
ABSTRACT
Several studies have demonstrated that NF-κB activation is common in lung cancer; however, the mechanistic links between NF-κB signaling and tumorigenesis remain to be fully elucidated. We investigated the function of NF-κB signaling in epidermal growth factor receptor (EGFR)-mutant lung tumors using a transgenic mouse model with doxycycline (dox)-inducible expression of oncogenic EGFR in the lung epithelium with or without a dominant inhibitor of NF-κB signaling. NF-κB inhibition resulted in a significant reduction in tumor burden in both EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant tumors. However, NF-κB inhibition did not alter epithelial cell survival in vitro or in vivo, and no changes were detected in activation of EGFR downstream signaling pathways. Instead, we observed an influx of inflammatory cells (macrophages and neutrophils) in the lungs of mice with oncogenic EGFR expression that was blocked in the setting of NF-κB inhibition. To investigate whether inflammatory cells play a role in promoting EGFR-mutant lung tumors, we depleted macrophages and neutrophils during tumorigenesis and found that neutrophil depletion had no effect on tumor formation, but macrophage depletion caused a significant reduction in tumor burden. Together, these data suggest that epithelial NF-κB signaling supports carcinogenesis in a non-cell autonomous manner in EGFR-mutant tumors through recruitment of pro-tumorigenic macrophages.
KEYWORDS:
Abbreviations
| BAL | = | Bronchoalveolar lavage |
| CCSP | = | Clara cell secretory protein |
| CT | = | Computed tomography |
| DN-IκB | = | Dominant negative IκBα |
| Dox | = | Doxycycline |
| EGFR | = | Epidermal growth factor receptor |
| EGFRL/T | = | EGFRL858R+T790M |
| EV | = | Empty vector |
| IP | = | Intraperitoneal |
| IT | = | Intratracheal |
| RT-PCR | = | Reverse transcriptase polymerase chain reaction |
| TKI | = | Tyrosine kinase inhibitor |
| WT | = | Wild-type |
Introduction
EGFR is a receptor tyrosine kinase that is mutated in approximately 15% of lung adenocarcinomas.Citation1,2 Mutations in EGFR destabilize the auto-inhibitory conformation of the kinase domain,Citation3 resulting in constitutive EGFR kinase activation as well as constitutive activation of downstream pathways, such as PI3K, MAPK, and JAK/STAT. Targeted therapies, such as TKIs, have been developed to treat lung cancer patients with EGFR-mutant tumors. However, these therapies have not significantly impacted overall patient survival because the tumors develop resistance, most commonly through the EGFR T790M mutation.Citation4,5 This lack of effective therapy for long-term survival demonstrates the need for identification of new pathways that can be targeted alone or in combination with current treatment methods.
NF-κB is a transcription factor family that regulates genes involved in a variety of processes, including cytokines, chemokines, receptors, adhesion molecules, and enzymes, as well as cell cycle, apoptosis, and survival regulators. NF-κB family members exist as dimers sequestered in the cytoplasm by inhibitor molecules or IκBs. When the pathway is activated, IκBs are phosphorylated and undergo rapid degradation by the proteasome, releasing NF-κB dimers to translocate into the nucleus and bind enhancer motifs found in the promoters and introns of target genes. NF-κB can be activated through either the classical or alternative pathways. Classical NF-κB signaling is associated with nuclear localization of a heterodimer consisting of p65 and p50, while alternative NF-κB pathway signaling involves p52 and RelB.
In lung cancer, NF-κB activation is associated with a poor prognosis.Citation6 High levels of nuclear p65 have been observed in lung cancer specimens relative to normal tissue, with increasing levels of nuclear p65 in advanced stage adenocarcinomas.Citation7 Lung adenocarcinoma precursor lesions also have increased nuclear p65 staining that progresses in intensity with increasing histological severity,Citation7,8 suggesting that NF-κB signaling is activated early in tumor formation. NF-κB activation in lung tumors is associated with mutations in common lung cancer driver genes, including KRAS and EGFR.Citation7 In KRAS-mediated lung tumorigenesis, as well as in mouse models of chemical carcinogen-induced lung cancer, NF-κB plays an important role in tumorigenesis.Citation9-12 EGFR signaling is known to activate NF-κB in a variety of tumor types,Citation13,14 and NF-κB signaling has been shown to facilitate resistance to EGFR inhibitor therapies in lung cancer.Citation15-18 However, the importance of the NF-κB pathway during oncogenic EGFR-driven lung tumorigenesis has not been examined.
To investigate the role of NF-κB signaling in EGFR-mutant lung tumors, we inhibited NF-κB signaling in mouse models of lung tumorigenesis by inducible expression of a dominant NF-κB inhibitor in the same epithelial cell population that expressed mutant EGFR. In the setting of NF-κB inhibition, we observed a dramatic reduction in lung tumor formation and a decrease in lung inflammation without changes in EGFR-regulated downstream signaling pathways. Together, these studies demonstrate that NF-κB-dependent signaling is important for generating a pro-tumorigenic inflammatory microenvironment in EGFR-mutant tumors.
Results
Inhibition of epithelial NF-κB signaling reduces EGFRL858R-mediated tumorigenesis
To investigate the requirement for epithelial NF-κB signaling in EGFR-driven lung tumor formation, we developed a model in which NF-κB signaling could be inhibited in vivo specifically in airway epithelial cells expressing oncogenic EGFR. In this model, transgenic mice harbor a dox-inducible human EGFRL858R construct and the Clara cell secretory protein (CCSP) promoter-driven reverse tetracycline transactivator (CCSP-rtTA; tet-O-EGFRL858R) so that EGFRL858R is expressed specifically in the airway epithelium when given dox (designated EGFRL858R mice).Citation19 To inhibit epithelial NF-κB signaling in the lungs of these mice, EGFRL858R mice were mated to transgenic mice that express a dox-inducible dominant negative IκBα (DN-IκB).Citation20 This DN-IκB is unable to be phosphorylated and degraded, thereby blocking translocation of the NF-κB heterodimers into the nucleus and preventing target gene transcription. Upon dox administration, these triple transgenic mice (designated EGFRL858R DN-IκB mice) express EGFRL858R with simultaneous inhibition of NF-κB signaling in airway epithelial cells.
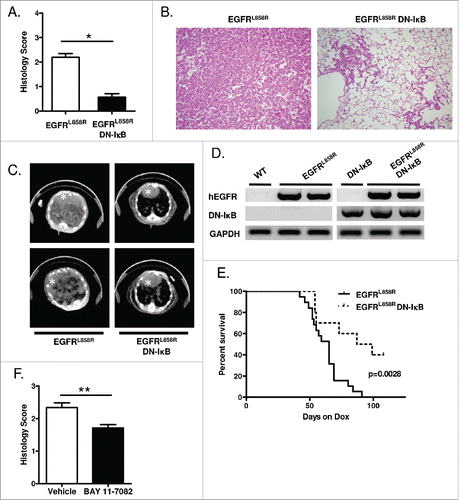
To assess the effect of NF-κB inhibition on EGFR-mediated tumor formation, mice were administered dox for 5 weeks, and tumor burden was assessed by histological scoring of lung sections. After 5 weeks of dox treatment, EGFRL858R DN-IκB mice demonstrated a marked decrease in lung tumor burden compared to EGFRL858R mice (). In addition, fewer tumors were observed on computed tomography (CT) scan images from EGFRL858R DN-IκB mice compared to EGFRL858R mice (). Transgene expression was validated in these mice using reverse transcriptase PCR (RT-PCR) to measure expression of human EGFRL858R as well as DN-IκB in the lungs of mice administered dox for 5 weeks (). To further investigate the impact of reduced tumor formation in EGFRL858R DN-IκB mice, we performed a survival analysis, which revealed that EGFRL858R DN-IκB mice had significantly prolonged survival compared to EGFRL858R mice following dox treatment (). To validate these findings using a different method of NF-κB inhibition, we employed BAY 11–7082, an inhibitor of IκBα phosphorylation.Citation21 EGFRL858R mice received intraperitoneal (IP) injections of BAY 11–7082 or vehicle control from the time of dox initiation. After 6 weeks of dox administration, BAY 11–7082 treatment led to a significant reduction in tumor burden (). Together, these data show that epithelial NF-κB inhibition can reduce tumor burden and prolong survival, implicating epithelial NF-κB signaling as a critical factor for EGFR-mediated tumorigenesis.
Figure 1. Epithelial NF-κB inhibition reduces EGFRL858R-mediated tumorigenesis. (A) Histological scoring of tumor burden, (B) representative H&E-stained lung sections (20x magnification), and (C) representative CT images from EGFRL858R and EGFRL858R DN-IκB mice administered dox for 5 weeks (n = 13–14 mice/group; *p < 0.0001). White asterisk designates the heart in CT images. (D) RT-PCR for EGFRL858R (hEGFR) and DN-IκB transgenes using mRNA isolated from lungs of WT, DN-IκB, EGFRL858R, and EGFRL858R DN-IκB mice administered dox for 5 weeks. (E) Kaplan–Meier survival curve of EGFRL858R and EGFRL858R DN-IκB mice after dox administration (for EGFRL858R n = 19 mice, for EGFRL858R DN-IκB n = 10; log rank test p = 0.0028). (F) Histological scoring of tumor burden in EGFRL858R mice administered dox and treated with BAY 11–7082 or vehicle control (n = 8/group; **p < 0.01).
Epithelial NF-κB inhibition does not affect epithelial cell survival or activation of EGFR-regulated signaling pathways
To investigate the function of epithelial NF-κB signaling in lung tumors, we tested whether NF-κB inhibition results in apoptosis of lung epithelial cells expressing oncogenic EGFR. In in vitro studies, we transfected wild-type (WT) and EGFRL858R-expressing immortalized human bronchial epithelial cells Citation22,23 with control empty vector (EV) or a myc-tagged DN-IκB expression vector and isolated nuclear and cytoplasmic protein extracts. Successful transfection was verified by western blotting for the myc-tagged transgene (). Reduced nuclear p65 was observed in DN-IκB-transfected cells, indicating reduced NF-κB activation (). However, no change in cell survival or increase in cleaved caspase-3 was observed as a result of NF-κB inhibition (). In vivo, we assessed cleaved caspase-3 by western blotting lysates from an enriched lung epithelial cell population generated from lungs of EGFRL858R and EGFRL858R DN-IκB mice administered dox for 5 weeks. Similar to in vitro studies, no differences in cleaved caspase-3 were detected (data not shown). In addition, we performed TUNEL staining on lung sections from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks (prior to appearance of tumors) and 5 weeks (when tumors were present). We observed no increase in TUNEL-positive cells in the lungs of EGFRL858R DN-IκB lungs compared to EGFRL858R at either time point (data not shown), suggesting that NF-κB activity is not required for survival of EGFRL858R-expressing cells.
Figure 2. NF-κB inhibition in airway epithelial cells does not alter apoptosis or EGFR-regulated signaling pathways. (A) Western blot of cytoplasmic and nuclear protein extracts from WT and EGFRL858R-mutant immortalized human bronchial epithelial cells transfected with DN-IκB expression vector or control empty vector. β-actin and TATA-binding protein (TBP) were probed as cytoplasmic and nuclear loading controls, respectively. (B) Western blot of EGFR-regulated signaling pathways using protein extracts from WT and EGFRL858R-mutant immortalized human bronchial epithelial cells transfected with DN-IκB expression vector or control empty vector. (C) Western blot and densitometry of EGFR-regulated signaling pathways (D) phospho-Akt, (E) phospho-ERK, and (F) phospho-STAT3 using lung lysates from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks.
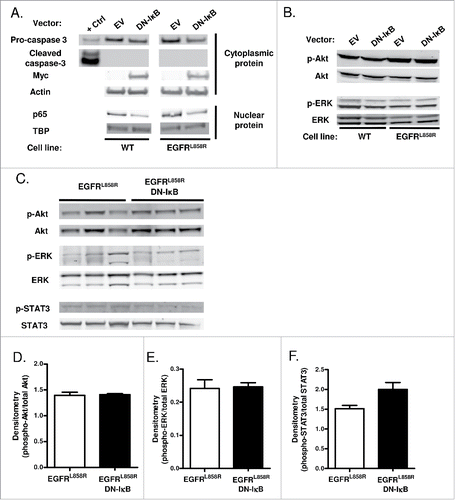
We also assessed activation of key EGFR-regulated downstream signaling pathways in the setting of NF-κB inhibition in vitro and in vivo. For in vitro measurements, we analyzed Akt, ERK, and STAT3 phosphorylation in lysates from WT and EGFRL858R-expressing immortalized human bronchial epithelial cells with and without NF-κB inhibition by western blotting. Phosphorylated STAT3 was below the level of detection, and no differences in Akt or ERK phosphorylation were observed (). Akt, ERK, and STAT3 signaling were also assessed in vivo by immunostaining on lung sections from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 5 weeks, but minimal phosphorylated Akt, ERK, and STAT3 was detected in both groups of mice. Western blots were performed using whole lung lysates obtained from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks as well as enriched lung epithelial cell lysates generated from mice administered dox for 5 weeks revealing no changes in Akt, ERK, or STAT3 phosphorylation as a result of NF-κB inhibition (; data not shown). Together, these data suggest that NF-κB inhibition does not affect downstream EGFR pathway signaling in EGFR-mutant cells.
NF-κB inhibition reduces macrophage and neutrophil influx into the lungs of mice expressing mutant EGFR
Since NF-κB is a central mediator of the inflammatory response, we next evaluated the effects of NF-κB inhibition on inflammatory cell recruitment in the lungs of mice expressing mutant EGFR. Total inflammatory cells were counted in bronchoalveolar lavages (BAL) from EGFRL858R, EGFRL858R DN-IκB, DN-IκB (CCSP-rtTA; tet-O-IκBα-DN), and WT mice treated with dox for 2 or 5 weeks. Prior to tumor formation, a ≥3-fold increase in BAL inflammatory cells was observed in EGFRL858R mice administered dox for 2 weeks compared to control groups, and this inflammatory cell influx was not present in EGFRL858R DN-IκB mice (). When tumors were present at 5 weeks of dox treatment, a ≥10-fold increase in inflammatory cells was observed in EGFRL858R mice compared to control groups (). This dramatic increase was abrogated by inhibition of NF-κB in the epithelium. Together, these data suggest that epithelial NF-κB signaling is important for recruiting inflammatory cells in this model, even prior to tumor formation.
Figure 3. Inhibition of epithelial NF-κB signaling decreases inflammatory cells in the lung during carcinogenesis. (A) Total BAL inflammatory cell numbers from WT, DN-IκB, EGFRL858R, and EGFRL858R DN-IκB mice administered dox for 2 weeks or 5 weeks (for 2 week n = 6–8/group, for 5 week n = 6–14; *p < 0.05, **p < 0.01, ***p < 0.001). (B) Quantitation and (C) representative lung photomicrographs (40x magnification) of CD68 macrophage immunostaining of lung sections from EGFRL858R and EGFRL858R DN-IκB mice administered dox for 2 weeks or 5 weeks (for 2 week n = 4/group, for 5 week n = 5/group; **p < 0.01). (D) Flow cytometry analysis of Ly6G+ cells from CD11b+CD45+ cells in lung single cell suspensions from WT, DN-IκB, EGFRL858R, and EGFRL858R DN-IκB mice administered dox for 2 weeks or 5 weeks (for 2 week n = 3–4/group; for 5 week n = 2–5/group; *p < 0.05). (E) Flow cytometry analysis for CD4+ and CD8+ T cells using lung single cell suspensions from WT, DN-IκB, EGFRL858R and EGFRL858R DN-IκB mice administered dox for 2 weeks (n = 3–4/group).
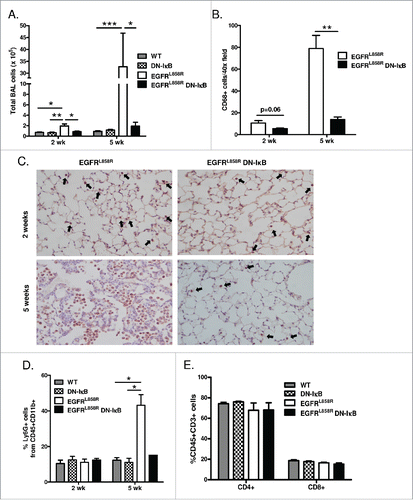
To identify inflammatory cell subpopulations altered as a result of epithelial NF-κB inhibition, we performed CD68 immunostaining and quantified CD68+ macrophages on lung sections from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks or 5 weeks. At both time points, lungs from EGFRL858R DN-IκB mice contained fewer macrophages compared to EGFRL858R mice (). To analyze changes in other lung inflammatory cell populations, we performed flow cytometry on lung single cell suspensions generated from EGFRL858R, EGFRL858R DN-IκB, DN-IκB, and WT mice. In mice administered dox for 5 weeks, NF-κB inhibition led to reduced neutrophils (CD45+CD11b+Ly6G+) in the lungs () but no differences in T lymphocyte populations (). Together, these data demonstrate that NF-κB inhibition results in reduced neutrophils when tumors are present and reduced macrophages throughout tumorigenesis.
NF-κB inhibition decreases lung tumors in mice expressing a TKI-resistant EGFR mutation
To assess whether epithelial NF-κB inhibition affects lung tumor burden and inflammatory cell numbers in a TKI-resistant model of EGFR-mediated tumorigenesis, we used transgenic mice with dox-inducible expression of human EGFRL858R+T790M (CCSP-rtTA; tet-O-EGFRL858R+T790M). Similar to the EGFRL858R model, these mice express EGFRL858R+T790M in the airway epithelium when given dox (designated EGFRL/T mice). However, the presence of the T790M mutation renders tumors in these mice insensitive to treatment with first generation TKIs erlotinib and gefitinib.Citation24 To inhibit epithelial NF-κB signaling in the lungs of these mice, EGFRL/T mice were mated to transgenic DN-IκB mice (designated EGFRL/T DN-IκB).
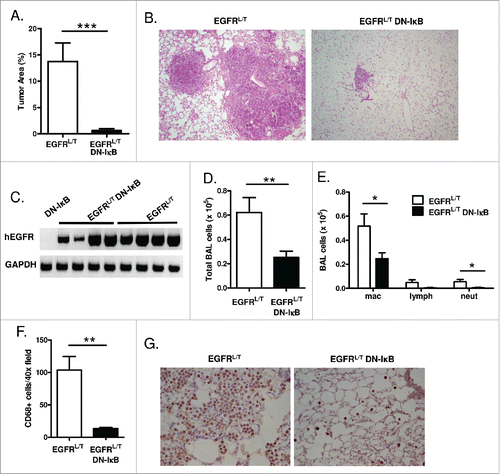
To determine the effect of epithelial NF-κB inhibition on EGFR TKI-resistant EGFRL/T tumors, mice were treated with dox for 8 weeks, and tumor burden was assessed histologically. Similar to the EGFRL858R model, EGFRL/T DN-IκB mice demonstrated a dramatic reduction in lung tumor burden compared to EGFRL/T mice (). RT-PCR for EGFR transgene expression verified that EGFRL/T DN-IκB mice expressed the EGFR transgene after 8 weeks of dox administration (). In addition to decreased tumor burden, EGFRL/T DN-IκB mice had significantly fewer inflammatory cells in BAL fluid (). Inflammatory cell subpopulations were analyzed on BAL cytospins as well as by CD68 staining for macrophages on lung sections from EGFRL/T and EGFRL/T DN-IκB mice, revealing a significant reduction in macrophages and neutrophils when NF-κB signaling was inhibited (). These data were consistent with our findings in the EGFRL858R model and suggest that epithelial NF-κB signaling functions similarly in EGFR TKI-sensitive and TKI-resistant tumors.
Figure 4. NF-κB inhibition reduces tumor burden and inflammatory cells in the TKI-resistant EGFRL858R+T790M lung carcinogenesis model. (A) Histological quantitation of lung tumor burden and (B) representative images from H&E-stained lung sections (10x magnification) from EGFRL/T and EGFRL/T DN-IκB mice treated with dox for 8 weeks (n = 16 mice/group; ***p < 0.001). (C) RT-PCR for EGFRL/T transgene (hEGFR) expression using mRNA isolated from lungs of EGFRL/T and EGFRL/T DN-IκB mice treated with dox for 8 weeks. (D) Total BAL inflammatory cells and (E) differential cell counts from EGFRL/T and EGFRL/T DN-IκB mice treated with dox for 8 weeks (n = 16 mice/group; **p < 0.01, *p < 0.05). (F) Quantification and (G) representative lung photomicrographs (40x magnification) of CD68 macrophage immunostaining of lung sections from EGFRL/T and EGFRL/T DN-IκB mice treated with dox for 8 weeks (n = 6–8/group; **p < 0.01).
NF-κB signaling promotes macrophage and neutrophil chemotaxis
To investigate whether epithelial NF-κB inhibition affects macrophage and neutrophil recruitment to the lungs, we measured chemotaxis induced by BAL fluid or lung lysates using a modified Boyden chamber assay. Significantly more neutrophils migrated toward BAL fluid from EGFRL858R mice than BAL fluid from EGFRL858R DN-IκB mice (). To identify NF-κB-regulated mediator(s) responsible for neutrophil chemotaxis in the setting of EGFR-mutant lung tumors, we measured mRNA expression of neutrophil chemokines by quantitative real-time PCR in lungs of mice treated with dox for 5 weeks, the time point at which we observed a significant difference in lung neutrophil numbers between EGFRL858R and EGFRL858R DN-IκB mice. KC and MIP-2 mRNA levels were reduced in EGFRL858R DN-IκB lungs compared to EGFRL858R (). KC and MIP-2 protein was also reduced in BAL from EGFRL858R and EGFRL858R DN-IκB mice (), suggesting that NF-κB signaling promotes neutrophil migration to the lungs through regulation of CXC chemokines.
Figure 5. NF-κB signaling promotes macrophage and neutrophil migration. (A) In vitro Boyden chamber chemotaxis assay for neutrophils using BAL fluid from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 5 weeks (n = 8/group; ***p < 0.001). (B) Quantitative real-time PCR for KC and MIP-2 using mRNA from lungs of EGFRL858R and EGFRL858R DN-IκB mice administered dox for 5 weeks (n = 5–9/group; *p < 0.05; all EGFRL858R DN-IκB MIP-2 measurements below threshold). (C) KC and MIP-2 ELISA measurements using BAL fluid from WT, DN-IκB, EGFRL858R, and EGFRL858R DN-IκB mice treated with dox for 5 weeks (n = 4–10 mice/group; *p < 0.05, **p < 0.01, ***p < 0.001). (D) Boyden chamber chemotaxis assay for macrophages using lung lysates from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 5 weeks (n = 5–8/group; **p < 0.01). (E) Quantitative real-time PCR for CCL2 and M-CSF using mRNA from lungs of EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks (n = 4/group). (F) Protein expression array for inflammatory mediators using whole lung lysates from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks.
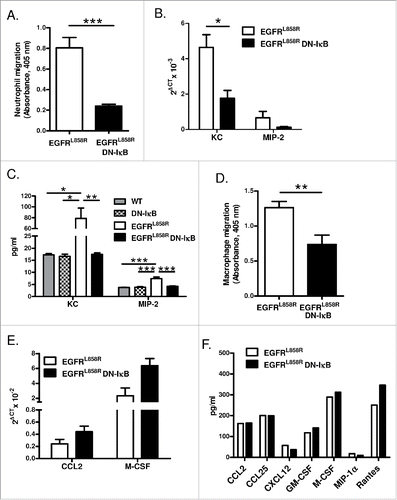
Similarly, more macrophages migrated toward lung lysates from EGFRL858R mice than from EGFRL858R DN-IκB mice (). By quantitative real-time PCR, we measured mRNA expression of the macrophage chemokine CCL2 and M-CSF, but neither of these factors was significantly reduced in lungs of EGFRL858R DN-IκB mice after 2 weeks of dox treatment (). To look more broadly for differences in potential macrophage chemotactic factors, we analyzed protein expression of a panel of inflammatory mediators (including known macrophage chemoattractants) in lung lysates from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks or 5 weeks by protein array ( and data not shown). However, we observed no significant reduction in any of these factors in EGFRL858R DN-IκB mice, suggesting that non-traditional or non-protein mediators may impact macrophage migration in this model.
Macrophages promote EGFR-mediated tumorigenesis
In both TKI-sensitive and resistant models of EGFR-mediated lung tumorigenesis, we identified increased neutrophils and macrophages in the lungs, and those inflammatory cell types were reduced in the setting of NF-κB inhibition, suggesting the importance of these cells for EGFR-driven lung cancer. Therefore, we next investigated whether neutrophils and/or macrophages contribute to EGFR-mediated tumorigenesis. To determine whether neutrophils play a role in tumorigenesis in EGFRL858R mice, we used Ly6G antibodies to deplete neutrophils throughout tumorigenesis. Ly6G depletion antibodies or IgG isotype control antibodies were administered by IP injection to EGFRL858R mice. Neutrophil depletion was verified by counting neutrophils in peripheral blood smears as well as by flow cytometry for neutrophils in lung single cell suspensions from mice on dox for 5 weeks (). Even though we observed successful neutrophil depletion, no difference in tumor burden was identified between EGFRL858R mice receiving Ly6G depletion antibodies and mice receiving isotype control antibodies (). These data indicate that neutrophils are not a critical cell type required for EGFR-mediated tumor formation in this model and suggest that neutrophils are not the primary cell type through which epithelial NF-κB signaling impacts tumorigenesis.
Figure 6. Macrophages promote EGFR-mediated lung tumorigenesis. (A) Flow cytometry analysis of lung single cell suspensions (n = 4/group; *p < 0.05), (B) quantification of neutrophils as a percentage of total white blood cells in peripheral blood smears (n = 7/group; **p < 0.01), and (C) histological analysis of tumors (n = 11–14/group) from EGFRL858R mice administered dox for 5 weeks and treated with Ly6G depletion or IgG isotype control antibodies. (D) Quantification of CD68 macrophage immunostaining (n = 3–5/group; *p < 0.05) and (E) histological analysis of tumors (n = 12–14/group; **p < 0.01) on lung sections from EGFRL858R mice administered dox for 5 weeks and treated with weekly IT injections of PBS or clodronate liposomes.
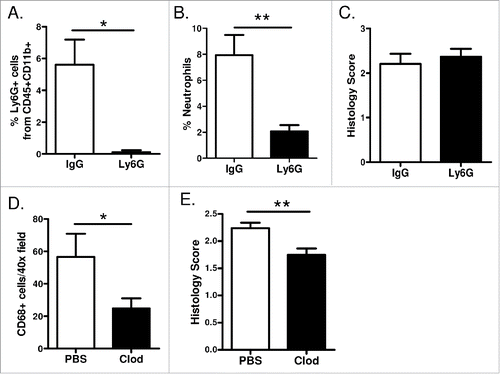
To test whether NF-κB-dependent recruitment of macrophages to the lungs is important for EGFR-mutant tumors, we depleted alveolar macrophages. Using a protocol that we previously developed, Citation25,26 macrophages were depleted by repeated dosing of liposomal clodronate, which causes selective macrophage apoptosis. Liposomal clodronate or control PBS liposomes were delivered intratracheally (IT) to EGFRL858R mice weekly, beginning 3 days prior to starting dox treatment. CD68 immunostaining was performed on lung sections from mice treated with dox for 5 weeks, verifying macrophage depletion (). After 5 weeks of dox administration, tumor burden was significantly reduced in EGFRL858R mice administered IT liposomal clodronate when compared to mice receiving PBS liposomes (), indicating that recruitment of macrophages is important for EGFR-mediated tumor formation.
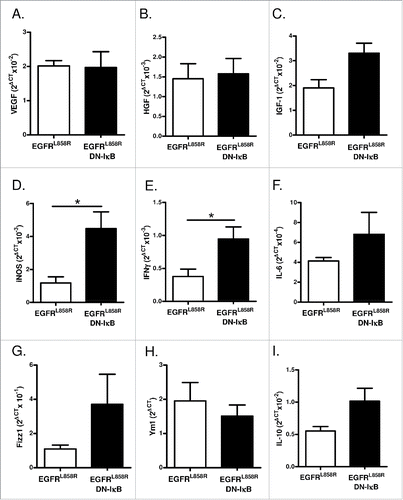
To investigate the mechanism by which macrophages promote EGFR-mediated lung tumorigenesis, we measured expression of macrophage mediators by quantitative real-time PCR in a macrophage-enriched cell population generated from lungs of EGFRL858R and EGFRL858R DN-IκB mice administered dox for 2 weeks. No difference in expression of the angiogenic factor VEGF or growth factors HGF or IGF-1 were observed (), and EGF was below the limit of detection. We also measured expression of M1 macrophage markers iNOS, IFNγ, IL-6, and IL-12p35 and found increased expression of iNOS and IFNγ in cells from EGFRL858R DN-IκB mice; however, this was not accompanied by reduced expression of M2 markers Arg1, Fizz1, Ym1, and IL-10, which were similar in both groups (IL-12p35 and Arg1 expression was below the limit of detection) (). Although the implications of increased M1 marker expression in EGFRL858R DN-IκB macrophages are not entirely clear, this finding may provide a clue regarding differential functions of macrophages in EGFRL858R and EGFRL858R DN-IκB mice.
Figure 7. Macrophages from EGFRL858R DN-IκB mice have increased expression of M1 markers. Quantitative real-time PCR analysis for (A) VEGF, (B) HGF, (C) IGF-1, (D) iNOS, (E) IFNγ, (F) IL-6, (G) Fizz1, (H) Ym1, and (I) IL-10 expression in macrophage-enriched cell population isolated from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks (n = 4/group). *p < 0.05.
Discussion
These studies demonstrate that epithelial NF-κB signaling promotes lung tumorigenesis through recruitment and/or activation of macrophages. In transgenic mouse models of EGFR TKI-sensitive and TKI-resistant lung tumorigenesis, epithelial NF-κB inhibition reduced tumor burden; however, NF-κB inhibition did not affect survival of EGFR-mutant airway epithelial cells or alter activation of known EGFR-mediated signaling pathways. Instead, in the settings of both TKI-sensitive and TKI-resistant lung tumors, epithelial NF-κB signaling was important for recruiting inflammatory cells to the lung, namely macrophages and neutrophils. In additional studies, we observed that neutrophil depletion did not impact EGFR-mediated tumorigenesis, but macrophage depletion resulted in a reduction in tumor burden. Taken together, these data suggest that classical NF-κB signaling functions primarily in a non-cell autonomous manner to promote EGFR-mediated lung tumorigenesis.
NF-κB is one of several potential mechanisms proposed to be important for mediating TKI resistance, which develops over time in almost all lung cancers.Citation14-18,27 De et al. demonstrated that stimulation of NF-κB signaling in vitro enhanced resistance of lung cancer cells to erlotinib treatment.Citation14 Furthermore, NF-κB inhibition in conjunction with erlotinib treatment has been shown to promote apoptosis of erlotinib-resistant lung cancer cells in vitro and reduce tumor growth in xenograft models.Citation15,17 More recently, NF-κB activation was shown to drive resistance to a mutant-selective EGFR inhibitor in T790M mutant cells.Citation16 While these prior studies indicate that NF-κB signaling supports resistance of EGFR-mutant tumors to TKIs, our findings indicate that NF-κB signaling plays a broader role in EGFR-driven lung carcinogenesis.
Inflammation has been linked to cancer promotion through two pathways: the extrinsic pathway, characterized by a pre-existing inflammatory condition, and the intrinsic pathway, driven by oncogenic mutations that lead to an inflammatory response.Citation28 In our transgenic mouse models, expression of oncogenic EGFR in the lungs leads to a significant intrinsic inflammatory response that is dependent upon NF-κB signaling. Similarly, transgenic mice with expression of oncogenic KRAS also exhibit an inflammatory influx characterized by increased macrophages and neutrophils.Citation29 Oncogenic KRAS has been shown to activate NF-κB signaling,Citation12 and NF-κB inhibition in KRAS-mutant tumors reduces tumor burden,Citation11,12 suggesting that NF-κB signaling functions analogously to promote intrinsic inflammation in both EGFR- and KRAS-mutant lung tumors.
We observed no effect on survival of EGFR-mutant tumor cells as a result of NF-κB inhibition, yet in other models of lung cancer, canonical NF-κB signaling has been implicated in cell survival. In mouse models of urethane-induced and KRAS-driven lung cancers, inhibition of canonical NF-κB increased apoptosis and/or reduced epithelial cell proliferation,Citation10-12,30 although whether these findings are a direct or indirect effect of NF-κB inhibition is unclear. Supporting our findings, others have reported that NF-κB inhibition did not affect proliferation of human lung cancer cell lines.Citation31 Our studies do not rule out a direct role for NF-κB signaling in the survival and proliferation of lung cancer cells, but instead suggest that induction of intrinsic inflammation may be the most important function of canonical NF-κB signaling in supporting EGFR-mutant tumors. However, when considering the translational importance of these findings it is important to keep in mind that mouse models with widespread over-expression of oncogenic EGFR (or KRAS) in the lungs may result in more exuberant inflammation than occurs during lung cancer development in humans, thus potentially overestimating the importance of NF-κB induced inflammation for supporting tumorigenesis.
Even though we observed no effect on tumorigenesis after neutrophil depletion, neutrophils have been shown to contribute to carcinogenesis in other contexts. Neutrophils can produce a variety of growth factors, reactive oxygen species, and proteases that promote tumor angiogenesis, proliferation, and invasion,Citation32 and NF-κB signaling in tumors has been shown to regulate expression of chemokines leading to neutrophil recruitment.Citation33 In human lung cancer, neutrophil influx is associated with a poor prognosis,Citation34 and in a transgenic mouse model of KRAS-driven lung cancer, depletion of neutrophil elastase caused significantly fewer lung tumors due to reduced tumor cell proliferation.Citation35 While we saw enhanced neutrophil recruitment in our EGFR-driven lung cancer models, this increase in neutrophils was modest compared to the increase in macrophages and was only observed at later time points, suggesting that the contributions of the neutrophils to lung tumorigenesis may have been overshadowed by the pro-tumorigenic effect of the macrophage influx.
Our findings indicate that epithelial NF-κB signaling recruits macrophages to promote lung carcinogenesis and suggest that therapeutic targeting of macrophages should be explored in EGFR-mutant lung cancer. Because macrophages can promote tumors through a variety of mechanisms, a diverse array of strategies are being explored to therapeutically target them. The major approach has focused on preventing macrophage recruitment by inhibiting the M-CSF receptor or CCL2 signaling.Citation36 However, in our studies, we did not measure significant differences in M-CSF or CCL2, suggesting a different mediator may be the dominant macrophage chemotactic factor in our model. Whether these inhibitors would reduce macrophage recruitment and consequently tumor burden in EGFR-driven pre-clinical models or in patients with EGFR-mutant lung cancer is uncertain. In addition to M-CSF and CCL2, NF-κB regulates a variety of other chemokines that promote macrophage recruitment, but we did not observe changes in known macrophage chemokines upon NF-κB inhibition. Although it is possible that novel protein mediators induce macrophage recruitment in our models, we speculate that non-protein mediators may account for the NF-κB-dependent influx of macrophages in mice expressing mutant EGFR. Future studies to identify the NF-κB-regulated mechanism of macrophage recruitment in the EGFRL858R and EGFRL/T lung cancer models may reveal additional potential therapeutic targets for development of novel treatment strategies for patients with EGFR-mutant lung cancers.
Materials and methods
Animal models
All animal care and experimental procedures were approved and conducted according to guidelines of the Vanderbilt University Institutional Animal Care and Use Committee. tet-O-EGFRL858R mice,Citation19 tet-O-EGFRL858R+T790M miceCitation24 (gifts from Dr. William Pao, Vanderbilt University), tet-O-IκBα-DN mice,Citation20 and CCSP-rtTA miceCitation37 and genotyping procedures have all been previously described. EGFRL858R and EGFRL/T transgenic mice were generated by mating tet-O-EGFRL858R or tet-O-EGFRL858R+T790M mice to homozygous CCSP-rtTA mice. Triple transgenic EGFRL858R DN-IκB or EGFRL/T DN-IκB mice were generated by mating EGFRL858R or EGFRL/T mice to homozygous tet-O-IκBα-DN mice. All mice were on an FVB background. Age- and sex-matched transgenic mice along with appropriate genotype-negative littermate controls (designated WT) were used in experiments. Transgene expression was activated by administering dox at a concentration of 0.5 g/L for EGFRL858R experiments or 1 g/L for EGFRL/T experiments in 2% sucrose drinking water, and water was replaced twice weekly.
For tissue harvest, lungs were lavaged as previously described,Citation9 and the left lung was tied off and frozen. The right lung was perfused and fixed by inflating with 10% neutral-buffered formalin. Total and differential BAL cell counts were determined as previously described.Citation10 For survival analysis, EGFRL858R and EGFRL858R DN-IκB mice were weighed on the day of dox initiation and at least twice weekly. Twenty percent weight loss was considered a mortality endpoint at which time mice were euthanized.
Tumor histological analysis
Hematoxylin and eosin staining was performed on 5 µm sections for quantification of tumor burden. Three sections were analyzed per mouse, and each section was separated by 50 µm. For EGFRL858R tumor analysis, 20 non-overlapping 20x fields were scored on each section based on lesion formation using a scale of 0 to 4 (0 = normal, 1 = <10% of alveoli have hyperplastic proliferative lesions, 2 = 10–50% of alveoli have proliferative lesions, 3 = <50% of alveoli have proliferative lesions, 4 = formation of tumor focus). For EGFRL858R+T790M tumor analysis, tumor area was measured and normalized to total lung area using Image-Pro Plus software (Media Cybernetics).
Immunostaining
For CD68 immunostaining, lung sections were stained with a CD68 antibody (ab125212; Abcam), and positive cells were enumerated on 20 40x-fields. Mean scores were calculated for each animal.
CT imaging
CT images were acquired using a Bioscan NanoSPECT/CT system. All imaging was performed by the Vanderbilt University Institute of Imaging Science.
Cell lines
HBEC3-KT (WT) and HBEC3-KTGmSRZ (EGFRL858R-mutant) immortalized human bronchial epithelial cellsCitation22,23 (gifts from Dr. John Minna, The University of Texas Southwestern Medical Center) were maintained at 37°C 5% CO2 in Keratinocyte-SFM supplemented with epidermal growth factor 1–53 and bovine pituitary extract (Thermo Scientific). Cells were transfected with myc/His-IκB-DN pEF4Citation20 or EV pEF4 using Xfect transfection reagent (Clontech).
Epithelial/macrophage enrichment
Epithelial enrichment was performed using dispase and negative selection for leukocyte and monocyte populations as described by others.Citation38 Briefly, mice were euthanized, and the lungs were perfused with 10 mL of sterile saline. 3 mL of dispase (Invitrogen) was rapidly instilled into the trachea followed immediately by 0.5 mL of warm 1% low melting point agarose (Sigma). The chest cavity was filled with ice for 2 min to harden the agarose before the lungs were dissected out and incubated at room temperature in 2 mL of dispase for 45 min. Lungs were then placed in a culture dish containing media and 100 U/mL DNase I (Sigma) and teased apart. The tissue was filtered through a series of strainers to generate single cell suspensions, which were then placed on tissue culture plates coated in CD45 and CD32 antibodies (BD Biosciences). After incubation for 1 h at 37°C in 5% CO2, cells that remained in suspension were collected by centrifugation for immediate protein isolation as the epithelial-enriched fraction. mRNA was isolated from adherent cells as the macrophage-enriched fraction as described below.
RNA isolation, RT-PCR, and quantitative real-time PCR
Total mRNA was isolated from enriched cells and lung tissue using the RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. RT-PCR was performed to measure EGFRL858R and EGFRL/T expression as previously described.Citation19,24 RT-PCR was also performed for DN-IκB and GAPDH using the following primers: DN-IκB (F: CCTGGCTGTTGTCGAATACC; R: GGTGATGGTGATGATGACCGG), GAPDH (F: CCTGCACCACCAACTGCTTAG; R: GTGGATGCAGGGATGATGTTC). Quantitative real-time PCR was performed using Sybr Green PCR Master Mix (Applied Biosystems) and the following primer sets: CCL2 (F: TTAAAAACCTGGATCGGAACCAA; R: GCATTAGCTTCAGATTTACGGGT), M-CSF (F: GTGTCAGAACACTGTAGCCAC; R: TCAAAGGCAATCTGGCATGAAG) (PrimerBank ID: 166064045c1),Citation39-41 KC (F: CCGAAGTCATAGCCACACTCAA; R: GCAGTCTGTCTTCTTTCTCCGTTAC), MIP-2 (F: ACTCTCAAGGGCGGTCAAAA; R: GGCACATCAGGTACGATCCA), iNOS (F: CACCTTGGAGTTCACCCAGT; R: ACCACTCGTACTTGGGATGC), IFNγ (F: GCGTCATTGAATCACACCTGA; R: CTCGGATGAGCTCATTGAATGC), IL-6 (F: TCCTCTGGTCTTCTGGAGTA; R: CTTAGCCACTCCTTCTGTGA), Fizz1 (F: TCCCAGTGAATACTGATGAGA; R: CCACTCTGGATCTCCCAAGA), Ym1 (F: GGGCATACCTTTATCCTGAG; R: CCACTGAAGTCATCCATGTC), IL-10 (F: ACCTGCTCCACTGCCTTGCT; R: GGTTGCCAAGCCTTATCGGA), VEGF (F: CTGCACCCACGACAGAAG; R: ACACAGGACGGCTTGAAGAT), IGF-1 (F: CACATCATGTCGTCTTCACACC; R: GGAAGCAACACTCATCCACAATG) (Primer Bank ID: 163659888c1), HGF (F: ACTTCTGCCGGTCCTGTTG; R: CCCCTGTTCCTGATACACCT) (Primer Bank ID: 170172521c1), GAPDH (F: TGAGGACCAGGTTGTCTCCT; R: CCCTGTTGCTGTAGCCGTAT). For each sample, expression was normalized to GAPDH. Threshold values greater than 33 were considered below the limit of detection.
Western blot analysis
Whole lung lysates were prepared using RIPA buffer, separated by SDS-PAGE gel, transferred to nitrocellulose membranes, and probed with the following antibodies: p-Akt (Ser473) (4060; Cell Signaling), total Akt (9722; Cell Signaling), p-ERK (T202/Y204) (9101; Cell Signaling), total ERK (9102; Cell Signaling), p-STAT3 (Y705) (9131; Cell Signaling), total STAT3 (9132; Cell Signaling), or β-actin (SAB2100037, Sigma). Nuclear and cytoplasmic protein fractions were prepared from immortalized human bronchial epithelial cells using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher). Western blots were probed using the following antibodies: myc (M4439; Sigma), caspase-3 (9622; Cell Signaling), actin (A2066, Sigma), p65 (C-20, Santa Cruz), or TATA-binding protein (TBP) (N-12, Santa Cruz). Jurkat cell lysates treated with cytochrome c were included as a positive control for cleaved caspase-3 (Cell Signaling; 9663). Immunodetection was performed using the corresponding AlexaFluor-conjugated antibodies and the Odyssey Infrared Imaging System (LI-COR Biosciences). All images were converted to grayscale.
Flow cytometry
Lung single-cell suspensions were generated and stained as previously described.Citation9,26 To reduce non-specific staining, single-cell suspensions were incubated with Fc receptor block (BD Biosciences). For detection of T cell populations, cells were incubated with the following antibodies: CD3-PE-Cy7 (145–2C11) and CD8a-AF700 (53–6.7) from BioLegend, CD4+-FITC (RM 4–5) from eBioscience, and CD45-AP-Cy7 (30-F11) from BD Biosciences. For detection of neutrophil populations, cells were incubated with CD11b-A450 (M1/70) and Ly6G-PerCP-Cy5.5 (1A8) from BD Biosciences and CD45-AP-Cy7. Dead cells were excluded using a Live/Dead Fixable Blue Dead Cell Stain Kit (Life Technologies). Cells were analyzed using a LSR II flow cytometer (BD Biosciences), and data analysis was performed using FlowJo software (Tree Star, Inc.).
In vitro migration assays
Mouse bone marrow neutrophils were isolated from WT FVB mice as described by others.Citation42 Neutrophil chemotaxis was assessed using a modified Boyden chamber MBA 96 (Neuroprobe) with a 3 µm pore size polycarbonate filter.Citation43 Cell-free BAL supernatant was diluted 1:1 with 0.2% BSA/RPMI serum-free and phenol red-free media (final BSA concentration of 0.1%) and placed in the bottom chamber as the chemoattractant. Each BAL sample was run in triplicate. 2 × 105 neutrophils in 0.1% BSA/RPMI were loaded into the top chamber and incubated for 1 h at 37°C, 5% CO2.
Mouse bone marrow-derived macrophages were isolated from WT mice (FVB background) and matured as described by others.Citation44 Macrophage chemotaxis was assessed as for neutrophils with a 5 µm pore size polycarbonate filter. 250 ug of whole lung lysates from 5 week dox EGFRL858R and EGFRL858R DN-IκB mice was diluted in DMEM supplemented with 1% FBS and placed in the bottom chamber as the chemoattractant. Each lysate was run in triplicate. 1.5 × 105 macrophages in DMEM supplemented with 1% FBS were loaded into the top chamber and incubated for 5.5 h at 37°C, 5% CO2.
The trans-migrated cells were collected by centrifuging the 96 well plate, washed three times, and resuspended in 100 µL Hank's Balanced Salt Solution for neutrophils or 100 µL PBS for macrophages. 60 µL NAG solution (4mM NAG, 25mM citric acid, 25 mM sodium citrate, and 0.25% Triton X-100, pH 5.0) was added to each well and incubated overnight at room temperature in the dark. 100 µL stop solution (50 mM glycine and 5 mM EDTA, pH 10.4) was added to each well, and the absorbance was measured at 405 nM.
Neutrophil depletion
For neutrophil depletion experiments, mice received either 100 μg of anti-Ly6G antibodies (Clone 1A8, BioLegend) or IgG2a isotype control antibodies (BioLegend) by IP injection. Mice were injected on day 7, 9, 12, and 14 after dox initiation and twice weekly thereafter until they were sacrificed at day 35 of dox. Neutrophil depletion was confirmed by lung flow cytometry and Wright-Giemsa staining of peripheral blood smears.
Clodronate macrophage depletion
Clodronate (Sigma) or PBS-containing liposomes were prepared as previously described.Citation25 100 µL of clodronate or control PBS lipsomes was delivered IT 3 d prior to dox initiation, on the day of dox initiation, and weekly thereafter until mice were harvested at day 35 of dox. For liposome delivery, mice were anesthetized and intubated with a 1-mL syringe with a 6-mm-long, 22-gauge over-the-needle catheter (Abbocath-T; Venisystems). Macrophage depletion was confirmed by CD68 staining of lung sections.
BAY 11–7082 treatment
BAY 11–7082 (10 mg/kg body weight, Cayman Chemical) was delivered by IP injection twice weekly as described.Citation45
Protein array
Whole lung lysates were prepared from EGFRL858R and EGFRL858R DN-IκB mice treated with dox for 2 weeks. To minimize the effects of mouse-to-mouse-variability, lysates were pooled from three mice for each sample, resulting in a total of four samples (two EGFRL858R and two EGFRL858R DN-IκB). Cytokine expression was determined using the Mouse Inflammation Array (AAM-INF-G1; RayBiotech, Inc.), which measures 40 different inflammation-associated analytes, including BLC (CXCL13), CD30L (TNFSF8), Eotaxin-1 (CCL11), Eotaxin-2 (CCL24), FasL (TNFSF6), Fractalkine (CX3CL1), G-CSF, GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-6, IL-9, IL-10, IL-12 p40/p70, IL-12 p70, IL-13, IL-17A, I-TAC (CXCL11), KC, Leptin, LIX, Lymphotactin, MCP-1 (CCL2), M-CSF, MIG (CXCL9), MIP-1α (CCL3), MIP-1γ, RANTES (CCL5), SDF-1α (CXCL12α), I-309 (TCA-3/CCL1), TECK (CCL25), TIMP-1, TIMP-2, TNF-α, TNFRI (TNFRSF1A), and TNFRII (TNFRSF1B).
ELISAs
KC and MIP-2 (R&D Systems) were measured in cell-free BAL supernatants. ELISAs were performed according to manufacturer's instructions.
Statistics
Data were analyzed using GraphPad Prism 5.0 software (GraphPad Software, Inc.). Unpaired student t-tests were performed for comparisons between two groups. To analyze differences among more than two groups, one-way ANOVA followed by a Tukey's post-test was used. For the Kaplan–Meyer survival analysis, a log-rank test was employed. Values are presented as the mean ± SEM, and p ≤ 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Vanderbilt University Institute of Imaging Science, supported by NCRR instrument grant 1S10 RR32784, for their technical assistance in imaging mice. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource, which is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
Funding
This work was supported by the Department of Veterans Affairs and NIH HL 119503.
References
- Li AR, Chitale D, Riely GJ, Pao W, Miller VA, Zakowski MF, Rusch V, Kris MG, Ladanyi M. EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J Mol Diagnostics 2008; 10:242-8; PMID:18403609; http://dx.doi.org/10.2353/jmoldx.2008.070178
- Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009; 361:958-67; PMID:19692684; http://dx.doi.org/10.1056/NEJMoa0904554
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007; 11:217-27; PMID:17349580; http://dx.doi.org/10.1016/j.ccr.2006.12.017
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:e73; PMID:15737014; http://dx.doi.org/10.1371/journal.pmed.0020073
- Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008; 105:2070-5; PMID:18227510; http://dx.doi.org/10.1073/pnas.0709662105
- Zhang Z, Ma J, Li N, Sun N, Wang C. Expression of nuclear factor-κB and its clinical significance in nonsmall-cell lung cancer. Ann Thorac Surg 2006; 82:243-8; PMID:16798222; http://dx.doi.org/10.1016/j.athoracsur.2006.01.049
- Tang X, Liu D, Shishodia S, Ozburn N, Behrens C, Lee JJ, Hong WK, Aggarwal BB, Wistuba II. Nuclear factor-κB (NF-κB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer 2006; 107:2637-46; PMID:17078054; http://dx.doi.org/10.1002/cncr.22315
- Tichelaar JW, Zhang Y, LeRiche JC, Biddinger PW, Lam S, Anderson MW. Increased staining for phospho-Akt, p65/RELA and cIAP-2 in pre-neoplastic human bronchial biopsies. BMC Cancer 2005; 5:155; PMID:16332260; http://dx.doi.org/10.1186/1471-2407-5-155
- Zaynagetdinov R, Stathopoulos GT, Sherrill TP, Cheng D-S, McLoed AG, Ausborn JA, Polosukhin VV, Connelly L, Zhou W, Fingleton B et al. Epithelial nuclear factor-κB signaling promotes lung carcinogenesis via recruitment of regulatory T lymphocytes. Oncogene 2011; 31:3164-76; PMID:22002309; http://dx.doi.org/10.1038/onc.2011.480
- Stathopoulos GT, Sherrill TP, Cheng DS, Scoggins RM, Han W, Polosukhin VV, Connelly L, Yull FE, Fingleton B, Blackwell TS. Epithelial NF-κB activation promotes urethane-induced lung carcinogenesis. Proc Natl Acad Sci U S A 2007; 104:18514-9; PMID:18000061; http://dx.doi.org/10.1073/pnas.0705316104
- Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-κB signalling in a mouse model of lung adenocarcinoma. Nature 2009; 462:104-7; PMID:19847165; http://dx.doi.org/10.1038/nature08462
- Bassères DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-κB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res 2010; 70:3537-46; PMID:20406971; http://dx.doi.org/10.1158/0008-5472.CAN-09-4290
- Alberti C, Pinciroli P, Valeri B, Ferri R, Ditto A, Umezawa K, Sensi M, Canevari S, Tomassetti A. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFkB pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012; 31:4139-49; PMID:22158046; http://dx.doi.org/10.1038/onc.2011.572
- De S, Dermawan JKT, Stark GR. EGF receptor uses SOS1 to drive constitutive activation of NFκB in cancer cells. Proc Natl Acad Sci U S A 2014; 111:11721-6; PMID:25071181; http://dx.doi.org/10.1073/pnas.1412390111
- Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, Moonsamy P, Dahlman K, Miller VA, Costa C et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011; 471:523-6; PMID:21430781; http://dx.doi.org/10.1038/nature09870
- Galvani E, Sun J, Leon LG, Sciarrillo R, Narayan RS, Tjin Tham Sjin R, Lee K, Ohashi K, Heideman DAM, Alfieri RR et al. NF-κB drives acquired resistance to a novel mutant-selective EGFR inhibitor. Oncotarget 2015; 6:42717-32; PMID:26015408; http://dx.doi.org/10.18632/oncotarget.3956
- Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, Hrustanovic G, Chan E, Lin L, Neel DS et al. NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep 2015; 11:98-110; PMID:25843712; http://dx.doi.org/10.1016/j.celrep.2015.03.012
- Lantermann AB, Chen D, McCutcheon K, Hoffman G, Frias E, Ruddy D, Rakiec D, Korn J, McAllister G, Stegmeier F et al. Inhibition of casein kinase 1 α prevents acquired drug resistance to erlotinib in EGFR-mutant non–small cell lung cancer. Cancer Res 2015; 75:1-12; PMID:26490646; http://dx.doi.org/10.1158/0008-5472.CAN-15-1113
- Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 2006; 20:1496-510; PMID:16705038; http://dx.doi.org/10.1101/gad.1417406
- Cheng DS, Han W, Chen SM, Sherrill TP, Chont M, Park GY, Sheller JR, Polosukhin VV, Christman JW, Yull FE et al. Airway epithelium controls lung inflammation and injury through the through the NF-κB pathway. J Immunol 2007; 178:6504-13; PMID:17475880; http://dx.doi.org/10.4049/jimmunol.178.10.6504
- Pierce J, Schoenleber R, Jesmok G, Best J, Moore S, Collins T, Gerritsen M. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem 1997; 272:21096-103; PMID:9261113; http://dx.doi.org/10.1074/jbc.272.34.21096
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res 2004; 64:9027-34; PMID:15604268; http://dx.doi.org/10.1158/0008-5472.CAN-04-3703
- Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, Ramirez RD, Sunaga N, Gazdar AF, Shay JW et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res 2006; 66:2116-28; PMID:16489012; http://dx.doi.org/10.1158/0008-5472.CAN-05-2521
- Regales L, Balak MN, Gong Y, Politi K, Sawai A, Le C, Koutcher JA, Solit DB, Rosen N, Zakowski MF et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One 2007; 2:e810; PMID:17726540; http://dx.doi.org/10.1371/journal.pone.0000810
- Everhart MB, Han W, Parman KS, Polosukhin VV, Zeng H, Sadikot RT, Li B, Yull FE, Christman JW, Blackwell TS. Intratracheal administration of liposomal clodronate accelerates alveolar macrophage reconstitution following fetal liver transplantation. J Leukoc Biol 2005; 77:173-80; PMID:15563581; http://dx.doi.org/10.1189/jlb.1203647
- Zaynagetdinov R, Sherrill TP, Polosukhin V V, Han W, Ausborn JA, McLoed AG, McMahon FB, Gleaves LA, Degryse AL, Stathopoulos GT et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J Immunol 2011; 187:5703-11; PMID:22048774; http://dx.doi.org/10.4049/jimmunol.1100558
- Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014; 26:207-21; PMID:25065853; http://dx.doi.org/10.1016/j.ccr.2014.05.019
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008; 454:436-44; PMID:18650914; http://dx.doi.org/10.1038/nature07205
- Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, Tonon G, McNamara K, Marconcini LA, Hezel A et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene 2006; 25:2105-12; PMID:16288213; http://dx.doi.org/10.1038/sj.onc.1209237
- Xia Y, Yeddula N, Leblanc M, Ke E, Zhang Y, Oldfield E, Shaw RJ, Verma IM. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat Cell Biol 2012; 14:257-65; PMID:22327365; http://dx.doi.org/10.1038/ncb2428
- Hopewell EL, Zhao W, Fulp WJ, Bronk CC, Lopez AS, Massengill M, Antonia S, Celis E, Haura EB, Enkemann SA et al. Lung tumor NF-κB signaling promotes T cell-mediated immune surveillance. J Clin Invest 2013; 123:2509-22; PMID:23635779; http://dx.doi.org/10.1172/JCI67250
- Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res 2011; 71:2411-6; PMID:21427354; http://dx.doi.org/10.1158/0008-5472.CAN-10-2583
- Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004; 6:447-58; PMID:15542429; http://dx.doi.org/10.1016/j.ccr.2004.09.028
- Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, Mayaud C, Milleron B, Baud L, Cadranel J. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol 1998; 152:83-92; PMID:9422526
- Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 2010; 16:219-23; PMID:20081861; http://dx.doi.org/10.1038/nm.2084
- Noy R, Pollard J. Tumor-associated macrophages: from mechanisms to therapy. Immunity 2014; 41:49-61; PMID:25035953; http://dx.doi.org/10.1016/j.immuni.2014.06.010
- Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem 2000; 275:11858-64; PMID:10766812; http://dx.doi.org/10.1074/jbc.275.16.11858
- Rice WR, Conkright JJ, Na CL, Ikegami M, Shannon JM, Weaver TE. Maintenance of the mouse type II cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol 2002; 283:L256-64; PMID:12114186; http://dx,doi.org/10.1152/ajplung.00302.2001
- Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res 2010; 38:D792-9; PMID:19906719; http://dx.doi.org/10.1093/nar/gkp1005
- Spandidos A, Wang X, Wang H, Dragnev S, Thurber T, Seed B. A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics 2008; 9:633; PMID:19108745; http://dx.doi.org/10.1186/1471-2164-9-633
- Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res 2003; 31:e154; PMID:14654707; http://dx.doi.org/10.1093/nar/gng154
- Boxio R, Bossenmeyer-Pourie C, Steinckwich N, Dournon C, Nu O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol 2004; 75:604-11; PMID:14694182; http://dx.doi.org/10.1189/jlb.0703340
- Sai J, Raman D, Liu Y, Wikswo J, Richmond A. Parallel phosphatidylinositol 3-kinase (PI3K)-dependent and Src-dependent pathways lead to CXCL8-mediated Rac2 activation and chemotaxis. J Biol Chem 2008; 283:26538-47; PMID:18662984; http://dx.doi.org/10.1074/jbc.M805611200
- Munder P, Modolell M, Hoelzl Wallach D. Cell propagation on films of polymeric fluorocarbon as a means to regulate pericellular pH and pO(2) in cultured monolayers. FEBS Lett 1971; 15:191-6; PMID:11945843; http://dx.doi.org/10.1016/0014-5793(71)80309-5
- Xue W, Meylan E, Oliver TG, Feldser DM, Winslow MM, Bronson R, Jacks T. Response and resistance to NF-κB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov 2011; 1:236-47; PMID:21874163; http://dx.doi.org/10.1158/2159-8290.CD-11-0073