation was also studied in the xenograft mouse model. In accordance to the in vitro studies, matched CD4+ T cells did not show reactivity against HRS cells and HL tumors were not rejected (). Nevertheless, matched CD4+ T cells were as functional as unmatched CD4+ T cells in terms of xeno-GvHD and induced dramatic weight loss in the recipients (). To ensure infiltration of the HL tumors by matched CD4+ T cells, histological analysis was performed. And indeed, matched matched CD4+ T cells were detectable within the CD30+ HL tumors (). Thus, matched CD4+ T cells are attracted by HRS cells and infiltrate HL tumors without developing an antitumor reaction, which is in striking contrast to unmatched CD4+ T cells.
Interaction with HLA-matched CD4+ T cells alters gene expression of HRS cells
To further characterize the interaction between matched CD4+ T cells and HRS cells, we performed microarray studies. L428 cells were co-cultured for 7 d with matched or unmatched CD4+ T cells and purified for RNA isolation by T-cell depletion. Gene expression profiles (GEP) of co-cultured L428 cells were compared to monocultures and to co-cultures treated with dexamethasone. Microarray studies revealed that co-incubation of L428 cells with matched as well as unmatched CD4+ T cells induced a similar GEP switch (, Fig. S4). In line with this, unsupervised hierarchical clustering or principal component analysis revealed dramatic alterations in GEP in monocultures compared to co-cultures. This switch in GEP was prevented by dexamethasone treatment (, Fig. S4A). Interestingly, L428-cell samples isolated from co-cultures with matched or unmatched CD4+ T cells clustered together in all analyses (, Fig. S4A). Most of the differentially expressed genes were upregulated in co-cultured L428 cells and belong to the group of inflammation-associated genes (GO cluster BioPro). Aicda as an exception was the most downregulated gene upon co-cultivation (). Next to the induction of pro-inflammatory genes, also some apoptosis-related genes (GO cluster BioPro) were altered in co-cultured L428 cells (Fig. S4B). Notably, focusing on a T-cell signature indicated some contamination in L428-cell samples co-cultured with unmatched CD4+ T cells, which may be due to much stronger interaction than with matched CD4+ T cells (Fig. S3C).
Figure 4. Gene expression profiling of co-cultures hints at interactions between MHC-II compatible CD4+ T cells and HRS cells. HLA-matched (mCD4+) or umatched (uCD4+) CD4+ T cells were co-cultured with HRS cells (HL cell line L428) in a ratio of 20:1 for 7 d. Co-cultures were separated in Dex-treated and non-treated groups. Monocultures of HRS cells served as controls. After 1 week of co-culture, HRS cells were purified by CD3-depletion and gene expression profiling (GEP) was performed, if amount of contaminating T cells was less than 5%. Controls were treated likewise. (A) Unsupervised hierarchical clustering shows differences between co-cultures and monocultures, whereas Dex-treated co-cultures clustered in one arm with monocultures. Interestingly, a random distribution of co-cultures with either mCD4+ (m) or uCD4+ (u) T cells could be observed. (B) Inflammation associated genes were significantly overexpressed by co-cultured L428 cells. (C) Gene set enrichment analysis (GSEA) showed up-regulation of genes associated with apoptosis in HRS cells after co-culture (left) and this effect was completely abolished by Dex-treatment (right). (D) Comparison of HRS cells co-cultured either with mCD4+ or uCD4+ T cells did not result in significant differences concerning apoptosis-related genes. ES, enrichment score; FDR, false discovery rate.
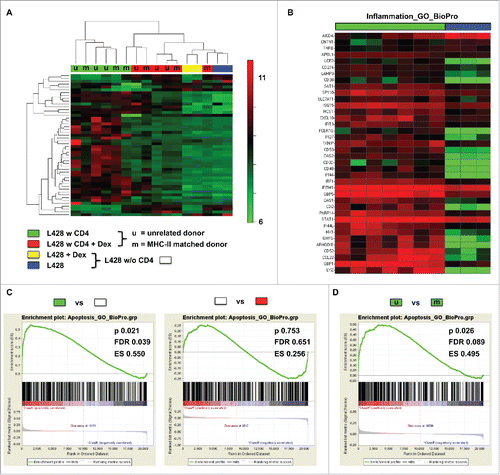
Gene set enrichment analysis (GSEA) of the relevant genes (GO clusters of inflammation and apoptosis) substantiated the differences in GEPs between monocultures and co-cultured L428 cells (, Fig. S4E), while again L428-cell samples after co-cultivation with matched or unmatched CD4+ T cells did not differ significantly (, Fig. S4F). Of note, dexamethasone treatment also abolished differences in GSEA without influencing GEP of L428 cells directly (, Fig. S4D). Taken together, microarray studies pinpoint to a fundamental interaction between matched CD4+ T cells with HRS cells, although an anti-tumor reaction is missing in co-culture experiments as well as in the xenograft model.
As a next step, we re-analyzed the published data by Tiacci et al. to arrange a probe set of the most differentially expressed genes between HL cell lines and microdissected primary HRS cells (Fig. S5A).Citation23 Then, we used this probe set to perform an unsupervised hierarchical cluster analysis with the microarray data generated in the presented study (Fig. S5B). And indeed, it became obvious that co-cultivation of HRS cells with T cells directly affects expression of the genes differentially expressed between HL cell lines and primary HRS cells.
An anti-CD30 CAR unleashes L428-cell killing potential of HLA-matched CD4+ T cells
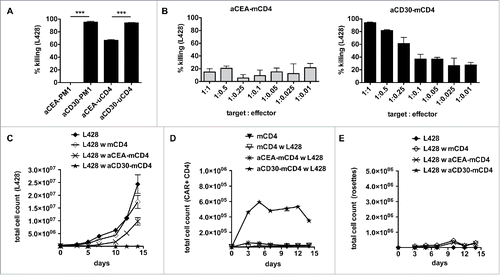
As HLA-matched CD4+ T cells lack L428-cell killing in vitro and in vivo, we asked whether bypassing the TCR/MHC interaction by a chimeric antigen receptor would reveal the cytolytic potential of matched CD4+ T cells. As proof of principle, we retrovirally transduced the human TCR-negative CD4+ T cell line PM1 with the anti-CD30 CAR or a CAR directed against carcinoembryonic antigen (CEA) as control. In co-culture with HL cell line L428, anti-CD30 CAR-expressing PM1 cells very efficiently eliminated the tumor cells whereas anti-CEA CAR-expressing control cells failed to do so (). As a next step, we asked to potentiate the antitumor reactivity of unmatched CD4+ T cells by a transgenic CAR. The L428-cell killing was increased by the anti-CD30 CAR compared to the anti-CEA CAR of irrelevant specificity (). Finally, we performed co-culture experiments with anti-CD30 CAR-expressing matched CD4+ T cells (). An antitumor cell response was obtained even at an effector to target ratio of a single anti-CD30 CAR-expressing matched CD4+ T cell to 10 tumor cells (). In addition, long-term co-cultures were analyzed for proliferation (). While L428 cells did not expand in the presence of anti-CD30 CAR-expressing matched CD4+ T cells (), there was a CAR-specific increase in anti-CD30 but not anti-CEA CAR-expressing matched CD4+ T cells (). Of note, HRS-cell killing was even faster than rosette formation bypassing the MHC/TCR interaction (). These results show that matched CD4+ T cells expressing an anti-CD30 CAR can be redirected to very efficiently kill HRS cells.
Figure 5. Gene-modified MHC-II compatible CD4+ T cells efficiently kill HRS cells. (A) TCR-negative PM1 cells were transduced with retroviral vectors encoding for anti-CEA (aCEA) or anti-CD30 (aCD30) chimeric antigen receptors (CARs) and co-cultured with GFP-expressing HRS cells (HL cell line L428). Killing efficacy was assessed after 48 h via flow cytometry. (B–E) CD4+ T cells were isolated from HLA-matched (mCD4+) or unmatched (uCD4+) donors and retrovirally transduced to express aCEA- or aCD30-CAR prior to co-culture with HRS cells. Experiments were performed in triplicates and repeated once. (B) Killing efficacy of different target (GFP-expressing HRS cells) to effector (CAR-positive CD4+ T cells) ratios was assessed after 96 h. Importantly, aCD30-mCD4+ T cells showed dose-dependent killing compared to aCEA-mCD4+ control. (C–E) Every 2–3 d T cells and HRS cells were counted by flow cytometry analysis after staining for CD3 and CAR (ratio was 1 aCD30-mCD4+ on 1 HRS cells). (C) Growth curves of HRS cells co-cultured with (w) untransduced, aCEA- or aCD30-mCD4+ T cells in comparison to a corresponding monoculture. (D) Frequency of CAR-positive mCD4+ T cells co-cultured with L428 cells in comparison to untreated or non-transduced control. (E) Frequency of CD3-positive HRS cells (rosettes) over time.
Discussion
Recent concepts in tumor biology consider the impact of the tumor microenvironment on tumor progression and therapy. Especially HL, with its low tumor cell content, provides compelling support for the notion that the surrounding non-neoplastic infiltrate may be a key player in the etiopathogenesis of the disease. Therefore, we investigated the interactions between HRS cells and T cells. In co-culture experiments we observed that CD4+ T cells but not CD8+ T cells strongly attached to HRS cells. This in turn prompted allogeneic CD4+ T-cell activation and a potent anti-tumor reaction (). The fierce T-cell rosette formation with HRS cells was diminished by dexamethasone treatment and by the administration of blocking antibodies, which were directed against the adhesion molecules known from immunological synapses (). In case of HL cell line L428, HRS cells were not affected by MHC-II matched CD4+ T cells, demonstrating that the observed anti-tumor reaction and rosette formation was most likely based on HLA incompatibility. The interaction is not exclusively mediated by MHC/TCR interaction since both cell types interact with each other even in the MHC-II compatible setting and matched CD4+ T cells infiltrated HL tumors in our mouse model (, ). Finally, matched CD4+ T-cell response was enforced toward HRS cells after retroviral transfer of an anti-CD30 CAR ().
It is commonly accepted that cytotoxic CD8+ T cells (CTLs) can get activated to react even against established tumors.Citation37,38 Currently, several clinical trials exploit tumor-specific CTLs in immunotherapy.Citation31,32,39-41 However, our study revealed that purified CD4+ T cells are able to kill HRS cells, even when CD8+ T cells are hindered most likely due to downregulation of MHC-I expression (). Although recent publications demonstrated that CD4+ T cells are able to acquire cytotoxic potential, the triggers mediating this unusual differentiation still remain obscure.Citation42-44 According to this, HRS cells can induce bidirectional differentiation of CD4+ T cells into a cytotoxic but also an immunosuppressive state.Citation15 Furthermore, GEP of HL-patient material revealed that HRS cells are mainly rosetted by CD4+ T cells of a Th2 or Treg phenotype.Citation8,45 Therefore, it is accepted that HRS cells shape an immunosuppressive microenvironment,Citation6,46,47 but the high immunogenicity of these cells, as revealed here, displays a so far neglected issue within co-culture studies using HL cell lines and allogeneic T cells.
To overcome the allogeneic response of CD4+ T cells against HRS cells, we treated co-cultures with dexamethasone what abolished the antitumor effect. Next to the fact that steroids are often implemented in HL-therapy regimes and treatment effect is still unclear, it was shown that dexamethasone reduces expression of the adhesion molecules CD2 and CD11a on T cells.Citation35 Both surface receptors are involved in immunological synapses, as these molecules are required to maintain a strong intercellular contact during APC-mediated T-cell activation.Citation48 Moreover, HRS cells express the corresponding interaction partners, CD54 and CD58, which are physiologically provided by APCs.Citation49 In fact, the mechanism how dexamethasone silenced the anti-HRS cell response was based on downregulation of CD2 and CD11a (). Accordingly, blocking of CD2 and CD11a as well as the corresponding interaction partners with specific antibodies further corroborated the hypothesis that HRS cells establish complex structures with CD4+ T cells (). Strong and prolonged binding to MHC-II, inducing constitutive activation of downstream signaling, might be a mechanism how CD4+ T cells kill HRS cells without the need of a cytotoxic reaction. In line with this, antibody-based cross-linking of MHC-II leads to HRS-cell death (Fig. S2). We speculate that in analogy to the observation that APCs, especially DCs, are driven into apoptosis upon T-cell activation by prolonged signaling through immunological synapses, HRS cells are susceptible to apoptosis by TCR-mediated binding to MHC-II.Citation50,51
So far, interaction studies between HRS cells and T cells were performed without regard to the HLA genotype of the T-cell donor.Citation14,15,29 Here, we excluded an allogeneic reactivity by using CD4+ T cells purified from blood of MHC-II compatible donors. Matched CD4+ T cells tolerated HRS cells of HL cell line L428 and no anti-tumor reaction evolved (). This observation does not exclude that there might be autologous T cells reactive against HRS cells in HL patients or other mechanisms of rejection. In case of HL cell line KMH2, matched CD4+ T cells showed an antitumor response underlining this hypothesis. However, as the matched setting with L428 cells can be seen as an ideal model system to investigate “true” HRS/T-cell interactions, the changed setting using KMH2 cells with compatible donors might rather serve as a model to study further anti-HRS cell immune response mechanisms. Although matched CD4+ T cells did not show interactions to HRS cells at first glance, we observed HL-tumor infiltration by matched CD4+ T cells indicating T-cell attraction by HRS cells and pinpointing to a potential crosstalk (). Interestingly, independent of the HLA genotype of co-cultured CD4+ T cells the gene expression pattern of HRS cells was altered in a similar manner when compared to monocultures. Moreover, it could be shown that co-cultivation drives GEP of HL cell line L428 toward the differentiation of primary HRS cells (, Fig. S5). This is of enormous interest, as primary HRS cells are extremely hard to maintain in vitro.Citation25 Taking these results together, we assume that matched CD4+ T cells either bind to HRS cells or produce soluble factors, which have a major impact on HRS-cell physiology.
In summary, in our experimental settings, CD4+ T cells demonstrated a very potent anti-HRS cell response and were attracted by HRS cells in vivo. Hence, our data clearly show that CD4+ T cells should be very well suited for the immunotherapy of HL and that our model system is heavily useful to test adoptive transfer approaches with genetically engineered T-cells ().
Materials and methods
Cell culture
HL cell lines KMH2, L428, L1236 and BL cell line Raji (DSMZ) were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (PAA), 1% L-glutamine (Gibco) and 1% penicillin/streptomycin (Gibco).
Magnetic activated cell sorting (MACS)
MACS procedures were performed according to the manufacturer´s instructions (Miltenyi Biotech) to isolate different T-cell subsets from PBMC for co-culture studies (negative isolation kits for bulk, CD4+ or CD8+ T cells) or deplete T cells from co-cultures for microarray studies (CD3 microbeads). T-cell isolation showed a purity of 95–98% and microarray samples were only further processed with contamination rates < 5%.
Flow cytometry
Cells were stained with fluorescently labeled antibodies for flow cytometric analysis or cell counting of co-cultures performed on a MACSquant analyzer (Miltenyi). The following antibodies were used: anti-CD2-PE (Miltenyi), anti-CD3-APC (Invitrogen), anti-CD3-FITC (Miltenyi), anti-CD4-PE-Cy7 (Miltenyi), anti-CD8-VioBlue (Miltenyi), anti-CD11a-PE (Biolegend), anti-CD30-PE (Miltenyi) and anti-IgG-FITC (Southern Biotech).
Lentiviral vectors and transduction of HL cell lines
Third generation self-inactivating lentiviral vectors were used for fluorescent cytosolic labeling of HL cell lines with green fluorescent protein (GFP).Citation52 HL cell lines were transduced with lentiviral particles at a multiplicity of infection (MOI) of 5–10 by spin-infection.
Retroviral vectors and transduction of primary T cells
Retroviral vectors were used for gene transfer of CARs directed against CD30 or CEA into human CD4+ T cells or T-cell line PM1.Citation47 T cells were stimulated for 2–5 d with CD3/CD28-coupled stimulation beads (Invitrogen) and transduced at a MOI of 5–10 by spin-infection.
Co-cultivation of HRS cells and T cells
1 × 106 Primary human T cells were co-cultured with 5 × 10Citation4 HRS cells per mL (ratio of 20:1) for up to 15 d. When indicated, 75 µM dexamethasone was added. Every 2–3 d, cells were stained for CD3 and CD30 to obtain growth curves. Next to discrimination, flow cytometry-based counting enabled detection of HRS/T-cell rosettes as CD3/CD30 double positive events. For cluster formation assay, co-cultures were photographed after 7 d using a phase contrast microscope (CKX41, Olympus). Cluster diameters were determined using ImageJ software.
Blocking antibodies
Adapted from the protocol of Fromm et al.,Citation34 co-cultures were incubated with antibodies against CD2 (5 µg, clone RPA-2.10), CD11a (12 µg, clone MHM23), CD40 (4 µg, clone MAB89), CD54 (10 µg, clone 84H10) and CD58 (10 µg, clone TS2/9). MHC-II blockade was performed likewise using an anti-HLA-DR/DP/DQ antibody from BD (2 µg, clone Tü39).
Fluorescence microscopy
Smears of 7 d-old co-cultures were stained against CD2/CD58 or CD11a/CD54 as described previously.Citation53 Blocking antibodies derived from mouse were used for staining of CD54 and CD58, whereas CD2 and CD11a were stained with rabbit antibodies (abcam) in combination with corresponding secondary antibodies (anti-murine Alexa-488, anti-rabbit Alexa-547).
Gene expression profiling (GEP)
RNA of previously co-cultured HRS cells was isolated using RNeasy Mini kit (Qiagen). Amplification (input: 40 ng) as well as cDNA labeling was done using standardized protocols (Ovation Pico WTA System V2 amplification kit and Encore Biotin Module labeling kit from NuGEN). Microarray hybridization to GeneChip Human Gene 1.0 ST V1 arrays (Affymetrix) and further procedures were performed according to the Affymetrix protocol. Statistical analysis in terms of bioinformatics concerning GEP data is described in detail within the supplement. GEO ID: GSE75267
Xenograft model
Non-obese diabetic severe combined immunodeficiency mice with γ-chain knockout (NSG) were first inoculated with 1 × 106 HRS cells i.p. or s.c. (right flank). After 1 week, 1 × 107 T cells were additionally transferred. Tumor size and body weight was measured twice weekly. In case of enlarged tumor size (> 0.8 ccm) or tremendous weight loss (> 15%), mice were sacrificed. Experiments were performed in accordance with the local animal experimentation guidelines and approved by the regional council (Regierungspräsidium, Darmstadt, Germany: F21/04).
Histological analyses
For histological analysis, sections of formalin-fixed and paraffin-embedded organs were stained with hematoxylin/eosin (HE) or for CD3, CD4, CD8, CD15 or CD30 expression, as previously described.Citation54
HLA genotyping
MHC class II (HLA-DRB1 and -DQB1) low resolution genotyping of the cell lines and healthy donors was performed by polymerase chain reaction (PCR) sequence-specific primers (BAG or InnoTrain) or PCR sequence-specific oligonucleotide probes (BAG) after DNA isolation (Qiagen).
Statistical analyses
Significance was calculated using Student's t-test (2 tailed, unpaired/unequal variances).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
B.R. designed and performed research, analyzed data and wrote the manuscript; F.S. designed and performed research, analyzed data and edited the manuscript; C.W., C.D., T.H., K.W. and P.B. performed research and analyzed data; R.W., S.K., E.W. and A.B. performed research; C.S., S.H. and H.A. analyzed data and edited the manuscript; R.K. analyzed data, edited the manuscript and contributed to research design; M.-L.H. and S.N. designed and supervised the project, analyzed data and wrote the manuscript.
KONI_A_1160186_supplementary_materials.zip
Download Zip (9.2 MB)Acknowledgments
The authors thank Smaro Soworka, Ekaterini Hadzoglou, Sabine Albrecht, Yvonne Michel and Ralf Lieberz for excellent technical assistance. B.R. is a MD/PhD candidate and the data presented here are part of his MD thesis.
Funding
R.K. and S.H. are supported by the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft) (KU1315/7-1 and HA6145/1-2) and S.N. as well as M.-L.H. are funded under NE1438/4-1 and HA1284/8-1 as part of the collaborative research group on mature T-cell lymphomas (CONTROL-T). The project was further supported by the “LOEWE Center for Cell and Gene Therapy Frankfurt,” Hessisches Ministerium für Wissenschaft und Kunst (CGT-TP:Newrzela). We also thank “Alfons und Gertrud Kassel-Stiftung” and the “Research Support Foundation Vaduz” for funding of the MACSQuant cytometer.
References
- Hodgkin T. On some Morbid Appearances of the Absorbent Glands and Spleen. Med Chir Trans 1832; 17:68-114; PMID:20895597; http://dx.doi.org/10.1177/095952873201700106
- Drexler HG. Recent results on the biology of Hodgkin and Reed-Sternberg cells. I. Biopsy material. Leuk Lymphoma 1992; 8:283-313; PMID:1337848; http://dx.doi.org/10.3109/10428199209051008
- Küppers R, Schwering I, Bräuninger A, Rajewsky K, Hansmann ML. Biology of Hodgkin's lymphoma. Ann Oncol 2002; 13(Suppl 1):11-8; PMID:12078890; http://dx.doi.org/10.1093/annonc/13.S1.11
- Rengstl B, Newrzela S, Heinrich T, Weiser C, Thalheimer FB, Schmid F, Warner K, Hartmann S, Schroeder T, Kuppers R et al. Incomplete cytokinesis and re-fusion of small mononucleated Hodgkin cells lead to giant multinucleated Reed-Sternberg cells. Proc Natl Acad Sci U S A 2013; 110:20729-34; PMID:24302766; http://dx.doi.org/10.1073/pnas.1312509110
- Rengstl B, Rieger MA, Newrzela S. On the origin of giant cells in Hodgkin lymphoma. Commun Integr Biol 2014; 7:e28602; PMID:25346790; http://dx.doi.org/10.4161/cib.28602
- Skinnider BF, Mak TW. The role of cytokines in classical Hodgkin lymphoma. Blood 2002; 99:4283-97; PMID:12036854; http://dx.doi.org/10.1182/blood-2002-01-0099
- Teruya-Feldstein J, Tosato G, Jaffe ES. The role of chemokines in Hodgkin's disease. Leuk Lymphoma 2000; 38:363-71; PMID:10830743; http://dx.doi.org/10.3109/10428190009087027
- Poppema S, Bhan AK, Reinherz EL, Posner MR, Schlossman SF. In situ immunologic characterization of cellular constituents in lymph nodes and spleens involved by Hodgkin's disease. Blood 1982; 59:226-32; PMID:7034810
- Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, Inagaki H, Ueda R. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res 2006; 66:5716-22; PMID:16740709; http://dx.doi.org/10.1158/0008-5472.CAN-06-0261
- Marshall NA, Christie LE, Munro LR, Culligan DJ, Johnston PW, Barker RN, Vickers MA. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood 2004; 103:1755-62; PMID:14604957; http://dx.doi.org/10.1182/blood-2003-07-2594
- Fischer M, Juremalm M, Olsson N, Backlin C, Sundstrom C, Nilsson K, Enblad G, Nilsson G. Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg cells and its possible role in the recruitment of mast cells into lymphomatous tissue. Int J Cancer 2003; 107:197-201; PMID:12949794; http://dx.doi.org/10.1002/ijc.11370
- van den Berg A, Visser L, Poppema S. High expression of the CC chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic T-cell infiltratein Hodgkin's lymphoma. Am J Pathol 1999; 154:1685-91; PMID:10362793; http://dx.doi.org/10.1016/S0002-9440(10)65424-7
- Ohshima K, Tutiya T, Yamaguchi T, Suzuki K, Suzumiya J, Kawasaki C, Haraoka S, Kikuchi M. Infiltration of Th1 and Th2 lymphocytes around Hodgkin and Reed-Sternberg (H&RS) cells in Hodgkin disease: Relation with expression of CXC and CC chemokines on H&RS cells. Int J Cancer 2002; 98:567-72; PMID:11920617; http://dx.doi.org/10.1002/ijc.10218
- Hartmann S, Jakobus C, Rengstl B, Doring C, Newrzela S, Brodt HR, Wolf T, Hansmann ML. Spindle-shaped CD163+ rosetting macrophages replace CD4+ T-cells in HIV-related classical Hodgkin lymphoma. Mod Pathol 2013; 26:648-57; PMID:23307058; http://dx.doi.org/10.1038/modpathol.2012.217
- Tanijiri T, Shimizu T, Uehira K, Yokoi T, Amuro H, Sugimoto H, Torii Y, Tajima K, Ito T, Amakawa R et al. Hodgkin's reed-sternberg cell line (KM-H2) promotes a bidirectional differentiation of CD4+CD25+Foxp3+ T cells and CD4+ cytotoxic T lymphocytes from CD4+ naive T cells. J Leukoc Biol 2007; 82:576-84; PMID:17545218; http://dx.doi.org/10.1189/jlb.0906565
- Cattaruzza L, Gloghini A, Olivo K, Di Francia R, Lorenzon D, De Filippi R, Carbone A, Colombatti A, Pinto A, Aldinucci D. Functional coexpression of Interleukin (IL)-7 and its receptor (IL-7R) on Hodgkin and Reed-Sternberg cells: Involvement of IL-7 in tumor cell growth and microenvironmental interactions of Hodgkin's lymphoma. Int J Cancer 2009; 125:1092-101; PMID:19391137; http://dx.doi.org/10.1002/ijc.24389
- Yamamoto R, Nishikori M, Kitawaki T, Sakai T, Hishizawa M, Tashima M, Kondo T, Ohmori K, Kurata M, Hayashi T et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood 2008; 111:3220-4; PMID:18203952; http://dx.doi.org/10.1182/blood-2007-05-085159
- Kim LH, Eow GI, Peh SC, Poppema S. The role of CD30, CD40 and CD95 in the regulation of proliferation and apoptosis in classical Hodgkin's lymphoma. Pathology 2003; 35:428-35; PMID:14555388; http://dx.doi.org/10.1080/00313020310001602567
- Huang X, van den Berg A, Gao Z, Visser L, Nolte I, Vos H, Hepkema B, Kooistra W, Poppema S, Diepstra A. Expression of HLA class I and HLA class II by tumor cells in Chinese classical Hodgkin lymphoma patients. PLoS One 2010; 5:e10865; PMID:20526359; http://dx.doi.org/10.1371/journal.pone.0010865
- Lee SP, Constandinou CM, Thomas WA, Croom-Carter D, Blake NW, Murray PG, Crocker J, Rickinson AB. Antigen presenting phenotype of Hodgkin Reed-Sternberg cells: analysis of the HLA class I processing pathway and the effects of interleukin-10 on epstein-barr virus-specific cytotoxic T-cell recognition. Blood 1998; 92:1020-30; PMID:9680372
- Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, Johnson NA, Zhao Y, Telenius A, Neriah SB et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011; 471:377-81; PMID:21368758; http://dx.doi.org/10.1038/nature09754
- Mathas S, Janz M, Hummel F, Hummel M, Wollert-Wulf B, Lusatis S, Anagnostopoulos I, Lietz A, Sigvardsson M, Jundt F et al. Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol 2006; 7:207-15; PMID:16369535; http://dx.doi.org/10.1038/ni1285
- Tiacci E, Döring C, Brune V, van Noesel CJ, Klapper W, Mechtersheimer G, Falini B, Küppers R, Hansmann ML. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood 2012; 120:4609-20; PMID:22955914; http://dx.doi.org/10.1182/blood-2012-05-428896
- Kamesaki H, Fukuhara S, Tatsumi E, Uchino H, Yamabe H, Miwa H, Shirakawa S, Hatanaka M, Honjo T. Cytochemical, immunologic, chromosomal, and molecular genetic analysis of a novel cell line derived from Hodgkin's disease. Blood 1986; 68:285-92; PMID:3013343
- Drexler HG. Recent results on the biology of Hodgkin and Reed-Sternberg cells. II. Continuous cell lines. Leuk Lymphoma 1993; 9:1-25; PMID:7682880; http://dx.doi.org/10.3109/10428199309148499
- Fisher RI B-BF, Sauder DN, Scala G, Diehl V. Neoplastic cells obtained from Hodgkin's disease are potent stimulators of human primary mixed lymphocyte cultures. J Immunol 1983; 130:2666-70; PMID:6222113
- Fisher RI BS, Bostick-Bruton F, Tuteja N, Diehl V. Neoplastic cells obtained from Hodgkin's disease function as accessory cells for mitogen-induced human T cell proliferative responses. J Immunol 1984; 132:2672-7; PMID:6609204
- Shafer JA, Cruz CR, Leen AM, Ku S, Lu A, Rousseau A, Heslop HE, Rooney CM, Bollard CM, Foster AE. Antigen-specific cytotoxic T lymphocytes can target chemoresistant side-population tumor cells in Hodgkin lymphoma. Leuk Lymphoma 2010; 51:870-80; PMID:20367572; http://dx.doi.org/10.3109/10428191003713968
- Ho WT, Pang WL, Chong SM, Castella A, Al-Salam S, Tan TE, Moh MC, Koh LK, Gan SU, Cheng CK et al. Expression of CD137 on Hodgkin and Reed-Sternberg cells inhibits T-cell activation by eliminating CD137 ligand expression. Cancer Res 2013; 73:652-61; PMID:23204227; http://dx.doi.org/10.1158/0008-5472.CAN-12-3849
- Fisher RI, Cossman J, Diehl V, Volkman DJ. Antigen presentation by Hodgkin's disease cells. J Immunol 1985; 135:3568-71; PMID:3876389
- Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009; 113:6392-402; PMID:19377047; http://dx.doi.org/10.1182/blood-2009-03-209650
- Ito A, Ishida T, Yano H, Inagaki A, Suzuki S, Sato F, Takino H, Mori F, Ri M, Kusumoto S et al. Defucosylated anti-CCR4 monoclonal antibody exercises potent ADCC-mediated antitumor effect in the novel tumor-bearing humanized NOD/Shi-scid, IL-2Rgamma(null) mouse model. Cancer Immunol Immunother 2009; 58:1195-206; PMID:19048251; http://dx.doi.org/10.1007/s00262-008-0632-0
- King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, Laning J, Fodor W, Foreman O, Burzenski L et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol 2009; 157:104-18; PMID:19659776; http://dx.doi.org/10.1111/j.1365-2249.2009.03933.x
- Fromm JR, Kussick SJ, Wood BL. Identification and purification of classical Hodgkin cells from lymph nodes by flow cytometry and flow cytometric cell sorting. Am J Clin Pathol 2006; 126:764-80; PMID:17050074; http://dx.doi.org/10.1309/7371XK6F6P7474XX
- Pipitone N, Sinha M, Theodoridis E, Goulding N, Hall M, Lanchbury J, Corrigall V, Panayi G, Pitzalis C. The glucocorticoid inhibition of LFA-1 and CD2 expression by human mononuclear cells is reversed by IL-2, IL-7 and IL-15. Eur J Immunol 2001; 31:2135-42; PMID:11449367; http://dx.doi.org/10.1002/1521-4141(200107)31:7%3c2135::AID-IMMU2135%3e3.0.CO;2-S
- Engert A, Plutschow A, Eich HT, Lohri A, Dorken B, Borchmann P, Berger B, Greil R, Willborn KC, Wilhelm M et al. Reduced treatment intensity in patients with early-stage Hodgkin's lymphoma. N Engl J Med 2010; 363:640-52; PMID:20818855; http://dx.doi.org/10.1056/NEJMoa1000067
- Miyazaki Y, Fujiwara H, Asai H, Ochi F, Ochi T, Azuma T, Ishida T, Okamoto S, Mineno J, Kuzushima K et al. Development of a novel redirected T-cell-based adoptive immunotherapy targeting human telomerase reverse transcriptase for adult T-cell leukemia. Blood 2013; 121:4894-901; PMID:23641014; http://dx.doi.org/10.1182/blood-2012-11-465971
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012; 490:412-6; PMID:23051752; http://dx.doi.org/10.1038/nature11538
- Savoldo B, Rooney CM, Di Stasi A, Abken H, Hombach A, Foster AE, Zhang L, Heslop HE, Brenner MK, Dotti G. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 2007; 110:2620-30; PMID:17507664; http://dx.doi.org/10.1182/blood-2006-11-059139
- Renner C, Bauer S, Sahin U, Jung W, van Lier R, Jacobs G, Held G, Pfreundschuh M. Cure of disseminated xenografted human Hodgkin's tumors by bispecific monoclonal antibodies and human T cells: the role of human T-cell subsets in a preclinical model. Blood 1996; 87:2930-7; PMID:8639913
- Renner C, Jung W, Sahin U, Denfeld R, Pohl C, Trumper L, Hartmann F, Diehl V, van Lier R, Pfreundschuh M. Cure of xenografted human tumors by bispecific monoclonal antibodies and human T cells. Science 1994; 264:833-5; PMID:8171337; http://dx.doi.org/10.1126/science.8171337
- Hombach A, Kohler H, Rappl G, Abken H. Human CD4+ T cells lyse target cells via granzyme/perforin upon circumvention of MHC class II restriction by an antibody-like immunoreceptor. J Immunol 2006; 177:5668-75; PMID:17015756; http://dx.doi.org/10.4049/jimmunol.177.8.5668
- Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009; 30:646-55; PMID:19464987; http://dx.doi.org/10.1016/j.immuni.2009.05.001
- Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, Reis BS, Huang Y, Lambolez F, Docherty M et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol 2013; 14:281-9; PMID:23334788; http://dx.doi.org/10.1038/ni.2523
- Ma Y, Visser L, Roelofsen H, de Vries M, Diepstra A, van Imhoff G, van der Wal T, Luinge M, Alvarez-Llamas G, Vos H et al. Proteomics analysis of Hodgkin lymphoma: identification of new players involved in the cross-talk between HRS cells and infiltrating lymphocytes. Blood 2008; 111:2339-46; PMID:18070985; http://dx.doi.org/10.1182/blood-2007-09-112128
- da Costa L, Renner C, Hartmann F, Pfreundschuh M. Immune recruitment by bispecific antibodies for the treatment of Hodgkin disease. Cancer Chemother Pharmacol 2000; 46 Suppl:S33-6; PMID:10950145; http://dx.doi.org/10.1007/PL00014047
- Hombach A, Muche JM, Gerken M, Gellrich S, Heuser C, Pohl C, Sterry W, Abken H. T cells engrafted with a recombinant anti-CD30 receptor target autologous CD30(+) cutaneous lymphoma cells. Gene Ther 2001; 8:891-5; PMID:11423937; http://dx.doi.org/10.1038/sj.gt.3301467
- Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol 2003; 3:973-83; PMID:14647479; http://dx.doi.org/10.1038/nri1245
- Küppers R. The biology of Hodgkin's lymphoma. Nat Rev Cancer 2009; 9:15-27; PMID:19078975; http://dx.doi.org/10.1038/nrc2542
- McLellan A, Heldmann M, Terbeck G, Weih F, Linden C, Brocker EB, Leverkus M, Kampgen E. MHC class II and CD40 play opposing roles in dendritic cell survival. Eur J Immunol 2000; 30:2612-9; PMID:11009095; http://dx.doi.org/10.1002/1521-4141(200009)30:9%3c2612::AID-IMMU2612%3e3.0.CO;2-G
- Leverkus M, McLellan AD, Heldmann M, Eggert AO, Brocker EB, Koch N, Kampgen E. MHC class II-mediated apoptosis in dendritic cells: a role for membrane-associated and mitochondrial signaling pathways. Int Immunol 2003; 15:993-1006; PMID:12882837; http://dx.doi.org/10.1093/intimm/dxg099
- Weber K, Bartsch U, Stocking C, Fehse B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol Ther 2008; 16:698-706; PMID:18362927; http://dx.doi.org/10.1038/mt.2008.6
- Benz AH, Renne C, Maronde E, Koch M, Grabiec U, Kallendrusch S, Rengstl B, Newrzela S, Hartmann S, Hansmann ML et al. Expression and functional relevance of cannabinoid receptor 1 in Hodgkin lymphoma. PLoS One 2013; 8:e81675; PMID:24349109; http://dx.doi.org/10.1371/journal.pone.0081675
- Hartmann S, Agostinelli C, Klapper W, Korkolopoulou P, Koch K, Marafioti T, Piccaluga PP, Patsouris E, Pileri S, Hansmann ML. Revising the historical collection of epithelioid cell-rich lymphomas of the Kiel Lymph Node Registry: what is Lennert's lymphoma nowadays? Histopathology 2011; 59:1173-82; PMID:22175897; http://dx.doi.org/10.1111/j.1365-2559.2011.04069.x