
ABSTRACT
There are numerous transcriptional, proteomic and functional differences between monocyte-derived dendritic cells (Mo-DC) and primary blood dendritic cells (BDC). The CMRF-56 monoclonal antibody (mAb) recognizes a cell surface marker, which is upregulated on BDC following overnight culture. Given its unique ability to select a heterogeneous population of BDC, we engineered a human chimeric (h)CMRF-56 IgG4 mAb to isolate primary BDC for potential therapeutic vaccination. The ability to select multiple primary BDC subsets from patients and load them with in vitro transcribed (IVT) mRNA encoding tumor antigen might circumvent the issues limiting the efficacy of Mo-DC. After optimizing and validating the purification of hCMRF-56+ BDC, we showed that transfection of hCMRF-56+ BDC with mRNA resulted in efficient mRNA translation and antigen presentation by myeloid BDC subsets, while preserving superior DC functions compared to Mo-DC. Immune selected and transfected hCMRF-56+ BDC migrated very efficiently in vitro and as effectively as cytokine matured Mo-DC in vivo. Compared to Mo-DC, hCMRF-56+ BDC transfected with influenza matrix protein M1 displayed superior MHC peptide presentation and generated potent antigen specific CD8+ T-cell recall responses, while Wilms tumor 1 (WT1) transfected CMRF-56+ BDC generated effective primary autologous cytotoxic T-cell responses. The ability of the combined DC subsets within hCMRF-56+ BDC to present mRNA delivered tumor antigens merits phase I evaluation as a reproducible generic platform for the next generation of active DC immune therapies.
Abbreviations
| APC | = | antigen-presenting cells |
| BDC | = | blood dendritic cells |
| CTL | = | cytotoxic T lymphocyte |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor |
| H | = | hour |
| IVT-mRNA | = | in vitro transcribed mRNA |
| mdc | = | myeloid dendritic cells |
| mo-dc | = | monocyte-derived dendritic cells |
| pdc | = | plasmacytoid dendritic cells |
Introduction
Therapeutic vaccination using tumor-associated antigen (TAA) loaded dendritic cells (DC) is an attractive concept. However, despite strong evidence of its utility in animal models and numerous clinical studies, there is limited evidence of widespread clinical efficacy.Citation1-4 Nonetheless, recent studies suggest that DC vaccination may have a place in treating both hematological and other malignancies; particularly if applied after a reduction in tumor burden following surgical resection, chemotherapy, or hematopoietic-stem-cell transplantation, when tumor immunosuppression is at its lowest.Citation5-7 Recent trials in acute myeloid leukemia (AML)Citation8-10 and multiple myelomaCitation7 investigating monocyte derived dendritic cell (Mo-DC) vaccination, after induction chemotherapy and transplantation, have demonstrated objective clinical and immunological responses. To build on this, major improvements in the DC product are needed, first, to address limitations in DC performance and secondly, to make DC vaccination practical. If applied after successful conventional induction or consolidation regimes, therapeutic DC vaccination has the much needed potential to induce immune antitumor memory and sustain long-term remission.Citation7,9,11
DC is divided into several subsets, each with different functional capabilities. Human blood DC (BDC), which are HLA-DR+ but lack specific lineage markers, account for approximately 1% of peripheral blood mononuclear cells (PBMC).Citation12 BDC have been classified into two major classes: myeloid (mDC; CD11c+ CD304−) and plasmacytoid (pDC; CD11c− CD304+). myeloid dendritic cells (mDC) can be subdivided into three populations: CD1c+, CD141+ (or XCR1+) and CD16+ mDC subsets.Citation12-15 Given the phenotypic and functional heterogeneity of BDC subsets,Citation16-18 it still remains unclear which might be best for therapeutic vaccination. The major myeloid CD1c+ DC have been isolated clinically using a two-step immune selection technology, but these isolations take a long time and the low yields limit the DC vaccination dose.Citation19,20 The minor CD141+ XCR1+ mDC subset generates excellent cytotoxic T lymphocyte (CTL) responses due to their strong capacity to cross present antigenCitation17,21 but their low frequency makes their purification unrealistic using current technologies. Citation2 Immune-selected pDC have also been explored in the context of vaccination, and have been shown to induce T-cell responses in patients with melanoma.Citation22
The practical difficulties in isolating sufficient BDC led to the predominant use of in vitro cultured “DC-like” Mo-DC in the vast majority of clinical studies. However, the properties of Mo-DC were shown to differ substantially from those of primary “in vivo” derived BDC,Citation23-25 and it is now well accepted that there are significant transcriptional and functional differences between Mo-DC and BDC.Citation12,14,Citation17,22,Citation25-30
A major concern regarding the use of Mo-DC for therapeutic vaccination has been their well-documented failure to migrate from sites of injection to draining lymph nodes (LN).Citation31,32 This suggests that the efficacy seen in some studies may relate to an adjuvant or antigen-transfer effect to host DC present at the site of injection. Replacing Mo-DC manufactured in vitro with primary BDC has two potential advantages: (1) BDC are likely to be superior antigen-presenting cells (APC), due to their specialized native antigen-presenting capacity and their potential to migrate more effectively than their in vitro counterparts;Citation1 (2) it simplifies the preparation, avoiding expensive long-term culture and regulatory processes.
The CMRF-56 monoclonal antibody (mAb) has been submitted to several human leucocyte differentiation antigen workshopsCitation18,33,Citation34 and identifies a 95 kDa cell surface molecule that is upregulated on BDC, some B cells and monocytes, after culturing PBMC for 12–16 h and is further upregulated on all BDC subsets following activation.Citation18,24,35-40 We have used the CMRF-56 mAb to develop a platform for BDC enrichment that is applicable to the clinical settinCitation38-40 and it has proven useful for the positive selection of heterogeneous BDC subsets in sufficient numbers for clinical application directly from aphaeresis collections following brief incubation using clinical gas permeable culture bags or tissue culture flasks.Citation24,39,40 To improve its clinical utility, we engineered a human IgG4 chimeric CMRF-56 mAb (hCMRF-56) for use in a single-step, clinical scale, magnetic bead-based immunoselection system.
We describe the generation of clinical hCMRF-56 and its ability to purify BDC, including the CD1c+, CD16+ and CD141+ myeloid subsets.Citation12 We also describe, for the first time, the use of in vitro transcribed (IVT)-mRNA to load viral and TAA into primary BDC and characterize their capacity to migrate and generate T-cell responses. The hCMRF-56 BDC selection and IVT-mRNA loading facilitates, (1) reproducible production of a heterogeneous population of BDC, (2) maintenance of DC migratory and stimulatory capacity and (3) delivery of TAA for presentation by diverse HLA types, making it feasible to use in generic protocols.
Results
hCMRF-56 selects for multiple human BDC subsets
The IgG4 hCMRF-56 antibody was produced by splice overlap extension PCR of the VH and VL regions from the original mouse anti-human CMRF-56 (clone: MMRI-0384) with a human IgG4 constant chain. The human chimeric mAb showed the same specificity as the mouse IgG1 mAb for the CMRF-56 antigen, which is present at low levels on APC within freshly isolated PBMC and is upregulated following overnight culture (Fig. S1A). The CMRF-56 antigen was upregulated on 0.4% (Range 0.1–0.7%) of PBMC following overnight culture. Single-step CMRF-56+ cell immune selection of recovered 43.9% (range 6.1–153.93) of the total potential BDC (defined as CD14−, CD19− & CD20−, hCMRF-56+cells) with an average BDC purity of 18.4% (range 6.1–45.6; ).
Table 1. Summary of hCMRF-56 BDC isolations.
The majority of CMRF-56+ cells were APC (p <0.001) with very few CD3+ T cells or CD56+ NK cells (). The hCMRF-56+ HLA-DR+ APC fraction contained all the described human blood mDC subsets,Citation12 with the majority being CD1c+ DC. However, both the major CD1c+ mDC and the rare CD141+ mDC were substantially enriched (46.0 ± 0.9 and 38.8 ± 5.7-fold, respectively). Few pDC, T cells and NK cells were recovered ().
Figure 1. hCMRF-56 immunoselects blood APC enriched for mDC. BDC were immunoselected from overnight cultured PBMC using hCMRF-56. The preparation was analyzed by flow cytometry to assess (A) the enrichment of BDC, (B) the leukocyte constituents, (C) the subsets of BDC enriched, (D) CD83 and (E) CD274 expression as a measure of DC activation. The BDC were identified as hCMRF-56+ HLA-DRhi cells. N = 4–7 independent donors; p < 0.001 (***), 1-way ANOVA with Tukey's test for multiple comparisons.
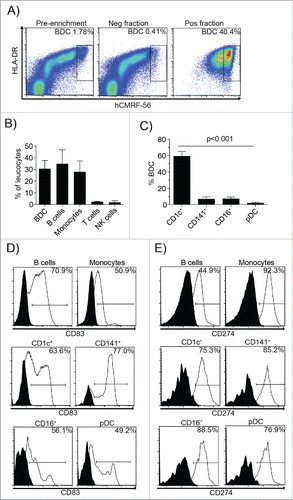
Purified CMRF56+ cells expressed varying levels of co-stimulatory CD83, with monocytes and B cells exhibiting consistently low expression and both CD1c+ and CD141+ BDC expressing uniformly high levels (), indicative of an activated DC phenotype. The ligand for PD1, namely CD274 (PDL1), was also upregulated as expected ().
To determine the relative contribution of BDCs, monocytes and B cells to T-cell priming CMRF-56 immune selected cells were by sorted into a lineage (CD14/CD19/CD20 ) negative HLA-DR+ BDC fraction and a lineage (CD14/19/20)+, B cells and monocyte, fraction. Both fractions were tested for their ability to generate specific CTL against tumor targets and compared to the complete product. Both fractions generated tumor specific CTL and lysed T2 targets, however, the bulk of CTL generation capacity was due to the BDC component of the CMRF-56+ preparation (Fig. S2.)
Optimized transfection targets IVT-mRNA to myeloid APC
We used the AMAXA Nucleofector 4D to transfect hCMRF-56 immunoselected cells with IVT-mRNA. We tested pre-set programs for primary cells, measuring transfection efficiency of EGFP mRNA and transfection related mortality of BDC. Cell death was minimal 24 h post-transfection using programsCA-138 and CU-110 (Figs. S3A–B). Program CU-110 was chosen for further experiments due to the increased viability and superior levels of EGFP expression. Transfection with IVT-mRNA resulted in high EGFP expression by BDC at 4 h, which was maintained for at least 48 h (Fig. S3C). EGFP was expressed predominantly by APC with the highest levels in mDC, notably the CD141+ and CD1c+ DC ().
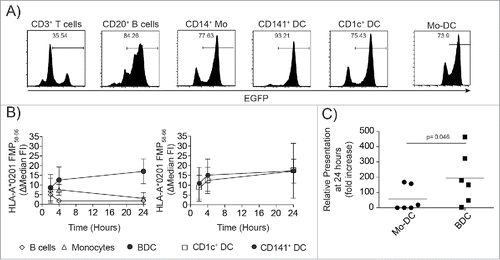
Figure 2. Primary BDC present mRNA antigens efficiently compared to other primary myeloid cells and in vitro generated Mo-DC. (A) CMRF-56 immunoselected APC subsets or in vitro derived Mo-DC showed comparable ability to translate EGFP-mRNA. EGFP expression was lowest in residual T cells, B cells and pDC, with greatest expression in primary blood CD14+ monocytes and CD141+ and CD1c+ DC (one of three experiments is shown). (B) Translated FMP-mRNA was detected as HLA-A*0201:FMP58–66 complexes on the cell surface using an anti-HLA-A*0201:FMP58–66 antibody by flow cytometry (three independent donors are shown). (C) The HLA-A*0201 presentation of FMP 58–66 on FMP–mRNA loaded CMRF-56+ BDC (n = 6) or immature Mo-DC (n = 6) was compared at 24 h post-transfection by flow cytometry (p = 0.046, Mann–Whitney test).
BDC process and present mRNA antigen more effectively than other APC
The ability of the CMRF-56+ BDC to process and present full-length FMP-IVT-mRNA encoded antigen by HLA class I molecules was assessed by monitoring cell surface HLA-A*0201:FMP58–66 peptide complexes after loading with mRNA using transfection conditions that resulted in similar translational efficiencies (). CMRF-56+ BDC presented FMP58–66 as early as 2 h post-transfection () peaking by 4 h and was still detected 24 h post-transfection. The other major CMRF-56+ APC populations present, CD20+ B cells and CD14+ monocytes, had different kinetics of antigen presentation. Both showed early processing and presentation of FMP, peaking at 4 h, but surface HLA-A*0201:FMP58–66 peptide complexes declined rapidly with little or no presentation observed at 24 h ().
We next tested whether in vitro derived Mo-DC used in current clinical protocols presented mRNA antigen equivalently to preformed BDC. Transfection of Mo-DC resulted in high levels of surface HLA-A*0201:FMP58–66 peptide complexes as early as 2 h, similar to CMRF-56+ BDC and primary monocytes (Data not shown). Cell surface HLA-A*0201:FMP58–66 peptide complexes persisted in BDC with a 194-fold increase in antigen presented at 24 h compared to 2 h post-transfection (). In contrast, surface HLA-A*0201:FMP58–66 peptide complexes on Mo-DC were significantly reduced compared to BDC after 24 h and similar to that of monocytes.
hCMRF-56 BDC retain DC capacity for costimulation and migration
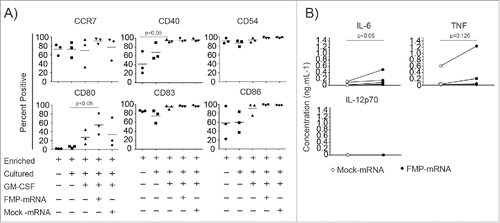
To determine whether mRNA transfection altered DC function, we assessed CMRF-56+ BDC cell surface phenotype associated with T-cell priming. Overnight PBMC culture was sufficient to induce upregulation of a number of costimulatory, chemotactic and adhesion molecules including CD83, CD86, CCR7 and CD54 () on BDC. CD54, which is used as a marker of activation in other primary BDC preparations,Citation45,41 was present in high density on CMRF-56+ BDC and was not increased by further stimulation. Additional exposure of CMRF-56+ BDC to GM-CSF for 2 h, which we previously demonstrated to promote efficient BDC: T-cell priming,Citation24 resulted in modest upregulation of CD40 and CD80 (). Transfection of FMP-IVT-mRNA into GM-CSF activated BDC led to significant upregulation of CD80 compared to mock transfected cells but not the other molecules tested. Transfection of CMRF-56+ BDC with FMP-mRNA resulted in significant IL-6 production and a small amount of TNF that was dependent on the presence of IVT-mRNA ().
Figure 3. Effect of transfection on CMRF-56+ BDC surface molecules and cytokine secretion. CMRF-56+ BDC were cultured overnight with or without GM-CSF, and either transfected with 10 μg of polyadenylated FMP IVT-mRNA or in the absence of mRNA (Mock). (A) Upregulation of BDC cell surface activation markers was examined using flow cytometry after 20 h with GM-CSF and FMP-mRNA or Mock transfection (n = 3 independent donors; *p < 0.05 Kruskal-Wallis test.) (B) Cell culture supernatants from GM-CSF stimulated transfected (FMP-mRNA or Mock) CMRF-56+ BDC were assessed for IL-6, TNF and IL-12 by cytometric bead arrays after 20 h (n = 5; paired t-test).
The relatively poor migration of Mo-DC from injection sites to draining LNs reported in clinical studiesCitation31,32 prompted us to test the migration capacity of CMRF-56+ APC including BDC, B cells and monocytes. In vitro transwell chemotaxis assays established that transfection did not impair BDC migration to CCL21 and perhaps slightly increased their migratory capacity (). Only the CMRF-56+ BDC, which expressed higher levels of CCR7, and not the B cells and monocytes migrated to CCL21 in vitro (p < 0.001).
Figure 4. Minimally manipulated CMRF-56+ BDC maintain the capacity to migrate in vitro and in vivo. (A) Expression of CCR7 on CMRF-56+ cells isolated from PBMC cultured for 15 h and stimulated with GM-CSF for 2 h was assessed by flow cytometry. A representative plot is shown. (B) Total CMRF-56+ cells that were activated with GM-CSF and either FMP–mRNA or Mock transfected were allowed to migrate in transwell plates to CCL21 for 4 h. Migrated cells were harvested, stained for lineage markers (CD14, CD19, CD20) and hCMRF-56. BDC, residual monocytes and B cells were enumerated by flow cytometry and absolute numbers determined. Migration of BDC was compared to that of residual B cells and monocytes within individual wells. (n = 4; one-way ANOVA with Dunnett's multiple comparisons test). (C) Human Mo-DC activated with clinical cytokine cocktail (IL-1β, IL-6, TNF, PGE2) or CMRF-56+ BDC activated with GM-CSF alone were administered intra-dermally into the tail base of NK cell depleted SCID mice. Cells were allowed to migrate for 6 h after which the inguinal LN was harvested and the presence of human Mo-DC or CMRF-56+ BDC assessed by flow cytometry. (D) The recovery of BDC in inguinal LN was calculated as the number of recovered BDC or Mo-DC as a proportion of the number of DC transferred based on the purity of each preparation as determined by flow cytometry. The combined data from three independent Mo-DC preparations and three primary CMRF-56+ BDC preparations in individual mice is shown.
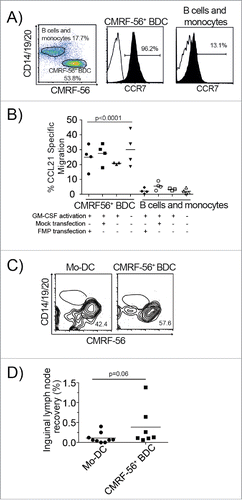
To investigate whether this might reflect better BDC migration in vivo, and since the CMRF-56 antigen is not present in mice, we established that human BDC migrated to mouse CCL19 in vitro (data not shown) prior to comparing migration of hCMRF-56 GM-CSF stimulated BDC to cytokine activated Mo-DC using a SCID mouse model.Citation42 Flow cytometric analysis of inguinal LNs, 6 h after intra-dermal injection of CMRF-56 BDC into the tail base, revealed that migrating human CD45+ cells were exclusively hCMRF-56 BDC and not CD14+, CD19+ or CD20+ cells (). Unlike the monocytes within the CMRF-56+ cells, cytokine activated Mo-DC treated with PGE2, as required for their migration, were recoverable to varying degrees from draining LN. The direct comparison of Mo-DC migration versus CMRF56+ BDC migration to the draining LN resulted in a trend toward an increased number of BDC migrating to the LN, but this fell short of statistical significance (p = 0.06) (), possibly because of other molecular compatibilities which limited human cell migration.
IVT-mRNA loaded BDC generate primary anti-viral and antitumor T-cell responses
To assess the ability of mRNA-loaded BDC to stimulate T-cell responses, we first tested the ability of FMP IVT-mRNA loaded HLA-A*0201+ hCMRF-56+ BDC to induce autologous recall antigen-specific responses. Intracellular IFNγ was readily observed in T cells stimulated with FMP-mRNA but not in transfected hCMRF-56+ BDC in the absence of mRNA. Interestingly, IFNγ production was observed in both CD4+ and CD8+ T cells, despite an immunodominant CD8+ epitope being used to re-stimulate cells (), suggesting that HLA Class II epitopes had also been presented to CD4+ T cells.
Figure 5. mRNA-loaded CMRF-56+ BDC induce recall T-cell responses to Influenza viral antigens and primary T-cell responses response to the TAA, WT1. (A) CMRF-56+ BDC isolated from HLA-A*0201+ donors were activated with GM-CSF and either transfected with IVT-FMP mRNA or with vehicle alone (Mock). These were mixed at a 10:1 ratio of autologous CMRF-56− cells: BDC. The cells were supplemented with IL-2, IL-7 and IL-15 at day 3 and again with IL-2 at day 5. On day 7, cells were re-stimulated with FMP58–66 peptide for 6 h and Brefeldin A for the last 4 h and analyzed by flow cytometry. (B) CD4 and CD8 IFNγ responses were quantified compared to Mock controls (n = 4 independent donors). Mock or WT1 mRNA transfected, HLA-A*0201 CMRF-56+ BDC were used to prime autologous CMRF-56− cells for 7 d. (C and D) Antigen specific WT1126–134 peptide specific IFNγ production (n = 7, paired t-test) and (E and F) extracellular CD107a expression (n = 3, paired t-test) was measured by flow cytometry following 6 h of peptide re-stimulation. (G and H) The frequency of dextramer+ WT1126–134-specific CD8+ T cells was increased following two additional rounds of expansion with WT1126–134 peptide for two donors and these cells had (I) the capacity to lyse T2 target cells pulsed with WT1126–134 but not control cells pulsed with KLK4 11–19 peptide. Effector: target ratio of 50:1 is shown. n = 3; one-tailed t-test.
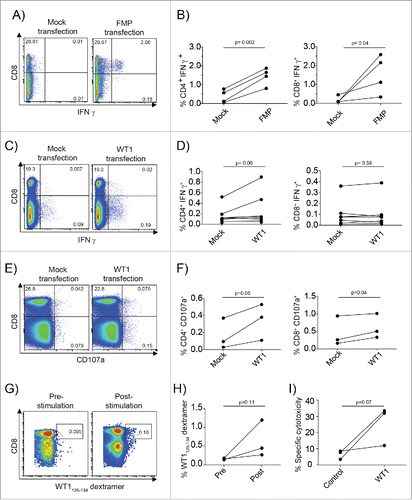
To test the ability of mRNA-loaded CMRF-56+ BDC to induce primary TAA-specific T-cell responses, we used the established AML TAA, Wilms tumor 1 (WT1). Transfection of HLA-A*0201+ healthy donor hCMRF-56+ BDC with IVT-mRNA encoding WT1 induced antigen specific IFNγ in both CD4+ and CD8+ T cells, days after a single priming event (). To confirm their cytotoxic capacity, we measured surface CD107a expression as a marker for cytotoxic granule exocytosis in peptide re-stimulated T cells. The mRNA-BDC primed CD4+ and CD8+ T cells significantly upregulated CD107a in response to WT1126–134 peptide re-stimulation (CD4+, p = 0.05; CD8+ p = 0.04), indicating their cytotoxic capacity (). To test cytotoxic potential, we subjected WT1-mRNA BDC primed T cells to two further rounds of peptide specific re-stimulation. Expanded specific WT-1126–134 − T cells () killed WT-1126–134, but not irrelevant KLK411–19 peptide loaded T2 cells ().
Discussion
The vast majority of therapeutic DC vaccination clinical trials have used in vitro-generated Mo-DC. These are both transcriptionally and functionally different to BDC and fail to migrate after injection to draining LNs, despite various manipulations to correct this deficit.Citation31,32 Given the potential of therapeutic DC vaccination for treating hematological and other malignancies, there is merit in investigating the postulated advantages of using primary BDC as an alternative to Mo-DC.Citation1 Initial clinical studies have shown that immune selected BDC preparations are safe and able to induce clinical responses in patients.Citation19,22,Citation43 The readily obtained CMRF-56+ BDC preparation described here was enriched for all myeloid BDC subpopulations and these were shown to have the expected intrinsic migration capacity, without the need for modulatory compounds such as PGE2. Furthermore, we found that these BDC were also superior to Mo-DC with respect to their ability to process and present mRNA antigens for MHC presentation to T cells.
The lack of suitable methodology to isolate primary BDC from leukapheresed PBMC has limited their use in clinical trials. The density gradient separation methodology used in Sipuleucel-TCitation44-46 enriched for low-density cells, but only 18% of these were CD54+ HLA-DR+ cells,Citation46 and of these, only a small proportion were BDC. Magnetic bead immune selection of individual populations of CD1c+ mDCCitation19,20 or CD304+ pDCCitation22 is starting to be explored in clinical trials. The generic platform used in this study built on previous work with the original mouse IgG1 CMRF-56 mAbCitation47 by developing a clinically suitable hCMRF-56 mAb to enrich for a similar heterogeneous BDC population. The platform can isolate sufficient BDC for a patient vaccination schedule and the peptide loaded CMRF-56+ BDC can generate tumor specific immune responses.Citation24,43,Citation24 The CMRF-56 selected cell preparation comprised over 80% APC and included both CD1c+ and CD141+ mDC subsets, which cannot be enriched readily using other technology. There are also significant advantages of a single-step mAb isolation platform over the alternative two or more mAb based selection methods. Notably, it reduces the time of the selection process and downstream effects on function, including product viability. Our experience using two-step isolation of CD1c DC from patient apheresis collections for our clinical trial made it clear that the processing time and the ultimate DC yield were practical issues.Citation19 Culturing PBMC without additional supplements in the apheresis bag for 12–16 h (done conveniently overnight) upregulated the CMRF-56 antigen sufficiently for hCMRF-56 mAb isolation. CD83, CD86 and CCR7 were induced and the use of GM-CSF as a stimulating agent supports their survival in vitro.Citation48 Transfection with mRNA further upregulated the costimulatory molecules. This BDC phenotype is optimal for cell migration, T-cell priming and generating cytotoxic T-cell responses.Citation24 CD54 was used as a surrogate DC activation marker to quality control Sipuleucel-T,Citation41 but its high expression on overnight cultured CMRF-56+ BDC makes it less useful in this context. However, CD80 and CD40 appear to be excellent biomarkers for monitoring the quality of clinical CMRF-56+ BDC preparations.
This is the first reported use of IVT-mRNA to load BDC and we show it is uniquely effective, with rapid processing and presentation of FMP peptides in HLA class I complexes within 2 h. BDC presentation was faster than that of B cells or monocytes, which expressed fewer cell surface HLA class I peptide complexes. The unique potential of BDC for vaccination was emphasized by the fact that HLA class I: FMP peptide complexes remained present for prolonged periods on their surface while disappearing rapidly on other APC. CMRF-56+ BDC (including both CD1c+ and CD141+ DC) exhibited enhanced capacity to present HLA class I peptide complexes compared to Mo-DC, which lost the bulk of their HLA-peptide complexes by 24 h, akin to the monocytes from which they were derived.
The migration of injected DC to the draining LN is necessary to engage T cells and initiate a response to TAA. Limited numbers of Mo-DC drain to the LN 24 h after injection and the vast majority remain at the site of injection.Citation31,32 Thus, we were particularly interested in how CMRF-56+ BDC migrated and whether their transfection with IVT-mRNA impacted this. The unique potential of BDC was once again emphasized by their specific migration to the LN homing chemokine CCL21, whereas there was little response by other APC, notably B cells and monocytes. Importantly, hCMRF-56+ BDC maintained their migration capacity in vitro irrespective of mRNA transfection. Our in vivo xeno-migration studies using a preclinical immunosuppressed mouse model suggested that human BDC, but not other APC, migrated to draining LNs. Cytokine activated, PGE2 treated Mo-DC migrated somewhat less effectively in this model. These are encouraging data and further optimization of BDC migration using alternative or additional activation stimuli might be investigated. Nevertheless, molecular incompatibilities between species limit this model and the use of primary BDC in a clinical trial with clinical imaging of their migration as the major endpoint is the next logical step.
The use of IVT mRNA to load TAA into CMRF-56+ BDC, combined with a short period of GM-CSF activation appears to generate an effective potential therapeutic DC vaccine that results in high levels of sustained antigen presentation, without compromising their enhanced migratory capacity. Mo-DC limitations demonstrated in these two key properties may go some way to explain the variable results seen using these cells in clinical trials to date. It also implies that the clinical benefits seen to date with Mo-DC may result from their reprocessing and presentation by host DC from the injection site. The results of therapeutic vaccination in AML are encouragingCitation9,10 and our initial results suggest that CMRF-56+ BDC can be generated from patients with AML in complete remission. The possibility that the use of BDC vaccination might improve these results merits further investigation. Using CMRF-56+ BDC to vaccinate AML patients against the TAA WT1 is therefore a logical path forward. The hCMRF-56 selection of BDC is clinically feasible and compatible with outpatient and hospital scheduling; nevertheless, further improvements in its efficacy are possible. It is likely, given the low inflammatory profile of mDC (including CD1c+ DC,Citation49 the major fraction of hCMRF-56 BDC), that improved stimulation of BDC (e.g. by CD40 ligation) might increase their migration or their T-cell priming potency.Citation50 The presence of PDL1 on the CMRF-56+ BDC confirmed our HLDA10 observationsCitation35 but, importantly, emphasizes combining BDC vaccination with co-incidental administration of PD1 blocking mAb may well enhance the antitumor T-cell response.
In summary, we have shown that the hCMRF-56 mAb can, in a single-step immune selection, isolate a heterogeneous population of blood APC enriched for BDC, including the important cross-presenting CD141+ mDC subset. Following transfection with IVT-mRNA, CMRF-56+ BDC present high levels of MHC class I peptide complexes for a sustained period and generate potent memory anti-viral and primary anti-TAA CTL responses. The cells maintain their inherent migratory capacity and migrate effectively in vivo. The CMRF-56+ BDC vaccine is easy to make and obviates the need to select HLA-compatible patients or antigen components. Documenting the migratory capacity of these BDC in humans using modern imaging techniques is now a priority.
Methods
Ethics
Venous blood was collected from healthy donors with informed consent under approval of the Sydney Local Health District (SLHD) Human Research Ethics Committee (HREC) consistent with the Helsinki declaration of 1975 (HREC/11/CRGH/61). Animal experiments were approved by the SLHD Animal Care and Ethics Committee (Protocol Number: 2011/029C).
Generation of human chimeric CMRF56 antibody
The heavy and light variable regions of CMRF-56 were amplified from cDNA prepared from the mouse hybridoma MMRI-0384, as described previously,Citation51 and cloned into pCR2.1 (Life Technologies). A 1017bp SmaI fragment representing the IgG4 heavy chain was cloned from mRNA derived from human B cells cultured in the presence of IL-4 (Life Technologies, PHC0044) and anti-CD40L (Biolegend). The hCMRF-56VH-IgG4 fragment was constructed from these clones by splice overlap extension PCR.Citation52 The PCR fragment was cloned into the KpnI and NotI sites of pBudCE4.1. The CMRF-56VL was cloned by splice overlap extension into SpeI and XbaI sites downstream of the CMV promoter of pBudCE4.1. The chimeric hCMRF-56 IgG4 was stably transfected into CHO cells and a master cell bank established. A batch of hCMRF-56 IgG4 GMP grade clinical mAb was produced and biotinylated by Q-Gen (QIMR-Berghofer Medical Research Institute).
DC isolation or generation
PBMC were isolated from fresh venous blood using Ficoll-Paque PLUS density gradient centrifugation (GE Healthcare, 17-1440-03). PBMC were cultured at 1 × 10Citation7 cells/mL in X-Vivo 15 (Lonza, 04-744Q) for 12–16 h in T75 tissue culture plates (Corning, FAL353136) at 37°C in 5% CO2. CMRF-56 BDC were isolated as described previously.Citation24 Cultured PBMC were stained with 1.25 μg/mL hCMRF-56 biotinylated antibody (DendroCyte Biotech), followed by anti-biotin microbeads (Miltenyi Biotec, 130-090-485) and positively selected using the Posseld2 program on an AutoMACS Pro Separator (Miltenyi Biotec).
Mo-DC were generated by culturing monocytes, selected using CD14 microbeads (Miltenyi Biotec, 130-050-021), with 800 U/mL GM-CSF (Miltenyi Biotec, 130-095-372) and 1,000 U/mL IL-4 (Life Technologies, PHC0044) for 7 d as described previously.Citation53 Maturation of Mo-DC was achieved by addition of a cytokine cocktail containing 5 ng/mL IL-1β (R&D Systems, 201-LB-005) 150 ng/mL IL-6 (R&D Systems, 206-IL-010), 5 ng/mL TNFα (R&D Systems, 210-TA-005) and 1 μg/mL PGE2 (Sigma-Aldrich, P0409) between days 5 and 7.Citation54
Production and loading of IVT- mRNA
The mRNA expression vectors pGEM4Z/A64 containing full-length coding sequences for enhanced Green Fluorescent Protein (EGFP) (pGEM4Z/EGFP/A64) or Influenza matrix protein M1 (FMP) (pGEM4Z/FMP/A64) were kindly provided by Professor Eli Gilboa.Citation55 WT1 antigen IVT-mRNA was produced from pGEM4Z/WT1/A64 or purchased from CureVac.Citation8 Plasmids were linearized with SpeI (New England Biolabs, R0133S) and IVT was performed using a mMessage Machine T7 Ultra (Life Technologies, AM1345) as described previously.Citation56 The quantity and quality of mRNA were analyzed using a Bioanalyzer 2000 (Agilent). BDC or Mo-DC (1–2 × 10Citation6) were transfected with 10 µg IVT-mRNA mRNA using an Amaxa Nucleofector 4D with P3 primary cell buffer (Lonza V4XP-3024). Samples that were transfected in the absence of mRNA (mock) were used as described.
Specific FMP HLA-A*0201 antigen presentation was measured by transfecting cells with FMP IVT-mRNA or irrelevant WT1 IVT-mRNA. FMP antigen presentation was measured kinetically by staining cells with HLA-A*0201 FMP mAb. The median fluorescence intensity of the control IVT-mRNA was subtracted from the FMP-IVT transfected sample to determine specific MHC I FMP presentation.
Flow cytometry
Cells were stained with anti-human antibodies to the following specificities: CD3 (clone HIT3a; Biolegend or clone SP34-2; BD Biosciences), CD4+ (clone RPA-T4; BD Biosciences), CD8+ (clone RPA-T8, BD Biosciences), CD14 (clone M5E2, BD Biosciences), CD19 (clone HIB19, BD Biosciences), CD20 (clone 2H7, BD Bioscience), CD40 (clone 5C3, BD Biosciences), CD45 (clone 2D1, BD Bioscience), CD54 (clone HA58, BD Biosciences), CD56 (clone B159, BD Biosciences), CD80 (clone L307.4, BD Biosciences), CD107a (clone H4A3, BD Biosciences), CD141 (clone AD5-14H12, Miltenyi Biotec or clone 1A4, BD Biosciences), CD274 (PDL1; clone M1H1, BD Biosciences), IFNγ (clone B27, BD Biosciences), CD83 (clone HB15a, Beckman Coulter), CD86 (clone IT2.2, Biolegend), CCR7 (clone 150503, R&D Systems), CD1c (clone AD5-8E7, Miltenyi Biotec) and CD304 (clone AD5-17F6, Miltenyi Biotec). Presentation of FMP58–66 was detected using an anti-HLA-A2:FMP58–66 mAb (clone 405H1.01, Dendritics, DDX0270). Cell viability was determined with propidium iodide or 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies, ) or Zombie Aqua (Biolegend, 423102). Intracellular staining was performed using the FoxP3 Fix/Perm kit (eBioscience, 00-5523-00). Fluorescence minus one (FMO) controls were used to determine positivity. Soluble TNF (BD Biosciences, 560112), IL-6 (BD Biosciences, 558276) and IL-12p70 (BD Biosciences, 558283) from cultured hCMRF-56+ immunoselected cell supernatants were measured using a cytometric bead array with the human soluble buffer kit (BD Biosciences, 558264) as per the manufacturer's protocol. Flow cytometry data was acquired on Accuri or Influx cytometers (BD Biosciences, San Jose, USA) and analyzed using FlowJo V10 (Treestar).
T-cell expansion
Autologous T-cell stimulations were performed by co-culturing freshly isolated HLA-A*0201+ CMRF-56− responding cells with electroporated CMRF-56+ BDC activated with 1,000 IU/mL GM-CSF for 2 h at a ratio of 1 BDC stimulator: 10 responders.Citation24 Cells were cultured in RPMI 1640 (Life Technologies, 21870–092) supplemented with 10% AB serum (Sigma-Aldrich, H4522), Glutamax (Life Technologies, 35050–061), Penicillin/Streptomycin (Life Technologies, 15140–122), Non-essential amino acids (Life Technologies, 11140–050), Sodium pyruvate (Life Technologies, 11360–070) and β-Mercaptoethanol (Life Technologies, 21985–023). On day 3, cultures were supplemented with 25 U/mL IL-2 (Miltenyi Biotec, 130-093-930), 20 ng/mL IL-7 (Miltenyi Biotec, 130-093-937) and 40 ng/mL IL-15 (Miltenyi Biotec, 130-093-955), with IL-2 every 2 d thereafter. Cells were re-stimulated on day 10 with 10 μg/mL FMP58–66 (GILGFVFTL), WT-1126–134 (RMFPNAPYL) or MART1Citation25-35 (ELAGIGILTV) peptides (Mimotopes) or expanded using peptide loaded, mitomycin C (Sigma-Aldrich, M-4287-2MG) treated autologous PBMC as described.Citation24 WT1-specific T cells generated following T-cell expansion were monitored using HLA-A*0201:WT1126–134 dextramers (Immudex).
Cytotoxicity assays
Cytotoxicity was measured by the ability of expanded antigen specific T cells to lyse peptide pulsed T2 cells (174xCEM.T2; ATCC CRL_1992) labeled with calcein-AM (Life Technologies, C1430) or using chromium release where described.Citation24 T2 cells were pulsed with 10 μg/mL WT1126–134, Kallikrein 411−19 (KLK4; FLGYLILGV)) or MART126–35 peptide in X-Vivo 15 for 2 h and stained with 2.5 µM calcein-AM. Antigen specific cytotoxicity was measured after 3 h incubation at 37°C using a 50:1 effector:target ratio on an Enspire plate reader (Perkin Elmer) with an excitation at 485 nm and fluorescent emission at 520 nm. Cytotoxicity was calculated as described.Citation24,57
In vitro migration
CMRF-56 enriched cells were resuspended in X-Vivo 15 and loaded into a 5 µm transwell plate (Corning, COR3421) containing 100 ng/mL CCL21 (Miltenyi Biotec, 130-094-618). Cells were allowed to migrate at 37°C in 5% CO2 for 4 h. Migrated cells were stained with hCMRF-56, CD14, CD19 and CD20. CountBright Absolute Counting Beads (Life Technologies, C36950) were added prior to enumeration by flow cytometry. Specific CCL21 migration was calculated as a percentage using the formula (chemokine specific migrated cells – spontaneous migrated cells)/total cells.
In vivo DC migration in SCID mice
5 to 7 week-old SCID (C.B-17-Igh-1b-Prkdcscid) mice (Animal Resource Center, Perth) were injected intra-peritoneally with 200 μg anti-Asialo GM-1 (Wako Chemicals, 986–10001) to deplete NK cells, 24 h prior to adoptive transfer.Citation42,58 One million GM-CSF activated CMRF-56+ cells or cytokine matured Mo-DC were injected intra-dermally into the tail base. Inguinal LN were collected after 6 h, treated with collagenase type III (Worthington Biochemical Corporation, LS004180) as describedCitation35 and migrated BDC quantitated by flow cytometry using CountBright Absolute Counting Beads.
Statistical analysis
Statistical analyses were performed using Prism 6.0 (GraphPad Software). Standard error of the mean is shown unless otherwise stated. One-Way ANOVA, Mann–Whitney, Kruskal–Wallis with Tukey's testing for multiple hypothesis or a paired t-test were used as described. Differences with p <0.05 were considered significant.
Disclosure of potential conflicts of interest
D.N.J.H. and G.J.C. are the Directors of Dendro Cyte Biotech, which holds patents for the CMRF-56 antibody. The remaining authors declare no conflict of interest.
Author Contributions
P.D.F., M.S.P., J.L.H., N.V.K.L., N.D.V., T.H.L., P.A.S., C.E.B., R.L.P., P.V., T.N., S.M.M. and S.A.A. performed experiments; P.D.F., M.S.P., J.L.H., S.A.A., G.J.C., and D.N.J.H. analyzed results and made the figures. P.D.F., C.J.T., G.J.C., D.J.M., R.T.B., L.G.H., Z.B., K.F.B. and D.N.J.H. designed the experiments and P.D.F., M.S.P., G.J.C., R.T.B. and D.N.J.H. wrote the paper.
KONI_A_1168555_s02.docx
Download MS Word (1.4 MB)Acknowledgments
We would like to thank the healthy donors and patients for volunteering blood for research and Elizabeth Newman, Christine Fong and nurses at the Department of Hematology, Concord Repatriation General Hospital who managed the collection of samples.
Funding
This work was supported by an Australian National Health and Medical Research Council Program Grant (Program Grant I.D 543727) and Cancer Institute New South Wales Translational Program Grant (11/TPG/3-012) to D.N.J.H., K.F.B. and D.E.J., the Anthony Rothe Memorial Trust (RRE/700) and Cancer Australia Priority-Driven Collaborative Cancer Research Scheme (543713) to D.N.J.H., and a Sydney Catalyst Pilot and Seed Funding Grant to P.D.F., L.G.H. and D.N.J.H.. M.S.P. was supported by an Australian Postgraduate Award, and an Arrow Bone Marrow Transplantation PhD Scholarship. S.A.A. was a recipient of an Australian Endeavor award. C.E.B. was supported by an Enid Ng Fellowship.
References
- Wimmers F, Schreibelt G, Skold AE, Figdor CG, De Vries IJ. Paradigm shift in dendritic cell-based immunotherapy: from in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front Immunol 2014; 5:165; PMID:24782868; http://dx.doi.org/10.3389/fimmu.2014.00165
- Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014; 15:e257-67; PMID:24872109; http://dx.doi.org/10.1016/S1470-2045(13)70585-0
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10:909-15; PMID:15340416; http://dx.doi.org/10.1038/nm1100
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265-77; PMID:22437871; http://dx.doi.org/10.1038/nrc3258
- Fromm P, Gottlieb D, Bradstock KF, Hart DNJ. Cellular therapy to treat haematological and other malignancies: progress and pitfalls. Pathology 2011; 43:605-15; PMID:21897329; http://dx.doi.org/10.1097/PAT.0b013e32834b6b24
- Anguille S, Smits EL, Bryant C, Van Acker HH, Goossens H, Lion E, Fromm PD, Hart DN, Van Tendeloo VF, Berneman ZN. Dendritic cells as pharmacological tools for cancer immunotherapy. Pharmacol Rev 2015; 67:731-53; PMID:26240218; http://dx.doi.org/10.1124/pr.114.009456
- Rosenblatt J, Avivi I, Vasir B, Uhl L, Munshi NC, Katz T, Dey BR, Somaiya P, Mills H, Campigotto F et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res 2013; 19:3640-8; PMID:23685836; http://dx.doi.org/10.1158/1078-0432.CCR-13-0282
- Van Driessche A, Van de Velde AL, Nijs G, Braeckman T, Stein B, De Vries JM, Berneman ZN, Van Tendeloo VF. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy 2009; 11:653-68; PMID:19530029; http://dx.doi.org/10.1080/14653240902960411
- Van Tendeloo VF, Van de Velde A, Van Driessche A, Cools N, Anguille S, Ladell K, Gostick E, Vermeulen K, Pieters K, Nijs G et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms' tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci USA 2010; 107:13824-9; PMID:20631300; http://dx.doi.org/10.1073/pnas.1008051107
- Berneman ZN, Van de Velde AL, Willemen Y, Anguille S, Saevels K, Germonpre P, Huizing MT, Peeters M, Snoeckx A, Parizel P et al. Vaccination with WT1 mRNA-electroporated dendritic cells: report of clinical outcome in 66 cancer patients. Blood 2014; 124:310-310
- Lendvai N, Cohen AD, Cho HJ. Beyond consolidation: auto-SCT and immunotherapy for plasma cell myeloma. Bone marrow transplantation 2015; 50:770-780; PMID:25751647; http://dx.doi.org/10.1038/bmt.2015.5
- MacDonald KPA, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DNJ. Characterization of human blood dendritic cell subsets. Blood 2002; 100:4512-20; PMID:12393628; http://dx.doi.org/10.1182/blood-2001-11-0097
- Ding Y, Ju X, Azlan M, Hart DN, Clark GJ. Screening of the HLDA9 panel on peripheral blood dendritic cell populations. Immunology letters 2011; 134:161-6; PMID:20970455; http://dx.doi.org/10.1016/j.imlet.2010.10.010
- Kassianos AJ, Jongbloed SL, Hart DN, Radford KJ. Isolation of human blood DC subtypes. Methods Mol Biol 2010; 595:45-54; PMID:19941104; http://dx.doi.org/10.1007/978-1-60761-421-0_3
- Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 2010; 207:1273-81; PMID:20479115; http://dx.doi.org/10.1084/jem.20100348
- Kassianos AJ, Hardy MY, Ju X, Vijayan D, Ding Y, Vulink AJ, McDonald KJ, Jongbloed SL, Wadley RB, Wells C et al. Human CD1c (BDCA-1)(+) myeloid dendritic cells secrete IL-10 and display an immuno-regulatory phenotype and function in response to Escherichia coli. Eur J Immunol 2012; 42:1512-22; PMID:22678905; http://dx.doi.org/10.1002/eji.201142098
- Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 2010; 207:1247-60; PMID:20479116; http://dx.doi.org/10.1084/jem.20092140
- Clark GJ, Kupresanin F, Fromm PD, Ju X, Muusers L, Silveira PA, Elgundi Z, Gasiorowski RE, Papadimitrious MS, Bryant C et al. New insights into the phenotype of human dendritic cell populations. Clinical and Translational Immunology 2015; 5:e61; PMID:26900474; http://dx.doi.org/10.1038/cti.2015.40
- Prue RL, Vari F, Radford KJ, Tong H, Hardy MY, D'Rozario R, Waterhouse NJ, Rossetti T, Coleman R, Tracey C et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J Immunother 2014; 38:71-6; PMID:25658616; http://dx.doi.org/10.1097/CJI.0000000000000063
- Schreibelt G, Bol KF, Westdorp H, Wimmers F, Aarntzen EH, Duiveman-de Boer T, van de Rakt MW, Scharenborg NM, de Boer AJ, Pots JM et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin Cancer Res 2015; 22:2155-66; PMID:26712687; http://dx.doi.org/10.1158/1078-0432.CCR-15-2205
- Schreibelt G, Klinkenberg LJ, Cruz LJ, Tacken PJ, Tel J, Kreutz M, Adema GJ, Brown GD, Figdor CG, de Vries IJ. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 2012; 119:2284-92; PMID:22234694; http://dx.doi.org/10.1182/blood-2011-08-373944
- Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, Boerman OC, Croockewit S, Oyen WJ, van Rossum M et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res 2013; 73:1063-75; PMID:23345163; http://dx.doi.org/10.1158/0008-5472.CAN-12-2583
- Jefford M, Schnurr M, Toy T, Masterman KA, Shin A, Beecroft T, Tai TY, Shortman K, Shackleton M, Davis ID et al. Functional comparison of DCs generated in vivo with Flt3 ligand or in vitro from blood monocytes: differential regulation of function by specific classes of physiologic stimuli. Blood 2003; 102:1753-63; PMID:12738673; http://dx.doi.org/10.1182/blood-2002-12-3854
- Freeman JL, Vari F, Hart DN. CMRF-56 immunoselected blood dendritic cell preparations activated with GM-CSF induce potent antimyeloma cytotoxic T-cell responses. J Immunother 2007; 30:740-8; PMID:17893566; http://dx.doi.org/10.1097/CJI.0b013e31814fb2d6
- Osugi Y, Vuckovic S, Hart DN. Myeloid blood CD11c(+) dendritic cells and monocyte-derived dendritic cells differ in their ability to stimulate T lymphocytes. Blood 2002; 100:2858-66; PMID:12351396; http://dx.doi.org/10.1182/blood.V100.8.2858
- Robbins SH, Walzer T, Dembele D, Thibault C, Defays A, Bessou G, Xu H, Vivier E, Sellars M, Pierre P et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Gen Biol 2008; 9:R17; PMID:18218067; http://dx.doi.org/10.1186/gb-2008-9-1-r17
- Lindstedt M, Lundberg K, Borrebaeck CAK. Gene family clustering identifies functionally associated subsets of human in vivo blood and tonsillar dendritic cells. J Immunol 2005; 175:4839-46; PMID:16210585; http://dx.doi.org/10.4049/jimmunol.175.8.4839
- Hemont C, Neel A, Heslan M, Braudeau C, Josien R. Human blood mDC subsets exhibit distinct TLR repertoire and responsiveness. J Leukoc Biol 2013; 93:599-609; PMID:23341538; http://dx.doi.org/10.1189/jlb.0912452
- Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu YJ, Blazar BR, Chen W. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J Immunol 2004; 173:4433-42; PMID:15383574; http://dx.doi.org/10.4049/jimmunol.173.7.4433
- Tel J, Lambeck AJ, Cruz LJ, Tacken PJ, de Vries IJ, Figdor CG. Human plasmacytoid dendritic cells phagocytose, process, and present exogenous particulate antigen. J Immunol 2010; 184:4276-83; PMID:20304825; http://dx.doi.org/10.4049/jimmunol.0903286
- De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, Strijk SP, Ruers TJ, Boerman OC, Oyen WJ et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res 2003; 63:12-7; PMID:12517769
- Verdijk P, Aarntzen EHJG, Lesterhuis WJ, Boullart ACI, Kok E, van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JFM et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res 2009; 15:2531-40; PMID:19318472; http://dx.doi.org/10.1158/1078-0432.CCR-08-2729
- Clark G, Munster D, Yusuf S, Hart DN. Eighth leucocyte differentiation antigen workshop DC section summary. Cell Immunol 2005; 236:21-8; PMID:16168976; http://dx.doi.org/10.1016/j.cellimm.2005.08.005
- Mason D, Andre P, Bensussan A, Buckley C, Civin C, Clark E, de Haas M, Goyert S, Hadam M, Hart D et al. CD antigens 2001: aims and results of HLDA workshops. Stem Cells 2001; 19:556-62; PMID:11713348; http://dx.doi.org/10.1634/stemcells.19-6-556
- Vremec D. The isolation of mouse dendritic cells from lymphoid tissues and the identification of dendritic cell subtypes by multiparameter flow cytometry. Methods Mol Biol 2010; 595:205-29; PMID:19941115; http://dx.doi.org/10.1007/978-1-60761-421-0_14
- Clark G, Stockinger H, Balderas R, van Zelm MC, Zola H, Hart DNJ, Engel P. Nomenclature of CD molecules from the tenth human differentiation leucocyte antigen workshop. Clin Transl Immunol 2016; 5:e57; PMID:26900471; http://dx.doi.org/10.1038/cti.2015.38
- Ohradanova-Repic A, Machacek C, Fischer MB, Stockinger H. Differentiation of human monocytes and derived subsets of macrophages and dendritic cells by the HLDA10 monoclonal antibody panel. Clin Trans Immunol 2016; 5:e55; PMID:26900469; http://dx.doi.org/10.1038/cti.2015.39
- Radford KJ, Turtle CJ, Kassianos AJ, Vuckovic S, Gardiner D, Khalil D, Taylor K, Wright S, Gill D, Hart DN. Immunoselection of functional CMRF-56+ blood dendritic cells from multiple myeloma patients for immunotherapy. J Immunother 2005; 28:322-31; PMID:16000950; http://dx.doi.org/10.1097/01.cji.0000163592.66910.e4
- Lopez JA, Bioley G, Turtle CJ, Pinzûn-Charry A, Ho CSK, Vuckovic S, Crosbie G, Gilleece M, Jackson DC, Munster D et al. Single step enrichment of blood dendritic cells by positive immunoselection. J Immunol Methods 2003; 274:47-61; PMID:12609532; http://dx.doi.org/10.1016/S0022-1759(02)00429-5
- Vari F, Munster DJ, Hsu JL, Rossetti TR, Mahler SM, Gray PP, Turtle CJ, Prue RL, Hart DN. Practical blood dendritic cell vaccination for immunotherapy of multiple myeloma. Br J Haematol 2008; 143:374-7; PMID:18729856; http://dx.doi.org/10.1111/j.1365-2141.2008.07346.x
- Sheikh NA, Jones LA. CD54 is a surrogate marker of antigen presenting cell activation. Cancer Immunol Immunother 2008; 57:1381-90; PMID:18297282; http://dx.doi.org/10.1007/s00262-008-0474-9
- Wilson J, Cullup H, Lourie R, Sheng Y, Palkova A, Radford KJ, Dickinson AM, Rice AM, Hart DN, Munster DJ. Antibody to the dendritic cell surface activation antigen CD83 prevents acute graft-versus-host disease. J Exp Med 2009; 206:387-98; PMID:19171763; http://dx.doi.org/10.1084/jem.20070723
- Wilkinson R, Woods K, D'Rozario R, Prue R, Vari F, Hardy MY, Dong Y, Clements JA, Hart DN, Radford KJ. Human kallikrein 4 signal peptide induces cytotoxic T cell responses in healthy donors and prostate cancer patients. Cancer Immunol Immunother 2012; 61:169-79; PMID:21874303; http://dx.doi.org/10.1007/s00262-011-1095-2
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010; 363:411-22; PMID:20818862; http://dx.doi.org/10.1056/NEJMoa1001294
- Small EJ, Fratesi P, Reese DM, Strang G, Laus R, Peshwa MV, Valone FH. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol 2000; 18:3894-903; PMID:11099318
- Burch PA, Breen JK, Buckner JC, Gastineau DA, Kaur JA, Laus RL, Padley DJ, Peshwa MV, Pitot HC, Richardson RL et al. Priming tissue-specific cellular immunity in a phase I trial of autologous dendritic cells for prostate cancer. Clin Cancer Res 2000; 6:2175-82; PMID:10873066
- Hock BD, Fearnley DB, Boyce A, McLellan AD, Sorg RV, Summers KL, Hart DN. Human dendritic cells express a 95 kDa activation/differentiation antigen defined by CMRF-56. Tissue antigens 1999; 53:320-34; PMID:10323336; http://dx.doi.org/10.1034/j.1399-0039.1999.530402.x
- van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 2012; 119:3383-93; PMID:22323450; http://dx.doi.org/10.1182/blood-2011-11-370130
- Benlahrech A, Duraisingham S, King D, Verhagen L, Rozis G, Amjadi P, Ford T, Kelleher P, Patterson S. Human blood CD1c dendritic cells stimulate IL-12-independent IFN-gamma responses and have a strikingly low inflammatory profile. J Leukoc Biol 2015; 96:873-885; PMID:25765676; http://dx.doi.org/10.1189/jlb.1A0114-058RR
- Sittig S, de Vries I, Schreibelt G. Primary human blood dendritic cells for cancer immunotherapy—tailoring the immune response by dendritic cell maturation. Biomedicines 2015; 3:282; http://dx.doi.org/10.3390/biomedicines3040282
- Krebber A, Bornhauser S, Burmester J, Honegger A, Willuda J, Bosshard HR, Pluckthun A. Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J Immunol Methods 1997; 201:35-55; PMID:9032408; http://dx.doi.org/10.1016/S0022-1759(96)00208-6
- Jones ML, Barnard RT. Chimerization of multiple antibody classes using splice overlap extension PCR. BioTechniques 2005; 38:181-2; PMID:15727121; http://dx.doi.org/10.2144/05382BM01
- Vuckovic S, Fearnley DB, Mannering SI, Dekker J, Whyte LF, Hart DN. Generation of CMRF-44+ monocyte-derived dendritic cells: insights into phenotype and function. Experimental hematology 1998; 26:1255-64; PMID:9845382
- Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol 1997; 27:3135-42; PMID:9464798; http://dx.doi.org/10.1002/eji.1830271209
- Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res 2000; 60:1028-34; PMID:10706120
- Hsu AK, Kerr BM, Jones KL, Lock RB, Hart DN, Rice AM. RNA loading of leukemic antigens into cord blood-derived dendritic cells for immunotherapy. Biol Blood Marrow Transplantation 2006; 12:855-67; PMID:16864056; http://dx.doi.org/10.1016/j.bbmt.2006.05.004
- Lichtenfels R, Biddison WE, Schulz H, Vogt AB, Martin R. CARE-LASS (calcein-release-assay), an improved fluorescence-based test system to measure cytotoxic T lymphocyte activity. J Immunol Methods 1994; 172:227-39; PMID:7518485; http://dx.doi.org/10.1016/0022-1759(94)90110-4
- Seldon TA, Pryor R, Palkova A, Jones ML, Verma ND, Findova M, Braet K, Sheng Y, Fan Y, Zhou EY et al. Immunosuppressive human anti-CD83 monoclonal antibody depletion of activated dendritic cells in transplantation. Leukemia 2015; 30:692-700; PMID:26286117; http://dx.doi.org/10.1038/leu.2015.231