
ABSTRACT
Tumor–host interaction is determined by constant immune surveillance, characterized by tumor infiltration of myeloid and lymphoid cells. A malfunctioning or diverted immune response promotes tumor growth and metastasis. Recent advances had been made, by treating of certain tumor types, such as melanoma, with T-cell checkpoint inhibitors. This highlights the importance of understanding the molecular mechanisms underlying the crosstalk between tumors and their environment, in particular myeloid and lymphoid cells.
Our aim was to study the contribution of the myeloid PI3K/PTEN-signaling pathway in the regulation of tumor-immune surveillance in murine models of cancer. We made use of conditional PTEN-deficient mice, which exhibit sustained activation of the PI3K-signaling axis in a variety of myeloid cell subsets such as macrophages and dendritic cells (DCs).
In colitis-associated colon cancer (CAC), mice deficient in myeloid PTEN showed a markedly higher tumor burden and decreased survival. We attributed this observation to the increased presence of immune-modulatory conventional CD8α+ DCs in the spleen, whereas other relevant myeloid cell subsets were largely unaffected. Notably, we detected enhanced surface expression of PD-L1 and PD-L2 on these DCs. As a consequence, tumoricidal T-cell responses were hampered or redirected.
Taken together, our findings indicated an unanticipated role for the PI3K/PTEN-signaling axis in the functional regulation of splenic antigen-presenting cells (APCs). Our data pointed at potential, indirect, tumoricidal effects of subclass-specific PI3K inhibitors, which are currently under clinical investigation for treatment of tumors, via myeloid cell activation.
Abbreviations
| AOM | = | azoxymethane |
| APC | = | antigen-presenting cell |
| CAC | = | colitis associated colon cancer |
| CEP | = | colon epithelium |
| CLP | = | colon lamina propria |
| CTL | = | cytotoxic T-cell |
| DC | = | dendritic cell |
| DSS | = | dextran sodium sulphate |
| LysM | = | lysozyme M |
| MDSC | = | myeloid derived suppressor cell |
| MHC | = | Major histocompatibility complex |
| MLN | = | mesenteric lymph node |
| Ova | = | ovalbumin |
| PD-L | = | programmed cell death ligand |
| PI3K | = | Phosphatidylinositol (3,4,5) trisphosphate kinase |
| PTEN | = | phosphatase and tensin homolog |
| SPL | = | spleen |
| TCR | = | T cell receptor |
Introduction
The PI3K/PTEN-signaling axis is an important pathway in regulating growth, cell cycle, migration and survival of cells. Hyper-activation of PI3K signaling by deletion or deactivation of the counteracting phosphatase, the tumor-suppressor PTEN (phosphatase and tensin homolog located on chromosome 10), was found in various late-stage human cancers.Citation1 So far, the role of this central pathway in the regulation of innate immune cell fate and activity has not been clarified.
We previously showed that deletion of PTEN in myeloid cells results in an anti-inflammatory phenotype, characterized by reduced expression of pro-inflammatory factors.Citation2,3 Accordingly, this anti-inflammatory phenotype (due to PTEN deletion) in macrophages led to a decreased inflammatory response and increased survival in a bacterial infection model.Citation4 Furthermore, livers of myeloid PTEN-deficient mice were protected against ischemia-reperfusion injury (IRI) due to increased production of IL-10,Citation5 and gut homing via CCL25/CCR9 signaling was increased by shRNA knock-down of PTEN.Citation6
The myeloid PI3K/PTEN-signaling pathway further has a role in DC development. Reizis and co-workers showed that deletion of PTEN in mouse haematopoietic cells promoted the development of FMS-like tyrosine kinase 3 ligand (Flt3L)-driven DCs in in vitro bone-marrow cultures. In vivo studies showed that DC-specific deletion of PTEN also increased DC numbers, in particular CD8α+ splenic DCs and CD103+ peripheral DCs. More specifically, downstream of PI3K signaling, mTOR mediated Flt3L-induced CD8α+DC and CD103+DC development, which could be inhibited by rapamycin.Citation7 Rapamycin affects DC development, maturation and function, since rapamycin-treated DCs reduced T-cell activation and led to anergy (reviewed in referenceCitation8). mTOR signaling further favored IL-10 over IL-12 production, which closely resembles the phenotype observed in PTEN-deficient DCs.Citation9,10 A recent publication found an involvement of rapamycin-sensitive mTOR signaling in the induction of IL-10, PD-L1 and PD-L2.Citation11
APCs have increased capacities for antigen-presentation than macrophages. Indeed some subsets are also able to present exogenous and endogenous antigens to T-cells, both via MHCI and MHCII (cross-presentation). The resulting response of T-cells further depends on co-stimulatory molecules (signal 2) and instructing cytokines (signal 3). Signaling via peptide-MHC TCR (signal 1) without co-stimulation (signal 2) results in anergic T-cells and tolerance induction. DCs are directly able to inhibit or even shut down T-cell responses via inhibitory members of the B7-family, PD-L1 and PD-L2, which seem to be—at least in part—differentially regulated, depending on the predominant T-helper cell subset. Th1-derived IFNγ leads to an increase in PD-L1, whereas Th2-derived IL-4 causes the upregulation of PD-L2.Citation12 Recently, PD-L1 was shown to be a direct target of HIF1α in MDSCs, which is also a downstream target of PI3K signaling.Citation13
Activated T-cells respond to co-stimulation via expression of CD40L, which binds CD40 on APCs leading to their full maturation. CD8α+ (lymphoid tissue) and CD103+ (peripheral tissue) DCs share functional (production of high levels of bioactive IL-12) and transcriptional (dependency on BATF3, Id2 and IRF8) similarities, but so far only CD8α+DCs have been shown to exhibit the unique ability to cross-present exogenous antigens via MHCI to CD8+-T-cells. This is the pathway that enables CD8α+DCs to present tumor- or virus-derived antigens. Depending on additional signals, presentation of self-antigens might result in cross-priming or cross-tolerance.Citation14
PTEN-deficiency in cells of myeloid origin gives rise to an immune-suppressive phenotype that is advantageous in models of acute infection and inflammation. In an autoimmunity animal model resembling human multiple sclerosis, myeloid PTEN-deficiency attenuated disease severity and progression suggesting that innate PI3K/PTEN signaling controls activation and intensity of adaptive immune responses.Citation10 Tumor-immune surveillance (mounting an immunological response against abnormally proliferating self-tissue) might therefore also be similarly affected and thus tumor-cell rejection and elimination by the adaptive immune system impaired. To address this question, we applied two different tumor models (orthotopically implanted B16 melanoma and CAC) in PTENfl/fl LysM cre (myPTEN−/−) and WT littermate (myPTEN+/+) control mice and found that myeloid PTEN-deficiency results in decreased tumor-immune surveillance caused by immune-checkpoint inhibition.
Material and methods
Animals
Animals were housed under SPF conditions; all experiments were approved by the Austrian Federal Ministry for Health. Floxed PTEN mice were a kind gift from Tak W. Mak and were crossed with LysM cre mice in house. All mice were backcrossed to C57BL/6 background for at least 10 generations. Mice were genotyped for floxed PTEN alleles and presence of cre-recombinase in a direct PCR reaction (GoTaq Polymerase; Promega) using tail- or ear-lysates (Proteinase K) as templates. The following primers were used for genotyping: cre-for: 5′-TCGCGATTATCTTCTATATCTTCA-3′; cre-rev: 5′-GCTCGACCAGTTTAGTTACCC-3′; fl-PTEN-for: 5′-CTCCTCTACTCCATTCTTCCC-3′; fl-PTEN-rev: 5′-ACTCCCACCAATGAACAAAC-3′.
Induction of colitis-associated colon cancer
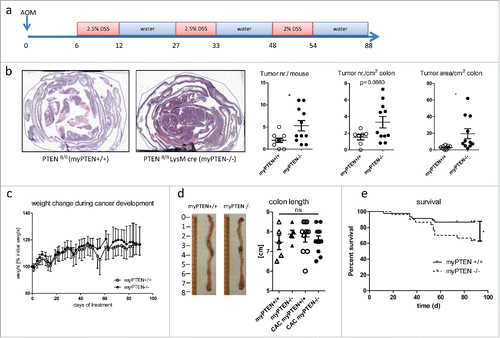
CAC was induced in 8–12 week old mice by injection of 12.5 mg/kg azoxymethane (AOM, Sigma-Aldrich) i.p. followed by three cycles of dextran sodium sulfate (DSS, MP Biomedicals) supplied in the drinking water (2.5% cycles 1 and 2, 2% cycle 3 for 6 d each with 15 d abstention in between, see ); mice were sacrificed at days 85–90 after AOM injection.
Figure 1. Myeloid PTEN-deficiency increases tumor burden and mortality. (A) timeline of AOM/DSS treatment to induce colitis-associated colon cancer (CAC), (B) Representative H&E-stained sections of swiss rolls (small and large intestines) of PTENfl/fl and PTENfl/fl LysM cre mice and quantification of tumor number and area, n = 7–11, *p <0.05. (C) Weight change during CAC development of one representative experiment n = 6–8, (D) representative pictures of unflushed colons and quantification of colon length (n = 5–12). (E) Kaplan–Meier estimator of survival during CAC development (n = 30–46), *p < 0.05.
B16-F10-Ova-Luc melanoma tumor model
The B16-F10-Ova melanoma cell line was a kind gift from Dr Gottfried Baier. B16-F10-Ova were transduced with a lentiviral construct containing a red-shifted firefly luciferase (Perkin–Elmer) and checked for luciferase activity. B16-F10-Ova-Luc were grown in DMEM supplemented with 10% FCS, 1% penicillin-streptomycin-fungizone, 1% L-glutamate and 5 µg/mL puromycin. Before injection, cells were trypsinized, washed with 1 × PBS and adjusted to 1 × 106 cells/mL in PBS on ice; 100 µL of the cell suspension was injected intra-dermally on the chest. To enable the comparison of in vivo tumor growth, 1 × 106 B16-F10 or B16-F10-Ova melanoma cells were injected per mouse.
Luciferase assay
For the in vitro killing assay, 5 × 105 B16-F10-Ova-Luc cells were plated one day prior to addition of LPS-activated and SIINFEKL (Bachem)-primed splenocytes (106 per well). After 1 h, cells were washed with PBS and lysis buffer (0.1 M KH2PO4, 0.1% Triton x-100 pH7.8) was added. Lysis was performed by freezing and thawing. Luciferase activity was measured in a Synergy H4 plate reader after injection of substrate (200 mM Tris-HCL pH 8.0, 15 mM MgSO4, 0.1 mM EDTA pH 8.0, 25 mM DTT, 1 mM ATP, 0.2 mM coenzyme A and 200 µM luciferin); values represent arbitrary luminescence units.
Histological analysis
Histological analysis of colon tumors were performed as described elsewhere.Citation15 In brief, mice were sacrificed; colons were removed and rinsed with PBS and fixed with 4% PFA (Roth). Tissues were further dehydrated and paraffin-embedded as “swiss rolls”. 7 µm sections were used for H&E staining. Stained tumor sections were scanned with a TissueFAXS microscope (TissueGnostics) and analyzed with TissueQuest (TissueGnostics).
Immunohistochemistry
Paraffin-embedded colons were cut at a thickness of 5 µm and left to dry overnight. Paraffin was removed with Xylene and a series of decreasing percentage alcohol solutions. Antigen retrieval was done using heat-induced antigen retrieval by gentle cooking for 10 min in a pH 9.0-adjusted TRIS buffer for CD3, or by applying a solution of 20 µg/mL Proteinase K for 10 min at 37°C for F4/80. Slides were then rinsed, permeabilized with a 0.2% solution of Triton x-100 and blocked with a solution of 10% donkey serum and 1% BSA for 2 h at RT. The same solution was also used as diluent for the primary and secondary antibody. The primary antibody was incubated overnight at 4°C. Slides were rinsed well and the secondary antibody was incubated for 1 h at RT. The slides were then counterstained with a 5 µg/mL Hoechst solution for 15 min at RT. Finally, they were rinsed thoroughly and mounted with DAKO fluorescent mounting medium. 1x TBS with 0.025% Triton x-100 was used as a diluent for all solutions. Stained sections were scanned with a Nikon A1 confocal laser scanning microscope.
Bioluminescence imaging
Bioluminescence imaging of B16-F10-Ova-Luc melanoma growth was recorded in an IVIS Lumina Series III pre-clinical imaging system (Perkin–Elmer). Before the measurement, mice were anesthetized with 4% Furane/O2 (Abott Laboratories Ltd.) in a gas chamber and then injected with 150 mg/kg bodyweight D-luciferin (BioVision) solution in 0.9% NaCl. During the measurement, mice were kept anesthetized with 2% Furane/O2. Several images were taken at an exposure of 2–30 sec, depending on tumor size; the maximal photon flux (p/s) after 8–12 min was used for estimation of tumor size.
Single-cell preparations from tissues
Single cells from colons were isolated by first incubating colons with 5 mM EDTA for 2 × 20 min, to gain intestinal epithelial cells (IELs), followed by enzymatic digestion with 10 mg collagenase VIII (Sigma) and 69.6U DNAseI (Worthington) per colon to isolate cells from colon lamina propria (CLP). Cells from spleens were isolated by enzymatic digestion of whole spleens with 30 mg collagenase D (Roche), 300U hyaluronidase (Sigma-Aldrich) and 30 ug DNAseI (Roche) followed by filtering through 70 µm cell strainers and lysis of erythrocytes. Mesenteric lymph nodes were removed and filtered through 70 µm cell strainers to obtain single-cells suspensions. Single cells isolated from these organs were further labeled with CD11c or CD11b antibodies (Miltenyi Biotech) for magnetic cell enrichment according to manufacturer's protocols.
Flow cytometry
For flow cytometry analysis, single-cell suspensions from colon LP and epithelium, mesenteric lymph nodes and spleen were stained using the following antibodies: CD45.2-APC-eFluor780 (eBioscience),
CD3ϵ-APC-eFluor780 (eBioscience), CD3-CF594 (BD Biosciences), CD4+-PerCP (BD Biosciences), CD4+-eFluor710 (eBioscience), CD8a-PE (eBioscience), CD8α-Brilliant Violet 510 (BioLegend, Brilliant Violet™ products are trademark of Sirigen Group Ltd.), CD8α-Alexa Fluor 700 (eBioscience, Alexa Fluor® products are trademark of Molecular Probes, Inc.), CD25-PerCP-Cy5.5 (eBioscience), CD69-PECy7 (eBioscience), PD1-AF700 (eBioscience), CD107a-PE (eBioscience), B220-Alexa700(eBioscience), B220- Brilliant Violet 510 (BioLegend), CD11b-APC (eBioscience), CD11c-eFluor450 (eBioscience), NK1.1-PE-Cy7 (eBioscience), NK1.1-APC (eBioscience), GR1-FITC (eBioScience), MHCII-FITC (eBioscience), CD274(PD-L1)-PE-Cy7 (eBioscience), CD273(PD-L2)-PerCP-eFluor710 (eBioscience), CD103- Brilliant Violet 510 (Biolegend), 7-AAD (Tonbo), SYTOX (Thermo Fisher Scientific). Flow cytometry acquisition was performed on a LSR Fortessa flow cytometer (BD Biosciences) or a Gallios flow cytometer (Beckman Coulter), Data were analyzed with FlowJo software version 9 (Treestar) or Kaluza (Beckman Coulter). For intracellular cytokine production analysis, splenocytes were isolated and re-stimulated with anti-CD3/CD28 dynabeads (Life Technologies) and PMA/Ionomycin (Sigma) treatment together with Golgi-Stop (BD Biosciences) for 4 h. The following antibodies were used for cytokine staining: IFNγ–PE (eBioscience), IL-17–PE-Cy7 (eBioscience), IL-2–eFlour 450 (eBioscience), IL-10–AF647 (eBioscience). CFSE proliferation dye was purchased from eBioscience and cells were labeled according to manufacturer's instructions.
RT-qPCR
RNA was isolated from cells and tissues using the mRNeasy Kit (Qiagen) or Trifast (PeqLab) and reverse-transcribed into cDNA using High Capacity cDNA Reverse Transcription Kit (Fermentas) according to manufacturer's protocols. Expression of mRNA was measured using FastSybrGreen Mix (Applied Biosystems) in a StepOne RT-PCR 7,500 system (Applied Biosystems). Ct-values of target genes were normalized to mHPRT and expressed as fold mean WT control using efficiencies calculated by LinReqPCRCitation16 for each amplification. The following primers were used:HPRT: for: CGCAGTCCCAGCGTCGTG; rev: CCATCTCCTTCATGACATCTCGAG; PTEN: for: ACACCGCCAAATTTAACTGC; rev: TACACCAGTCCGTCCCTTTC; IL-10: for: AGCTGAAGACCCTCAGGATG; rev: TGGCCTTGTAGACACCTTGG; Arg1: for: GGAAAGCCAATGAAGAGCTG; rev: GCTTCCAACTGCCAGACTGT; Fizz: for: CTGGATTGGCAAGAAGTTCC; rev: CCCTTCTCATCTGCATCTCC; Stab1: for: CCCTCCTTCTGCTCTGTGTC; rev: CAAACTTGGTGTGGATGTCG; IL-23a: for: ATGCTGGATTGCAGAGCAGT; rev: ACGGGGCACATTATTTTTAG; Ym1: for: TTTCTCCAGTGTAGCCATCCTT; rev: TCTGGGTACAAGATCCCTGAA.
Whole blood cell analysis
Blood samples were collected in tubes containing 500 mM EDTA and blood parameters were measured on a VET ABC Hematology Analyzer (Scil Animal Care Company).
Micro-array analysis
RNA of splenic CD11c+ cells was isolated using the mRNeasy Kit (Qiagen) and submitted for quality control (Agilent Bioanalyzer RNA 6000 Nano), labeling and measurement on a Mouse Gene 2.0 ST Array (Affymetrix) to the Core Facility Genomics at the Medical University of Vienna; labeling, quality control and array measurement were kindly performed by Markus Jeitler. Array data were normalized by RMA Sketch (Expression Console, Affymetrix) and analyzed by the using one-way between-subject ANOVA (unpaired) with a p value <0.05 defining significance. Sample comparison, hierarchical clustering and pathway enrichment analyses were carried out using Transcriptome Analysis Console software (Affymetrix). Gene Functional Annotation analysis was performed using DAVID Bioinformatics Resources 6.7Citation17,18
T-cell proliferation measurement
To assess T-cell proliferation, cells were cultured ex vivo in the presence of plate-bound anti-CD3ϵ for 3 d. Cells were then labeled with 3H-thymidine (1 µ Ci/well) and incubated for 20 h. Incorporation of labeled thymidine was measured on a scintillation counter. For the measurement of in vivo T-cell proliferation, CD8+ T-cells were isolated from spleens of OT-I transgenic mice, enriched using magnetic isolation (Miltenyi Biotech), CFSE labeled and i.v. transferred in to CAC mice. On the following day, mice were immunized with LPS (10 µg/mouse) and SIINFEKL (2 µg/mouse), T-cells from spleens and mesenteric lymph nodes were isolated and analyzed by flow cytometry on day 3 after i.v. transfer.
Statistical analysis
Data were analyzed for Gaussian distribution using GraphPad Prism 5.0 (GraphPad Software Inc.). Data with Gaussian distribution were checked for statistical significance using Student's t-test, data sets with unequal variances were analyzed using Student's t-test with Welch's correction, data with more than two groups were analyzed with one-way ANOVA, data with more than two groups and different conditions were analyzed using two-way ANOVA.
Results
Myeloid PTEN deficiency increases inflammation-driven colon cancer growth and reduces survival
WeCitation2-4 and othersCitation5 have previously shown that the conditional knock-out of PTEN in the myeloid cell lineage results in an increased activation of the PI3K pathway, thereby shifting the functional phenotype toward an “alternatively-activated” or “M2-like” state. Most strikingly, pro-inflammatory molecules such as TNFα, IL6 and KC/Gro1 are decreased in myeloid PTEN-deficient mice. This is accompanied by an increase in IL-10, which protects from damaging effects caused by acute inflammation or infection. This acute protective and anti-inflammatory phenotype might become detrimental during chronic inflammation and tumor development.
To address the question of whether this shifted phenotype in the myeloid compartment might have an influence on tumor development, we induced (CAC, ) chemically in PTENfl/fl LysM cre (myPTEN−/−) mice by AOM/DSS treatment and did a software-assistedCitation15 analysis of H&E stained histological sections of colon swiss rolls for tumor development. Myeloid PTEN−/− mice showed an increase in tumor incidence (assessed by tumor number per mouse and cm2; colon) and progression (assessed by tumor area per colon area) (). Weight loss during the AOM/DSS treatment period () and changes in colon length () were similar in myeloid PTEN-deficient mice and littermate controls suggesting that besides an increase in tumor development, AOM/DSS-induced formation of CAC (including inflammation and the course of disease) was similar in both genotypes. In a model of acute colitis, weight loss was not significantly different between myeloid PTEN-deficient and littermate-control mice (Fig. S1B). Male myPTEN−/− mice showed a significantly reduced survival rate during the development of CAC (); survival in female mice was not significantly altered (Fig. S1A). To rule out any influence of differences in the composition of the microbiome between genotypes, PTENfl/fl LysM cre and PTENfl/fl mice were co-housed. Blood cell counts did not show significant differences when we compared myeloid PTEN-deficient animals to wild types, although myPTEN−/− mice produced more granulocytes at the expense of lymphocytes after CAC development compared to untreated control KOs, and the ratio of granulocytes to lymphocytes was also significantly higher than in tumor-bearing WT mice (Fig. S1C). Platelet counts were decreased in CAC mice and MPV was increased–regardless of genotype–most probably due to the DSS treatment during which mice suffer from bleeding events. Hence, myPTEN+/+ and myPTEN−/− mice exhibited comparable colitis-associated inflammation leading to the development of colon cancer but the PTEN-deficiency in the myeloid cell compartment significantly attenuated tumor-immune surveillance.
Myeloid PTEN-deficient mice show enhanced growth of B16-F10 melanoma
To address the question of whether the observed enhanced tumor growth upon AOM/DSS treatment in myeloid-PTEN deficient mice might be restricted to endogenously grown tumors and to rule out that only tumor initiation is affected, we injected myPTEN−/− and myPTEN+/+ with B16-F10-Ova-Luc melanoma cells. These cells were previously transduced with a lentiviral construct encoding a red-shifted firefly luciferase to allow for a more precise estimate of tumor growth via measurement of bioluminescence. PTENfl/fl LysM cre mice showed an increased tumor growth in this orthotopically-implanted tumor model over 10 d (). We observed a rejection of tumors in some mice, leading to an almost complete elimination of the tumors 10 d after subcutaneous injection (). This was usually not the case with conventional B16-F10-melanomas, but a decrease in in vivo growth rate was already observed in ovalbumin-expressing B16-F10 cells (compared to an ova negative-cell line, Fig. S6B), indicating an increase in immunogenicity. Myeloid PTEN-deficient and control mice did not show any significant difference in weight change () or blood parameters (Fig. S2A), with the exception of the lymphocyte to granulocyte ratio. Similar to the changes observed in the CAC model (Fig. S1C), this ratio was decreased.
Figure 2. Myeloid PTEN-deficiency increases tumor growth of B16-F10 melanoma. (A) Representative bioluminescence images of myPTEN+/+ and myPTEN−/− mice on day 2, day 6 and day10 after s.c. B16-F10-Ova-Luc injection, (B) Quantification of tumor growth by bioluminescence imaging of luciferase activity, n = 10–14. (C) Weight change of B16-injected mice, values are expressed as percent initial weight before tumor inoculation, n = 4 – 7.
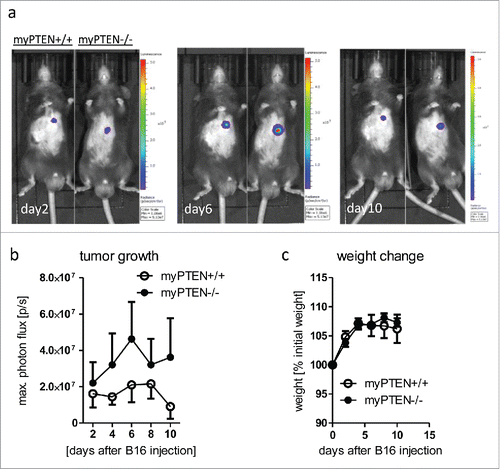
The increase in tumor load in the B16 melanoma model in myPTEN−/− mice indicated that, independent of tumor origin (endogenously grown or orthotopically implanted), myeloid PTEN-deficiency reduced tumor rejection.
Myeloid PTEN deficiency increases immune-regulatory CD8α+ DCs
Analysis of flow cytometry data of myeloid cell populations from PTENfl/fl LysM cre and cre-negative animals revealed an increase in CD11c+ cells in the spleens of myPTEN−/− mice (). Splenic CD11b+ and CD11c+CD11b+ myeloid cells were not significantly changed in numbers between genotypes (). Within the CD11c+ subset, we found CD8α+CD11c+ cells significantly increased in myPTEN−/− animals (). The corresponding DCs in the gut (CD103+DCs) were not altered in numbers in PTEN-deficient animals compared to wild types (). Within the DC compartment in the spleen, myPTEN−/− mice showed a significant increase in professional APCs (CD11c+MHCII+) expressing the immune-checkpoint molecules PD-L1 and PD-L2 (). Both of these inhibitory ligands were shown to bind to PD-1 on T-cells and inhibit proliferation and cytokine production.Citation19 CD11c+ CD8α+ DCs in the spleen that were increased in myPTEN−/− mice also expressed the immune checkpoint-inhibiting ligands PD-L1 and PD-L2 and this finding was independent of genotype (). There was no significant difference between the number of CD11c–CD11b+ cells expressing PD-L1 versus PD-L2. CD11b+CD11c+ cells expressing PD-L1 or PD-L2 did not significantly differ in number, but their numbers were lower than those of CD11b–(CD11c+) DCs, although the difference was less pronounced when considering PD-L2 expressing cells ( and Fig. S3A). The increase in CD11c+ dendritic cells in the spleen was also observed in healthy animals, as was the increase in the CD11c+CD8α+ subset, and could therefore be attributed to the PTEN knockout in dendritic cells,Citation7 which are also targeted by the LysM promoter.Citation20 Nevertheless, CD11c+ and CD11c+CD8α+ increased in numbers after CAC induction (0.9% to 2% CD11c+ in myPTEN+/+ and 1.7% to 4.5% CD11c+ in myPTEN−/− and 0.1% to 0.4% CD11c+CD8α+ in myPTEN+/+ and 0.8% to 1.8% CD11c+CD8α+ in myPTEN−/−, respectively. ). Healthy control mice of both genotypes did not show any significant difference in numbers of PD-L1 or PD-L2 expressing CD11c+MHCII+ APCs in the spleen (), but after CAC development their numbers increased about 4-fold (0.9% to 4.0% PD-L1+APCs and 0.4% to 1.9% PD-L2+APCs) in myPTEN−/− mice and remained almost unchanged in myPTEN+/+ animals (). We further found a non-significant trend toward an increase in MDSCs (CD11b+ GR1+) in spleen, colon lamina propria and colon epithelium (Fig. S3B) of myPTEN−/− mice. MDSCs were shown to be increased in the spleen and bone marrow of C57BL/6 mice by DSS treatment.Citation21 In the B16 melanoma model, myeloid PTEN-deficient mice exhibited an increase in splenic CD11b+, CD11b+C11c+ and CD11c+ cells (Fig. S2B). The highest increase in cell numbers between myPTEN+/+ and myPTEN−/− animals was observed in the CD11c+ CD8α+ compartment. Surprisingly, CD11b+CD11c+ cells expressing either PD-L1 or PD-L2, did not significantly differ in numbers, although there seemed to be a trend toward an increase of PD-L1 or PD-L2 expressing CD11c+ cells in myPTEN−/− mice that didn't reject the tumor, which was completely absent in myPTEN+/+ mice.
Figure 3. Myeloid PTEN-deficiency increases CD8a+DCs in the spleen. (A) Representative dot plots for identification of CD8α+DCs (gated on live, CD45+ CD3− cells). (B) Quantification of myeloid subsets (CD11c+CD11b+; CD11c+CD11b− and CD11c+CD8a+ cells in spleen-SPL, colon lamina propria-CLP and colon epithelium-CEP), values are expressed as % of live cells, n = 4–6, *p < 0.05, **p < 0.01. (C) Representative density plot for identification of CD11c+ MHCII+PDL-1+ and PDL-2+ cells, respectively (gated on live, CD45+CD11c+CD11b−). (D) Quantification of myeloid MHCII+ PDL1+ and PDL2+ cells (gated on live, CD45+) in the spleen, values are expressed as % of live cells, n = 4–6, *p < 0.05, **p < 0.01. (E) Splenic CD11c+ MHCII+APCs co-express CD8α and PDL-1 or PDL-2, respectively, (F) Myeloid subsets and immune checkpoint inhibitors in naive healthy mice, gating as in (A) or (C), respectively, values are expressed as % of live cells, n = 4–5, *p < 0.05.
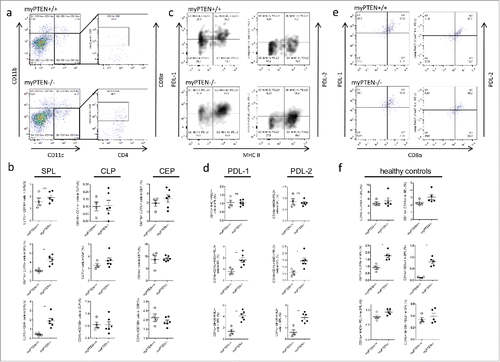
Taken together, myeloid PTEN-deficient mice showed increased numbers of CD11c+ and more specifically CD11c+CD8α+ cells in the spleen, which also express either PD-L1 or PD-L2, after CAC development. Both of these inhibitory surface proteins become upregulated during tumor progression and PD-L1 or PD-L2 expressing APCs are even more abundant in myPTEN−/− mice, indicating that immune-checkpoint inhibition by PD-Ls attenuated tumor-immune surveillance in myeloid PTEN deficiency.
Gene-expression profile of splenic PTEN-deficient DCs of CAC mice
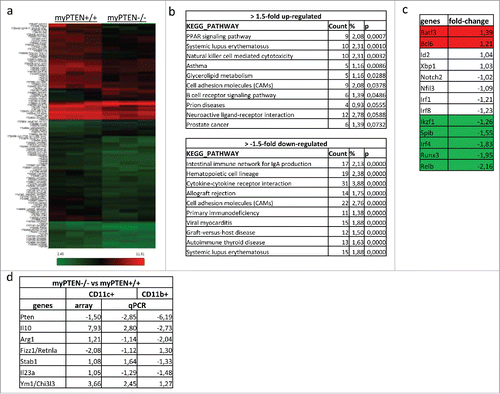
Since deletion of PTEN in myeloid cells resulted in an increase in immune-suppressive myeloid cells, we were interested in the genetic basis that accounted for the observed tumor-promoting phenotype of professional APCs (). mRNA array-expression analysis of enriched CD11c+ cells revealed an increased expression of BatF3 and Bcl6 as well as a concomitant decrease of Irf4, Runx3, Relb and PU.1 (also known as Spib), in myPTEN−/− compared to myPTEN+/+ mice. The relative changes in the levels of these transcription factors (TF) resemble their changes during CD8α+ DC development ().Citation14 The most highly upregulated TF was Batf3, which has been shown to be indispensable for CD8α+ DC development.Citation22 This present finding clearly showed that DCs isolated from myPTEN−/− were predominantly CD8α+ DCs. Next, we were interested in relevant pathways for DC function that were affected most significantly by deletion of PTEN. Therefore, we analyzed all genes with a log2-expression value of more than four in at least one sample, so as to focus only on highly expressed genes in CD11c+ cells. Furthermore, we concentrated on genes that were at least 1.5-fold differentially regulated and analyzed the pathways containing these genes. Using the KEGG-collection, we found 14 pathways affected by upregulated genes and 26 pathways influenced by downregulated genes ( and Fig. S4A). The expression of some genes known to be affected by myeloid PTEN-deletion was further validated by qPCR and their fold changes were compared to the mRNA array data. All analyzed genes showed similar trends for up or downregulation (). We further compared the qPCR expression data from isolated CD11c+ cells with isolated CD11b+ cells. Different expression of IL-10, Fizz-1 and Stab-1 clearly showed that these two cell populations were distinct, with different transcriptional and phenotypical properties, although both cell populations were deficient for PTEN (). These findings further supported the notion that CD8α+DCs from myPTEN−/− mice were immature and not efficient in T-cell activation.
Figure 4. Transcriptome analysis of enriched splenic CD11c+ cells. (A) Heatmap of differentially expressed genes considering a p value of <0.05 as significantly changed. (B) Selection of most significantly changed Kegg pathways containing differentially expressed genes in splenic CD11c+ cells from myPTEN−/− and myPTEN+/+ mice; list is sorted according to p value. (C) Fold changes of transcription factors involved in DC development and function. (D) Fold changes of selected genes know to be differentially regulated between myPTEN−/− and myPTEN+/+ in CD11b+ and CD11c+ cells.
T-cells from CAC myPTEN−/− mice are rendered hypo-responsive
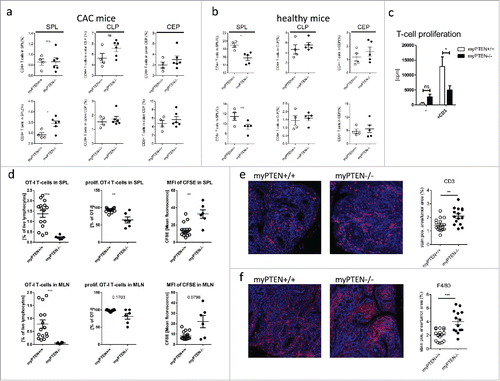
Considering the observed increase in PD-L+CD8α+ DCs in myPTEN−/− animals, we were interested in the functional phenotype of T-cells in these mice. First, relative numbers of CD4+ T-helper (Th) and CD8+ cytotoxic T-cells (CTL) were analyzed by flow cytometry. Except for an increase in CD8+ CTLs in the spleens of myPTEN−/−, there was no significant difference in the numbers of B-, T- or NK-cells, between genotypes in any analyzed organ (). All cell populations in mesenteric lymph nodes and spleen were decreased in CAC mice compared to healthy controls, except splenic B-cells ( and data not shown). Quite strikingly, cell populations in the colon (lamina propria and epithelium) were almost unchanged between CAC and healthy animals, with the exception of CD4+ T-helper cells in colonic epithelium that were increased in CAC mice (1.3% to 2% in myPTEN+/+ and 1.5% to 3% in myPTEN−/−, ), which was more pronounced in myPTEN−/− mice. This finding could also be confirmed by IHC staining for CD3 on sections from colon swiss rolls of CAC mice, showing that myPTEN−/− mice disclosed higher numbers of infiltrating T-cells per tumor (). Relative numbers of CD4+ and CD8+ T-cells in the lamina propria and epithelium of the colon further showed that the separation of both compartments was as anticipated, since CD4+ T-cells are primarily found in the lamina propria and CD8+ T-cells in the colon epithelium.Citation23 The proliferative capacity of splenic T-cells from CAC myPTEN−/− animals was reduced compared to myPTEN+/+ when total splenocytes were cultured ex vivo for 3 d in the presence of activating anti-CD3ϵ (). Consistently, analysis of flow cytometry data of intracellularly stained splenocytes revealed an increase in IL-10+ in CD8+ CTLs (Fig. S5B and Fig. S5C). In vivo data obtained from flow cytometry analysis of CFSE-labeled i.v. transferred OT-I CD8+ T-cells showed a reduction in percentage of OT-I T-cells as well as percentage of proliferating OT-I T-cells in spleens and mesenteric lymph nodes of tumor-bearing myPTEN−/− mice. This impairment of CD8+ T-cell proliferation was further substantiated by a higher MFI of CFSE in myPTEN−/− mice (). Additionally, a higher infiltration of F4/80 monocytes was found in tumors of myPTEN−/− compared to myPTEN+/+ mice (). Some of these cells were also positive for Arg1, while others were negative, regardless of genotype. Flow-cytometry analysis revealed an increase of CD80 and CD86 positive and negative F4/80 monocytes in myeloid PTEN-deficient mice. iNOS mRNA expression in cell suspensions of colon lamina propria and epithelium showed a decrease in cell from myPTEN−/− mice compared to WTs (data not shown). These findings indicated that the increased population of F4/80 monocytes in myPTEN−/− mice did not represent pro-inflammatory infiltrates.
Figure 5. Myeloid PTEN deficiency induces hypo-responsiveness in T-cells. (A) Quantification of flow cytometry analysis of (live, CD45+ CD3+) CD4+ T-helper cells and CD8+ CTLs in spleen (SPL), colon lamina propria (CLP) and colon epithelium (CEP) of CAC mice, values are expressed as % of live cells, n = 4–6, *p < 0.05; (B) quantification as in (A) of healthy animals, values are expressed as % of live cells, n = 4–5; (C) proliferation of total splenocytes of myPTEN+/+ and myPTEN−/− mice after ex vivo culture for 3 d in the presence of activating anti-CD3ϵ measured by the incorporation of radioactive 3H-thymidine, n = 8, *p < 0.05; (D) Proliferation of CFSE-labeled OT-I T-cells 3 d after i.v. transfer into myPTEN+/+ and myPTEN−/− CAC mice. Mice were immunized with LPS/Ova one day after i.v. transfer, proliferation was assessed by CFSE dilution and is shown as % OT-I T-cells in SPL and MLN, % proliferating OT-I T-cells and MFI of CFSE, n = 2–5, values were measured in triplicates, **p < 0.01, ***p < 0.001; (E) representative CD3-IHC and (F) F4/80 staining of paraffin-embedded sections of colon swiss rolls from myPTEN+/+ and myPTEN−/− CAC mice and quantification of positive stained areas per tumor area, values are expressed as % of tumor area, n = 15–17, **p < 0.01, ***p < 0.001.
T-cells from B16 myPTEN−/− mice show reduced killing in vitro
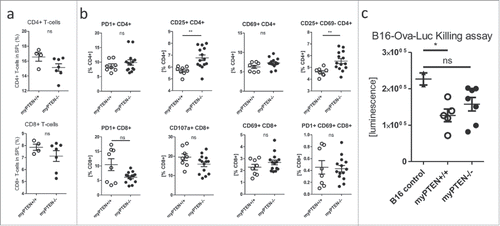
Analysis of CD4+ and CD8+ T-cells from myPTEN+/+ and myPTEN−/− mice injected with B16-Ova-Luc melanoma cells showed a slight, although not significant, reduction in CD4+ T-helper cells and CD8+ CTLs in spleens of myeloid PTEN-deficient mice (). Furthermore, a higher percentage of splenic CD4+ T-cells expressed CD25 () and a higher percentage of CD25+CD4+ T-cells from myPTEN−/− mice were negative for the activation marker CD69 (). In the CD8+ T-cell subset, we found that the percentage of activated PD-1+ CD8+ T-cells tended to be reduced in myPTEN−/− mice compared to WT controls after B16 inoculation. PD-1 is usually upregulated upon T-cell activation, enabling the negative feedback loop through inhibitory PD-L1 or PD-L2 binding.Citation12 CD107a is a marker for degranulation, as it is located in the membrane near sites of cytolytic granule release after membrane fusion.Citation24,25 CD107a was found to be only slightly, but not significantly, reduced in myPTEN−/− mice (). These results indicated that CD8+ T-cells were not sufficiently primed and activated for an appropriate antitumor response. To determine whether the observed lack of T-cell activation marker expression also had biological significance, we tested LPS-activated and Ova peptide-primed splenocytes in an in vitro assay for their cytolytic activity toward B16 Ova-melanoma cells (). While activated splenocytes from myPTEN+/+ led to a significant reduction in B16-Ova-Luc melanoma cells in vitro, myPTEN−/− splenocytes did not. These results suggest that PTEN-deficient dendritic cells did not efficiently prime T-cells, which resulted in decreased elimination of tumor cells in vitro.
Figure 6. Myeloid PTEN deficiency induces regulatory T-cells. (A) Quantification of flow cytometry analysis of (live, CD45+ CD3+) CD4+ T-helper and CD8+ CTLs in the spleens of myPTEN+/+ and myPTEN−/− mice 10 d after s.c. B16 melanoma injection, values are expressed as % of live cells, n = 4–7; (B) Quantification of flow cytometry analysis of activation markers PD-1, CD25, CD69 and CD107a in CD4+ T-helper and CD8+ CTLs in the spleens of myPTEN+/+ and myPTEN−/− mice 10 d after s.c. B16 melanoma injection, values are expressed as % of CD4+ or CD8+, respectively, n = 8–14, *p < 0.05, **p < 0.01; (C) in vitro killing of B16-Ova-Luc cells was assayed by measurement of remaining luciferase activity specific for transduced melanoma cells after co-culture for 1 h with LPS/SIINFEKL-primed splenocytes from B16-injected myPTEN+/+ or myPTEN−/− mice as in (A) and (B), respectively, values represent arbitrary luminescence values; n = 2–7, *p < 0.05, **p < 0.01.
Discussion
The PI3K-signaling pathway induces anti-inflammatory reactions in cells of myeloid origin. It is counter-regulated by PTEN, a lipid and protein phosphatase that acts at the plasma membrane as well as in the nucleus.Citation1 The precise role of this central pathway in the regulation of myeloid-cell development and function is not fully understood. We previously showed that the activation of the myeloid PI3K signaling pathway leads to a shift toward increased production of anti-inflammatory cytokines.Citation4,26 Furthermore, we also showed that the PI3K pathway upregulates the enzyme Arginase-1 in macrophages, which is secreted and exerts autocrine and paracrine immune-suppressing effects.Citation27 One of those effects is the suppression of effector T-cell responses resulting in amelioration of experimental autoimmune encephalitis as well as inflammatory arthritis in myeloid PTEN-deficient mice.Citation10,27,28 In contrast, the opposite effect was observed in lymphocytes. Here PTEN-deficiency led to hyper-proliferation in affected T-cells, auto-immunity (reviewed in referenceCitation29), reduced germinal center formation and impaired immunoglobulin class switching in B-cells.Citation30 Additional findings in innate immune cells suggest that inhibition or deletion of positive integrators in the PI3K-Akt-mTOR pathway in these cells has immune-stimulatory effects and the inhibition or deletion of negative regulators has immune-inhibitory effects (reviewed in referenceCitation31). In some cases, the effect of deletion of a signaling member depends on the cell type (TSC1-deletion in macrophages or DCsCitation31) or the affected isoform (Akt1- or Akt2-deficiency in macrophagesCitation32). Suppression of innate and adaptive immune responses, which might be beneficial for limiting tissue destruction and prolonging overall survival in models of acute infection and inflammation, might be detrimental during tumor development. To address the question of whether an anti-inflammatory phenotype exerted by cells of the innate immune system has an influence in cancer development and progression, we used two different tumor models in PTENfl/fl LysM cre (myPTEN−/−) mice: the endogenous CAC model and the orthotopically implanted B16-melanoma model. CAC myPTEN−/− mice showed an increase in tumor incidence as well as in tumor progression. Colon inflammation was similar between myPTEN−/− and myPTEN+/+ mice, as we could not find any differences in weight loss or colon length. Furthermore, the weight change was also similar in an acute model of colitis. Analysis of blood parameters showed no differences between the genotypes with the exception of the ratio of lymphocytes to granulocytes, which was decreased in myPTEN−/− mice in the CAC model compared to healthy controls. Myeloid PTEN-deficient mice showed increased mortality, which unfortunately precluded the analyses of severely affected mice. Surprisingly, this effect was not seen in female mice, indicating an impact of sex on the development of CAC in this protocol, probably due to increased stress in cages containing male animals.Citation33 To avoid any influence of the microbiome, which clearly has an impact on the development and course of inflammation associated diseases,Citation34-37 experiments with myPTEN−/− and myPTEN+/+ mice were performed in a littermate-controlled and co-housed way. Taken together, this indicated that the pathologic changes observed in the myPTEN−/− could be attributed to malfunctioning tumor immune-surveillance mechanisms. Indeed, in the second tumor model, where B16 melanoma cells were subcutaneously injected, myPTEN−/− mice also showed an increase in tumor load. This tumor model differed from CAC by an important parameter, namely immunogenicity. Due to two additional neo-antigens—chicken ovalbumin and firefly luciferase—the B16-melanoma cell line we used seemed to elicit an immune response, which led to increased rejection of tumors. Nevertheless, the average tumor load was higher in myeloid PTEN-deficient animals compared to controls. As observed in the CAC model, the ratio of lymphocytes to granulocytes in the B16 model was also decreased in myPTEN−/− mice, indicating that due to PTEN deficiency, hematopoiesis favored innate cell production to lymphocytes. To investigate the dysregulated tumor-immune surveillance in greater detail, we analyzed various innate and adaptive cell subsets. We found splenic CD11c+ cells as well as CD11c+CD8α+ cells to be increased in myPTEN−/− animals in the CAC as well as in the B16 melanoma model. This finding was independent of disease status, as healthy control animals also showed an increase in this specific innate cell subset. This increase was observed just in CD11c+ cells but not in CD11c− cells, probably due to a higher dependence on a single cytokine (namely Flt3L) which is indispensable for DC development and requires downstream signaling through the PI3K-Akt-mTOR axis.Citation7 CD8α+ CD11c+ DCs are professional APCs in the spleen capable of cross-presenting exogenous antigens to CD8+ cytotoxic T-cells,Citation22,38-40 thereby activating or preventing T-cell-mediated responses in a context-dependent manner (cross-priming vs. cross-tolerization).Citation38 This cell population was shown to be more efficient at phagocytosis than CD8α− cellsCitation41-43 and at inducing regulatory T-cells (Tregs).Citation40 An increase in CD11c+CD8α+ cells in myeloid PTEN-deficient animals has already been shown.Citation7 Although the CD11c-cre mouse was used in this study, the LysM-cre mouse also targeted splenic APC, as shown by Gwendalyn Randolph's group and others.Citation20,44 Development of splenic CD8α+DCs depends on the hematopoietin Flt3L, which acts on its receptor, Flt3, to activate downstream signaling via PI3K.Citation7 In vitro development of CD8α+DCs was further shown to be abrogated by rapamycin treatment, indicating that signaling is mTORC-dependent, which is downstream of PI3K. We were interested in the immune regulatory properties of APCs in the spleen. Since recent data showed regulation of the immune inhibitory ligand PD-L1 by signaling downstream of PI3K,Citation11,13 we analyzed the expression of PD-1 ligands, PD-L1 and PD-L2, on splenic APCs that were dependent on PTEN. PD-L1 and PD-L2-expressing professional APCS (CD11c+MHCII+) were strongly and significantly increased in myPTEN−/− mice compared to myPTEN+/+ littermates. Some myeloid cells expressing PD-L1 or PD-L2 were also positive for CD11b+. When we further analyzed the expression of these inhibitory ligands on CD8α+DCs, we found that these cells expressed PD-L1 and PD-L2. Both of these inhibitory ligands were shown to bind PD-1 on T-cells and inhibit proliferation and cytokine production at low antigen concentrations when CD28-mediated co-stimulation was insufficient. Binding of PD-Ls to PD-1 induced cell cycle arrest in T-cells and phosphorylation of SHP-2.Citation19 Selective PD-L1 upregulation was recently shown to be dependent on HIF1α expression and binding to HRE sequences in the PD-L1 promotor region in a MDSC cell line.Citation13 An overall increase in splenic CD8α+DCs expressing the inhibitory ligands PD-L1 and PD-L2 therefore indicates an increase in self-tolerance, resulting in attenuation of tumor-immune surveillance in the CAC model. In the B16 melanoma model, we did not observe an increase in PD-L1 or PD-L2 expressing myeloid cells, but an increase of PD-L1 or PD-L2 expressing cells was seen in myPTEN−/− mice, which did not reject tumors. This led us to suggest a correlation between lack of tumor rejection and higher levels of PD-L expression. This tendency of increased PD-L-expressing DCs was only seen in myPTEN−/− mice but not in myPTEN+/+ animals.
The relative increase in CD8α+DCs was further validated at the transcriptional level. Analysis of mRNA-array data revealed that transcription factors important for CD8α+DC development and function were upregulated in CD11c+ cells from myPTEN−/− mice. The most highly upregulated TF in myPTEN−/− cells was Batf3 that specifically drives CD8α+DC development.Citation22 Pathway enrichment analysis showed an increase in genes involved in PPARγ signaling, glycerolipid metabolism and cell adhesion. Furthermore FC-γ receptors 1 (high affinity) and 4 (low affinity), complement components, MMP9 and IL-10, were all upregulated thereby affecting pathways involved in systemic lupus erythematosus, asthma, prion diseases and prostate-cancer development. Pathways affected by downregulated genes mostly contained the co-stimulatory molecules CD86 and CD80 as well as the DC activating molecule CD40, ICOS-ligand and Fas. Most interestingly, IL-12b (IL12p40) representing the shared binding partner of IL-12a and IL-23, was downregulated. Comparison of the gene expression in CD11b+ and CD11c+ cells from CAC mice revealed that, despite both populations being deficient in PTEN, the transcriptional effect was quite different in functionally related cells. Furthermore, among the most upregulated genes in CD11c+ cells from myPTEN−/− mice were S100A8 (3.85-fold) and S100A9 (3.98-fold). S100A8 and S100A9 together form heterodimers and were shown to bind TLR4 acting as DAMPs.Citation45 Gabrilovich and co-workers showed that overexpression of S100A9 blocks DC maturation and induces MDSC development.Citation46 Mice lacking S100A9 reject implanted tumors quite efficiently, which could be reversed by transplantation of WT MDSCs.Citation46 Co-stimulatory molecules like CD80 and CD86 as well as CD40, the most prominent signal for DC maturation, were downregulated in myPTEN−/− CD11c+ cells. It is noteworthy that PD-L1 and PD-L2 were not upregulated at the transcriptional level. These data supported the finding that CD8α+DC development was increased in myPTEN−/− mice and that these DCs were not very efficient inducers of T-cell responses due to a decrease in co-stimulatory molecules and IL-12b.
Since we found that immune-checkpoint inhibitors on APCs increased in myeloid-PTEN deficient animals, we were interested in the T-cell responses in these mice. By analyzing numbers of CD4+ and CD8+ T-cells, we found only CD8+ T-cells significantly increased in the spleens of myPTEN−/− CAC mice, although we could observe a trend of increased CD4+ T-cells in the colon of myPTEN−/− CAC mice. B- and NK-cells did not differ in number between genotypes after CAC induction. When comparing healthy mice, we observed a reduction in T-cells in the spleens of myPTEN−/− mice compared to myPTEN+/+. Surprisingly, in colon lamina propria and colon epithelium there were no striking differences in numbers of leukocyte populations between healthy and CAC animals, only CD4+ T-cells seemed to be increased after CAC development, leading us to suggest that there was a flux of, at least some, T-helper cells from the spleen to the tumor-bearing colon or local expansion. This increase in colonic CD4+ T-cells was more pronounced in myPTEN−/− than in myPTEN+/+ mice, which was also seen in IHC staining for CD3 of colon swiss rolls. Analysis of T-cells in an ex vivo culture of whole splenocytes in the presence of activating anti-CD3ϵ antibody revealed a decreased proliferative potential in T-cells from myPTEN−/− mice, indicating that inhibitory dendritic cells present in the spleen indeed block T-cell proliferation. This finding coincided with an increase in IL-10+ CD8+ splenic T-cells in myPTEN−/− mice. Flow cytometric analysis of intracellular T-helper cytokines revealed a slight increase in IL-10+ and IFNγ+ CD4+ T-helper cells in myPTEN−/− animals. The increase of splenic CD8+ T-cells in myPTEN−/− mice could be due to increased proliferation or a block in homing to other organs, but CD8+ CTLs were not reduced in MLN, CLP or CEP of myPTEN−/− animals, indicating that the latter was not the case. Nevertheless, CD8+ T-cells showed a slight, but not significant increase in IL-2 production as well as a trend toward downregulation of the gut-homing receptors CCR9 and α4β7-integrin (data not shown). The observed increase in CD8α+ DCs in the spleens of myPTEN−/− mice would also explain the increase of splenic CD8+ T-cells and IFNγ+ CD4+ T-cells as especially CD8α+ DCs produce high levels of bioactive IL-12p70, which induces Th1 and CTL responses after CpG detection via TLRs.Citation40 However, we did not find any difference in the ability to produce IL-12p40 ex vivo upon CpG stimulation between myPTEN−/− mice and their corresponding controls (data not shown). In an in vivo approach, we transferred ovalbumin-specific CD8+ T-cells into CAC mice and immunized them on the following day. Flow cytometry analysis showed an impairment of T-cell proliferation in myPTEN−/− animals, indicating that DCs capable of cross-presentation inhibit CD8+ T-cell activation. Taken together, these findings pointed toward a blockade of CD8+ CTL activation by engagement of PD-L1 and PD-L2 on CD8α+ DCs. This would consequently cause a higher tumor burden, as deficiencies in development or function of CTLs or Th1-cells results in increased cancer development and mice with combined immunodeficiencies in T- and NK-cells are shown to be more susceptible to cancer development.Citation47
In the B16 melanoma model, myPTEN−/− mice showed slightly reduced levels of T-cells in the spleen. Furthermore, a higher percentage of CD4+ T-helper cells expressed CD25, which is usually considered a marker for Tregs.Citation48 The percentage of CD25+CD69− cells was also increased in CD4+ T-helper cells of myPTEN−/−. CD69 is a C-type lectin usually upregulated in T- and NK-cells immediately after activation.Citation49-51 CD69− CD4+ T-cells were shown to exhibit defects in homing to the bone marrow, thereby providing less help to B-cells for plasma-cell differentiation.Citation52 Expression of CD69—at least in vitro—is dependent on antigen exposure.Citation51 Therefore, an increase in CD4+ CD25+ CD69− in myPTEN−/− animals indicated that these T-helper cells lacked activation, were still naive and might even have a regulatory phenotype.
As a secondary lymphoid organ, the spleen serves primarily as a filter for the blood by removing aged erythrocytes and recycling their hemoglobin, as well as removing opsonized bacteria and generating immune responses. It is also a reservoir for blood and monocytes in case of injury and harbors B- and T-cells. Enlargements of the spleen are observed in leukemia and cancer patients as well as in various infectious and congenital diseases. In early mouse and rat models, splenectomy increased cancer risk in various settings, indicating that the spleen has a protective function during tumor development.Citation53-55 A recent follow-up study of 8,149 veterans who underwent splenectomy for up to 27 y found a 1.51-fold increased risk for any cancer, with a 1.33-fold increased risk for developing colon cancer and a 5.2-fold increased risk for any leukemia. Further, splenectomized patients showed an increased risk for cancer death.Citation56
In conclusion we could show for the first time that genetic deletion of PTEN in myeloid cells specifically increased splenic CD8α dendritic cells, which upregulated the immune checkpoint inhibitors PDL-1 and PDL-2 thereby attenuating T-cell mediated tumor-immune surveillance. Our study indicated that PI3K inhibitors, which are currently being tested for cancer treatment, might have further beneficial immune-modulating effects beyond tumor-cell targeting.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1164918_s02.zip
Download Zip (858.6 KB)Acknowledgments
We thank Dr. Aner Gurvitz and Dr. Rona Strawbridge for a careful and thorough review of the manuscript.
Funding
This study was funded by the Austrian Science Fund projects P24802 (to GS) and SFB-F54 (to GS and JS).
References
- Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002; 296:1655-7; PMID:12040186; http://dx.doi.org/10.1126/science.296.5573.1655
- Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol 2008; 180:4218-26; PMID:18322234; http://dx.doi.org/10.4049/jimmunol.180.6.4218
- Gunzl P, Bauer K, Hainzl E, Matt U, Dillinger B, Mahr B, Knapp S, Binder BR, Schabbauer G. Anti-inflammatory properties of the PI3K pathway are mediated by IL-10/DUSP regulation. J Leukoc Biol 2010; 88:1259-69; PMID:20884649; http://dx.doi.org/10.1189/jlb.0110001
- Schabbauer G, Matt U, Gunzl P, Warszawska J, Furtner T, Hainzl E, Elbau I, Mesteri I, Doninger B, Binder BR et al. Myeloid PTEN promotes inflammation but impairs bactericidal activities during murine pneumococcal pneumonia. J Immunol 2010; 185:468-76; PMID:20505137; http://dx.doi.org/10.4049/jimmunol.0902221
- Yue S, Rao J, Zhu J, Busuttil RW, Kupiec-Weglinski JW, Lu L, Wang X, Zhai Y. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J Immunol 2014; 192:5343-53; PMID:24771857; http://dx.doi.org/10.4049/jimmunol.1400280
- Miething C, Scuoppo C, Bosbach B, Appelmann I, Nakitandwe J, Ma J, Wu G, Lintault L, Auer M, Premsrirut PK et al. PTEN action in leukaemia dictated by the tissue microenvironment. Nature 2014; 510:402-6; PMID:24805236; http://dx.doi.org/10.1038/nature13239
- Sathaliyawala T, O'Gorman WE, Greter M, Bogunovic M, Konjufca V, Hou ZE, Nolan GP, Miller MJ, Merad M, Reizis B. Mammalian target of rapamycin controls dendritic cell development downstream of Flt3 ligand signaling. Immunity 2010; 33:597-606; PMID:20933441; http://dx.doi.org/10.1016/j.immuni.2010.09.012
- Wang Y, Chi H. mTOR Signaling and Dendritic Cell Biology. J Immunol Clin Res 2014.
- Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008; 29:565-77; PMID:18848473; http://dx.doi.org/10.1016/j.immuni.2008.08.012
- Sahin E, Brunner JS, Kral JB, Kuttke M, Hanzl L, Datler H, Paar H, Neuwinger N, Saferding V, Zinser E et al. Loss of Phosphatase and Tensin Homolog in APCs Impedes Th17-Mediated Autoimmune Encephalomyelitis. J Immunol 2015; PMID:26246144; http://dx.doi.org/10.4049/jimmunol.1402511
- Rosborough B.R, Raich-Regue DM, Matta BM, Lee K, Gan B, DePinho RA, Hackstein H, Boothby M, Turnquist HR, Thomson AW. Murine dendritic cell rapamycin-resistant and rictor-independent mTOR controls IL-10, B7-H1, and regulatory T-cell induction. Blood 2013; 121(18):3619-30; PMID:23444404; http://dx.doi.org/10.1182/blood-2012-08-448290
- Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A 2003; 100:5336-41; PMID:12697896; http://dx.doi.org/10.1073/pnas.0931259100
- Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 2014; 211:781-90; PMID:24778419; http://dx.doi.org/10.1084/jem.20131916
- Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity 2014; 40:642-56; PMID:24837101; http://dx.doi.org/10.1016/j.immuni.2014.04.016
- Crncec I, Pathria P, Svinka J, Eferl R. Induction of colorectal cancer in mice and histomorphometric evaluation of tumors. Methods Mol Biol 2015; 1267:145-64; PMID:25636468; http://dx.doi.org/10.1007/978-1-4939-2297-0_7
- Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 2009; 37:e45; PMID:19237396; http://dx.doi.org/10.1093/nar/gkp045
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/10.1038/nprot.2008.211
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13; PMID:19033363; http://dx.doi.org/10.1093/nar/gkn923
- Latchman Y, Wood C, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long A, Brown J, Nunes R et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001; 2(3):261-8; PMID:11224527; http://dx.doi.org/10.1038/85330
- Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J Exp Med 2008; 205:2839-50; PMID:18981237; http://dx.doi.org/10.1084/jem.20081430
- Zhang R, Ito S, Nishio N, Cheng Z, Suzuki H, Isobe KI. Dextran sulphate sodium increases splenic Gr1(+)CD11b(+) cells which accelerate recovery from colitis following intravenous transplantation. Clin Exp Immunol 2011; 164:417-27; PMID:21413942; http://dx.doi.org/10.1111/j.1365-2249.2011.04374.x
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, ER U, Diamond MS et al. Batf3 Deficiency Reveals a Critical Role for CD8a+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008; 322(5904):1097-100; PMID:19008445; http://dx.doi.org/10.1126/science.1164206
- Carrasco A, Mane J, Santaolalla R, Pedrosa E, Mallolas J, Loren V, Fernandez M, Fernandez-Banares F, Rosinach M, Loras C et al. Comparison of lymphocyte isolation methods for endoscopic biopsy specimens from the colonic mucosa. J Immunol Methods 2013; 389:29-37; PMID:23279944; http://dx.doi.org/10.1016/j.jim.2012.12.006
- Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 2003; 281:65-78; PMID:14580882; http://dx.doi.org/10.1016/S0022-1759(03)00265-5
- Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods 2004; 294:15-22; PMID:15604012; http://dx.doi.org/10.1016/j.jim.2004.08.008
- Schabbauer G, Luyendyk J, Crozat K, Jiang Z, Mackman N, Bahram S, Georgel P. TLR4/CD14-mediated PI3K activation is an essential component of interferon-dependent VSV resistance in macrophages. Mol Immunol 2008; 45:2790-6; PMID:18339426; http://dx.doi.org/10.1016/j.molimm.2008.02.001
- Sahin E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal AM, Chen L, Cheng P, Hoesel B, Einwallner E et al. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol 2014; 193:1717-27; PMID:25015834; http://dx.doi.org/10.4049/jimmunol.1302167
- Bluml S, Sahin E, Saferding V, Goncalves-Alves E, Hainzl E, Niederreiter B, Hladik A, Lohmeyer T, Brunner JS, Bonelli M et al. Phosphatase and tensin homolog (PTEN) in antigen-presenting cells controls Th17-mediated autoimmune arthritis. Arthritis Res Ther 2015; 17:230; PMID:26307404; http://dx.doi.org/10.1186/s13075-015-0742-y
- Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene 2008; 27:5398-415; PMID:18794876; http://dx.doi.org/10.1038/onc.2008.238
- Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, Sasaki T, Mak TW, Nakano T. Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med 2003; 197:657-67; PMID:12615906; http://dx.doi.org/10.1084/jem.20021101
- Weichhart T, Hengstschlager M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol 2015; 15:599-614; PMID:26403194; http://dx.doi.org/10.1038/nri3901
- Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, Ieronymaki E, Androulidaki A, Venihaki M, Margioris AN et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A 2012; 109:9517-22; PMID:22647600; http://dx.doi.org/10.1073/pnas.1119038109
- Perse M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol 2012; 2012:718617; PMID:22665990; http://dx.doi.org/10.1155/2012/718617
- Zhan Y, Chen PJ, Sadler WD, Wang F, Poe S, Nunez G, Eaton KA, Chen GY. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res 2013; 73:7199-210; PMID:24165160; http://dx.doi.org/10.1158/0008-5472.CAN-13-0827
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature 2013; 494:116-20; PMID:23334413; http://dx.doi.org/10.1038/nature11809
- Shim JO. Gut microbiota in inflammatory bowel disease. Pediatr Gastroenterol Hepatol Nutr 2013; 16:17-21; PMID:24010101; http://dx.doi.org/10.5223/pghn.2013.16.1.17
- Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Bohm J, Brunetti F, Habermann N et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol 2014; 10:766; PMID:25432777; http://dx.doi.org/10.15252/msb.20145645
- Belz GT, Behrens GMN, Smith CM, Miller JFAP, Jones C, Lejon K, Fathman CG, Mueller SN, Shortman K, Carbone FR et al. The CD8 + Dendritic Cell Is Responsible for Inducing Peripheral Self-Tolerance to Tissue-associated Antigens. J Exp Med 2002; 196:1099-104; PMID:12391021; http://dx.doi.org/10.1084/jem.20020861
- Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol 2013; 13:566-77; PMID:23827956; http://dx.doi.org/10.1038/nri3477
- Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev 2010; 234(1):18-31; PMID:20193009; http://dx.doi.org/10.1111/j.0105-2896.2009.00870.x
- Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol 2015; 33:445-74; PMID:25622193; http://dx.doi.org/10.1146/annurev-immunol-032414-112043
- Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med 2002; 196:1091-7; PMID:12391020; http://dx.doi.org/10.1084/jem.20021215
- Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y, Takahara K, Steinman RM, Inaba K. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med 2002; 195:1289-302; PMID:12021309; http://dx.doi.org/10.1084/jem.20020161
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, I F. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999; 8(4):265-77; PMID:10621974
- Ehrchen JM, Sunderkotter C, Foell D, Vogl T, Roth J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol 2009; 86:557-66; PMID:19451397; http://dx.doi.org/10.1189/jlb.1008647
- Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 2008; 205:2235-49; PMID:18809714; http://dx.doi.org/10.1084/jem.20080132
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Xu D, Liu H, Komai-Koma M, Campbell C, McSharry C, Alexander J, Liew FY. CD4+CD25+ regulatory T cells suppress differentiation and functions of Th1 and Th2 cells, Leishmania major infection, and colitis in mice. J Immunol 2003; 170:394-9; PMID:12496424; http://dx.doi.org/10.4049/jimmunol.170.1.394
- Borrego F, Robertson MJ, Ritz J, Pena J, Solana R. CD69 is a stimulatory receptor for natural killer cell and its cytotoxic effect is blocked by CD94 inhibitory receptor. Immunology 1999; 97:159-65; PMID:10447727; http://dx.doi.org/10.1046/j.1365-2567.1999.00738.x
- Ziegler SF, Ramsdell F, Alderson MR. The activation antigen CD69. Stem Cells 1994; 12:456-65; PMID:7804122; http://dx.doi.org/10.1002/stem.5530120502
- Fazekas de St Groth B, Smith A, Higgins C. T cell activation: in vivo veritas. Immunol Cell Biol 2004; 82(3):260-8; PMID:15186257; http://dx.doi.org/10.1111/j.0818-9641.2004.01243.x
- Shinoda K, Tokoyoda K, Hanazawa A, Hayashizaki K, Zehentmeier S, Hosokawa H, Iwamura C, Koseki H, Tumes DJ, Radbruch A et al. Type II membrane protein CD69 regulates the formation of resting T-helper memory. Proc Natl Acad Sci U S A 2012; 109:7409-14; PMID:22474373; http://dx.doi.org/10.1073/pnas.1118539109
- Hull CC, Galloway P, Gordon N, Gerson SL, Hawkins N, Stellato TA. Splenectomy and the induction of murine colon cancer. Arch Surg 1988; 123:462-4; PMID:3348737; http://dx.doi.org/10.1001/archsurg.1988.01400280072012
- Yamagishi H, Pellis NR, Kahan BD. Effect of splenectomy upon tumor growth: characterization of splenic tumor-enhancing cells in vivo. Surgery 1980; 87:655-61; PMID:6445603
- Soda K, Kawakami M, Takagi S, Kashii A, Miyata M. Splenectomy before tumor inoculation prolongs the survival time of cachectic mice. Cancer Immunol Immunother 1995; 41:203-9; PMID:7489562; http://dx.doi.org/10.1007/BF01516994
- Kristinsson SY, Gridley G, Hoover RN, Check D, Landgren O. Long-term risks after splenectomy among 8,149 cancer-free American veterans: a cohort study with up to 27 years follow-up. Haematologica 2014; 99:392-8; PMID:24056815; http://dx.doi.org/10.3324/haematol.2013.092460