
ABSTRACT
Engineering potent bispecific antibodies from single-chain variable fragments (scFv) remains difficult due to the inherent instability and insufficient binding of scFv's compared to their parental immunoglobulin format. Previously, we described a scFv-based bispecific antibody (scBA) against disialoganglioside (GD2) based on the anti-GD2 murine 5F11-scFv and the anti-CD3 huOKT3-scFv (5F11-scBA). In this study, we substituted the 5F11-scFv with the higher affinity (13-fold) hu3F8-scFv to form hu3F8-scBA. With this modification, hu3F8-scBA redirected T cells to kill GD2(+) cancer cell lines with up to 5,000-fold higher potency (femtomolar EC50) compared with 5F11-scBA (picomolar EC50) in cytotoxicity assays, even against target cells with low GD2 densities. Furthermore, hu3F8-scBA induced stronger T-cell activation than 5F11-scBA, as measured by Ca2+ flux and cytokine release. Additionally, in vivo, hu3F8-scBA suppressed tumor growth and prolonged mice survival much more effectively than 5F11-scBA, in both neuroblastoma and melanoma xenograft models. We conclude that the functional properties of scBA's can be increased substantially by relatively modest increases in antigen affinity.
Introduction
Classic antibody therapies often require Fc-mediated effector functions to achieve their antitumor effect.Citation21 As a result, cytotoxic T cells (CTLs) have been largely underutilized. By fusing T-cell binding domains (commonly anti-human CD3 epsilon) to tumor targeting domains, one can efficiently harness the incredible potency of polyclonal CTLs to target non-HLA restricted tumor-associated antigens (TAAs).Citation22 One example of this is the tandem single-chain variable fragment (scFv) bispecific antibody (scBA) which has been exploited to target various human malignancies, such as leukemia,Citation5,25 pancreatic cancer,Citation13 skin cancerCitation26 and brain cancer.Citation12 Just recently, in December 2014, blinatumomab became the first FDA approved T-cell engaging bispecific in the treatment of leukemia.Citation25 In addition to the scBA, other platforms have been developed, such as tandem diabodiesCitation17 or IgG-scFv fusions,Citation28 with many groups now testing these formats in the clinic. Thus, targeting CTLs to TAAs via bispecific antibodies provides a novel and powerful way to treat cancer; especially given not all tumors elicit endogenous T-cell responses.
Disialoganglioside (GD2) is a glycolipid, which is overexpressed on many cancers, including neuroblastoma, melanoma, osteosarcomas and even some cancer stem cells.Citation23 As a glycolipid, GD2 presents itself as a very attractive target, ranked as 12th among the most important TAAs.Citation7 Unlike protein antigens, GD2 is embedded into the membrane via its ceramide tail, with only a small pentasaccharide head group rising above the membrane surface. As a result of its extreme proximity to the cell membrane and small epitope footprint (which would be entirely engulfed by an antibody's CDR loops), it was predicted that targeting GD2 could mediate highly potent cell lysis via bispecific antibodies.Citation6 Additionally, given its value as a TAA, several anti-GD2 monoclonal antibodies have been generated, with two already in the clinic treating patients with high-risk neuroblastoma,Citation10 and with one (Unituxin™) getting FDA approval in the last year (March 2015).Citation30
Previously, we reported the arming of T cells using a chemical conjugate of anti-GD2 and anti-CD3 monoclonal antibodies to lyse GD2(+) neuroblastoma target cells.Citation29 In a follow-up report,Citation28 we described a genetically engineered, humanized, aglycosylated IgG-scFv bispecific antibody targeting GD2 on cancer cell lines with a femtomolar (fM) cytotoxic EC50. Furthermore, this platform activated CTLs in situ, drove intravenous T cells and monocytes to infiltrate tumor stroma, and ablated neuroblastoma and melanoma xenografts. To reduce the size of the bispecific antibody and potentially enhance tumor penetration, we developed an scBA format, where the anti-GD2 5F11-scFv was fused to the anti-CD3 huOKT3-scFv (5F11-scBA) and successfully redirected polyclonal CTLs against GD2(+) cancer cells in vitro and in vivo.Citation9 With these scBA platforms, we were able to improve the tumoricidal activity by introducing a disulfide bond to stabilize the 5F11-scFv, increasing the thermal stability by 10.8°C. However, the in vitro tumor cytotoxicity of CTLs in the presence of 5F11-scBA (pM to nM EC50) was not optimal when compared to our IgG-scFv bispecific format (fM EC50). Other investigators have reported that bispecific antibodies built from higher affinity scFv's could increase tumor retention and improve antitumor efficacy,Citation20 prompting us to create a higher affinity scBA.
In the current study, we evaluated the effect of a relatively modest increase in affinity against GD2 by developing a new anti-GD2 scBA, hu3F8-scBA; a tandem fusion of hu3F8-scFvCitation11 to huOKT3-scFv.Citation2 Hu3F8-scBA showed 13-fold higher affinity (KD) to GD2 than 5F11-scBA, but also a substantially lower thermal stability (Tm reduced by 20°C). Despite this, in vitro cytotoxicity assays showed that hu3F8-scBA was up to 5,000-fold more potent than 5F11-scBA achieving an EC50 in the fM range, even against target cancer cell lines with low GD2 densities. Additionally, hu3F8-scBA suppressed tumor growth in vivo and prolonged mice survival much more effectively than 5F11-scBA in both neuroblastoma and melanoma xenograft models.
Results
Designing and characterization of hu3F8-scBA
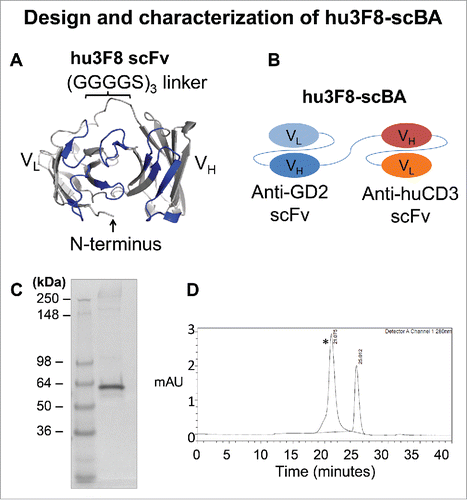
The hu3F8-scFv was designed based on the crystal structure of the original murine 3F8 antibody (PDB 3VFG), molecular docking simulations of 3F8:GD2,Citation4 and the sequence of the humanized 3F8 antibody (hu3F8)Citation11 (). The VL–VH orientation was chosen to preserve the free N-terminus of the VL domain, which was hypothesized to interact with the negatively charged head group of GD2. Utilizing identical linker and huOKT3-scFv sequences as previously reported for 5F11-scBA,Citation9 hu3F8-scBA was constructed and expressed in CHO-S cells (). After selection of high expressers from stable pools, supernatants were collected and purified by affinity chromatography. Under reducing conditions, hu3F8-scBA migrated at approximately 55 kDa (). By SEC-HPLC, it migrated as the major peak (>97%) with a retention time at 21 min confirming its molecular size (55 kDa) ().
Figure 1. Design and characterization of hu3F8-scBA. (A) Structural model showing a top down view of the antigen-biding site of hu3F8 scFv in the VL–VH orientation. CDR loops are colored in blue. A homology model was generated on Discovery Studio 4.1 (Dassault Systemes, San Diego, CA) using the crystal structure of murine 3F8 as a template (PDB 3VFG). The model was rendered in PyMol (Schrödinger LLC, New York, NY). (B) Diagram of hu3F8 scBA, with anti-GD2 scFv in the VL–VH format, and the anti-CD3 scFv in the VH–VL format. (C) Reduced SDS-PAGE analysis of hu3F8-scBA (D) HPLC profile of purified hu3F8-scBA. The peak with a retention time of 21 minutes (*) is hu3F8-scBA, while the peak with a retention time of 25 min is from absorbance of salt in the buffer (sodium citrate).
Stability and affinity of hu3F8-scBA versus 5F11-scBA
The best 5F11-scBA, (Y)5VHVLDS(15)BA, which used a VH–VL orientation and included both a stabilizing disulfide bond and an affinity maturation mutation in the 5F11-scFv, was used as a reference for these studies.Citation9 Using differential scanning fluorimetry (DSF), the melting temperature (Tm) of each scFv was measured (). The Tm for hu3F8 scFv (48.7°C) was much lower than that of 5F11 scFv (68.2°C). Interestingly, the Tm of huOKT3-scFv was also influenced by the N-terminal scFv; with an N-terminal 5F11-scFv, the Tm was 52°C, but with hu3F8-scFv it became only 48.7°C (Fig. S1). Additionally, hu3F8-scBA showed much stronger binding to GD2 by ELISA (). By surface plasmon resonance, the off rate (koff) for hu3F8-scBA was 25-fold slower (8.2 × 10−4) than it was for 5F11-scBA (2.0 × 10−2), and the binding affinity (KD), hu3F8-scBA was 13-fold higher (19 nM) than 5F11-scBA (250 nM) (Fig. S2 and ).
Figure 2. GD2 binding properties of hu3F8-scBA and 5F11-scBA. (A) Comparison of hu3F8-scBA and 5F11-scBA GD2 binding by ELISA. (B) Comparison of hu3F8-scBA and 5F11-scBA GD2 binding kinetics by SPR. Sensorgram depicts 1,000 nM run from each scBA binding to GD2, normalized to 100 RU.
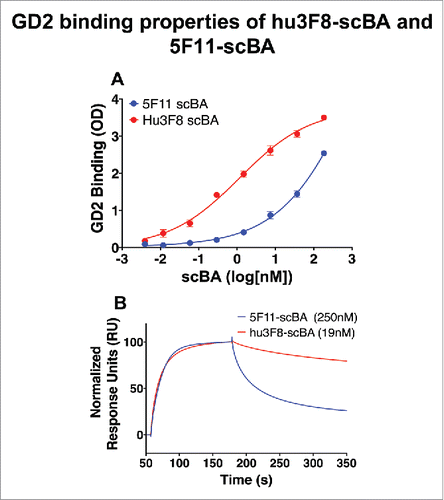
Table 1. Thermal Stability of 5F11-scBA and hu3F8-scBA. The thermal stabilities of hu3F8-scBA and 5F11-scBA were measured by differential scanning fluorimetry using the Protein Thermal Shift assay (Life Technologies).
Hu3F8-scBA induced stronger Ca2+ flux and cytokine release from T cells than 5F11-scBA
Despite the lower thermal stability, hu3F8-scBA was able to activate CTLs more potently than 5F11-scBA, as measured by both Ca2+ flux in T cells and cytokine release in PBMCs. On artificial lipid bilayers containing GD2, hu3F8-scBA induced more Ca2+ flux per T cell, over a 30-min time lapse, at both 10 and 2 nM concentrations as compared to the 5F11-scBA, indicative of more robust T-cell activation (). Additionally, the hu3F8-scBA induced higher cytokine release from human PBMCs than 5F11-scBA when incubated with GD2(+) cancer cell lines. In the presence of a melanoma derived cell line, M14, hu3F8-scBA induced significantly higher levels of IFNγ, IL-2 and IL-10 production (). Importantly, in the absence of target cells, levels of all four cytokines were barely detectable.
Figure 3. T-cell activation by hu3F8-scBA and 5F11-scBA. Human T cells were preincubated with three different concentrations of scBA (1–10 nM) and imaged on artificial lipid bilayers containing ICAM-1 and GD2. Ca2+ responses were measured using Fura-2AM. The Fura ratio was calculated during the plateau phase of the calcium response (15–30 min), from six to eight imaging fields per condition (∼100 cells/field). Each dot represents the mean Fura ratio of one field acquired over 15 min.
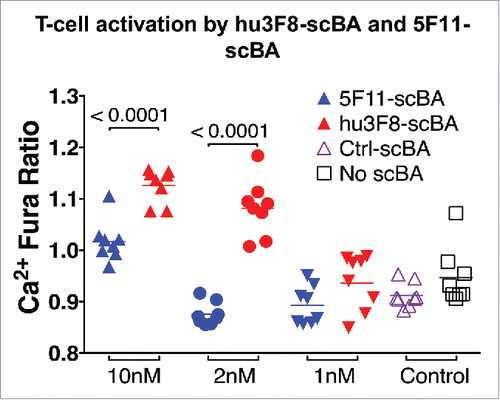
Table 2. Cytokine release from human PBMC in the presence of hu3F8-scBA and 5F11-scBA. PBMCs were isolated from a healthy donor and co-cultured with M14 cells (20:1) in the presence of 6.7 nM scBA at 37°C for 24 h. Cytokine concentrations in culture supernatant were measured by ELISA (OptEIA™ human cytokine set, BD Biosciences).
In vitro T-cell-dependent cytotoxicity mediated by hu3F8-scBA vs 5F11-scBA
The in vitro potency of hu3F8-scBA in redirecting CTLs to kill cancer cell lines was assessed by chromium release assay. Hu3F8-scBA induced highly efficient T-cell-dependent killing of both neuroblastoma derived cell lines (IMR-32) and melanoma derived cell lines (M14 and SKMEL-28) (). The EC50, as calculated from T-cell-mediated killing curves, was compared between hu3F8-scBA and 5F11-scBA, for each of the nine cancer cell lines assayed, including neuroblastoma, melanoma, osteosarcoma and ovarian carcinoma lines (). For high GD2 expressing cell lines (MFI between 1,000 and 1,500),Citation28 such as IMR-32, M14, LAN-1 and NMB-7, the EC50 for hu3F8-scBA (10–25 fM) was between 3,000- and 5,000-fold higher compared to that of 5F11-scBA (40–140 pM). For low GD2 expressing cell lines (MFI between 20 and 500) such as CRL1427, SKNBE(2)C, U2OS, SKMEL-28Citation28 EC50 for hu3F8-scBA (0.2–20 pM) was between 100- and 1,000-fold higher compared to 5F11-scBA (0.2–2.0 nM). For the GD2-negative cancer cell line SKOV3 (), neither hu3F8-scBA nor 5F11-scBA could mediate killing, confirming that both scBA's redirection of CTLs were GD2 specific.
Figure 4. Hu3F8-scBA and 5F11-scBA redirected T-cell cytotoxicity. (A) Neuroblastoma IMR-32. (B) Melanoma M14. (C) Melanoma SKMEL-28. (D) Ovarian Carcinoma SKOV3. T cells were incubated with 51Cr-labeled target cancer cells (10:1 E:T ratio) in the presence of a dilution series of scBA for 4 h. Cytotoxicity was measured by the amount of 51Cr released in the supernatant, counted by a γ-counter. Killing curves were analyzed with Graphpad Prism 6 using a nonlinear fit (log(agonist) vs response-Variable slope (four parameters)).
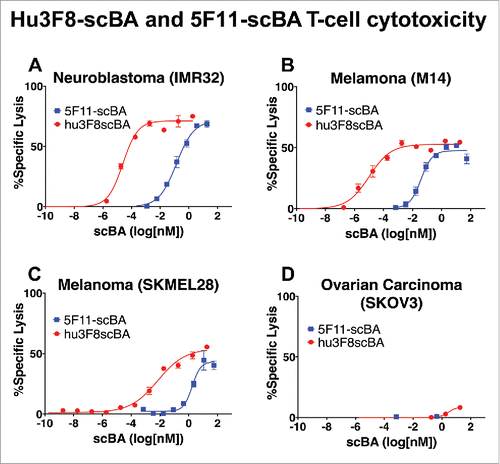
Table 3. EC50 of hu3F8-scBA and 5F11-scBA T-cell cytotoxicity against various GD2(+) cancer cell lines. EC50 was determined by 51chromium release assay. Each scBA was serially diluted to create a killing curve, and was analyzed using Graphpad Prism 6, using a nonlinear fitting algorithm (log(agonist) vs. response-Variable slope (four parameters)). GD2 expression was measured by FACS with a standard curve of counting beads (BD QuantiBRITE).
In vivo efficacy of hu3F8-scBA vs 5F11scBA
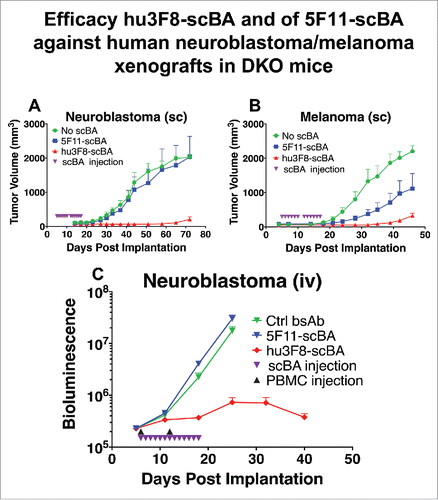
To compare the antitumor effects of hu3F8-scBA vs 5F11-scBA in vivo, two different establishedCitation28 xenograft mouse models were used: (1) human cancer cell lines were either mixed with human PBMCs and implanted subcutaneously (sc) () or (2) cancer cell lines were injected intravenously (iv) alone and once established, as determined by bioluminescence, human PBMCs were administered iv (). Treatment with 5F11-scBA or hu3F8-scBA was given daily. As shown in , in the sc model (neuroblastoma), daily injection of 180 picomoles of 5F11-scBA had negligible effect on tumor growth, while the same dosing of hu3F8-scBA suppressed tumor growth almost completely. Using another sc model (melanoma) (), 5F11-scBA treatments showed some antitumor effect while hu3F8-scBA again showed significantly better tumor suppression. For the iv model (), treatment was initiated on day 6, hu3F8-scBA again suppressed tumor growth effectively while 5F11-scBA treatment showed no appreciable effect. Survival of mice treated with hu3F8-scBA was substantially improved compared to those treated with 5F11-scBA (Fig. S3).
Figure 5. Efficacy of 5F11-scBA and hu3F8-scBA against human neuroblastoma/melanoma xenografts in DKO mice. (A) Neuroblastoma (IMR-32) or (B) melanoma (M14) and PBMCs were mixed (1:1) and coinjected subcutaneously on day 0. Treatment with scBA initiated on day 5, with 180 picomoles daily for 12 d (2 weeks). (C). Neuroblastoma (IMR32) cells and PBMCs were injected intravenously. 0.5 million neuroblastoma cells were injected on day 0, PBMCs were injected on days 6 and 12, and scBA treatment was initiated on day 6 at 180 picomoles daily for 13 d. n = 5 for each group. Bioluminescence is quantified as emitted photos per whole mouse per second.
Discussion
In this study, we successfully engineered a novel single chain bispecific antibody (hu3F8-scBA) by replacing a lower affinity anti-GD2 scFv (5F11-scFv) with a higher affinity one (hu3F8-scFv). Despite having significantly lower thermal stability, hu3F8-scBA could direct CTLs to kill cancer cells with exceptional potency, both in vitro and in vivo, without any loss in specificity.
Over the last two decades, substantial advances in our understanding of antibody structure and function have enabled the transition of bispecific antibodies from novel concepts to a clinically tested (and even approved) therapeutic agent. However, engineering the optimal scBA's for drug development remains challenging owing to the inherent in stability of these therapeutics. To improve the thermal stability of scFv's, various strategies have been applied: (1) disulfide bond engineering at the VH/VL interface;Citation16 (2) linker optimizationCitation18; (3) structure-based CDR grafting into more stable frameworks;Citation15 (4) stability screening of small scFv libraries encompassing rationally designed stabilizing mutations;Citation19 or (5) stability screening by the introduction of random mutations by yeast/phage display.Citation27 A stability-enhanced scFv usually demonstrated comparable antigen binding to the wild type scFv, although loss of antigen affinity could accompany these genetic engineering exercises. However, while affinity could be regained using phage or yeast display methods, thermodynamics-based engineering has also emerged as a promising technique for designing higher affinity scFv's.Citation24
The anti-GD2 scFv in hu3F8-scBA was derived from the sequence of hu3F8, which is currently undergoing phase I clinical trials (clinicaltrials.gov: NCT01419834, NCT01757626, and NCT01662804). Based on the mouse 3F8 crystal structure (PDB) and molecular modeling of hu3F8, the first residue of the light chain (E1) was predicted to be involved in antigen binding and should be free of further constraints – hence, the VL–VH orientation in hu3F8-scFv. In fact, recently we demonstrated experimentally that an E1K mutation could further enhance affinity at this position.Citation31 In our first generation scBA (5F11-scBA), both disulfide bond stabilization and affinity maturation were included to enhance stability and affinity. Expectedly, hu3F8-scBA had lower thermal stability than 5F11-scBA, given there was no disulfide bond stabilization. However, in terms of antigen binding, hu3F8-scBA showed higher GD2 binding than 5F11-scBA, with a 13-fold increase in affinity. This difference in affinity translated to an exceptional and unexpected increase in potency against a broad spectrum of human cancer cell lines that were GD2(+) (), mediating T-cell-mediated killing of high GD2 expressing cancer cell lines with a fM EC50 (10 fM = 0.55 pg/mL). When compared to 5F11-scBA, hu3F8-scBA demonstrated up to 5,000-fold higher potency. This affinity also led to improved T-cell activation as measured by Ca2+ flux and cytokine release. In vivo investigation using xenograft mouse models further supported that hu3F8-scBA was much more potent than 5F11-scBA in suppressing tumor growth. We are now working on enhancing hu3F8-scBA further by inserting disulfide stabilizations for both hu3F8-scFv and huOKT3-scFv, optimizing the linker size, and adding affinity maturation mutations.Citation31
In addition to the differences in affinity of hu3F8 and 5F11 to tumor antigen GD2, other factors may have contributed to the enhanced potency, such as subtle differences in binding geometries. The solvent accessible portion of the membrane bound GD2 molecule is a small penta-saccharide head group (approximately 1,500 daltons), which would be engulfed by the CDR loops of either antibody. Given the small size of the antigen, we expect that the epitopes of hu3F8 and 5F11 to be highly overlapping. Nevertheless, subtle differences in epitope may have contributed to the unexpectedly higher potency of the hu3F8 scBA. While it is widely believed that epitope distance can influence potency,Citation6 it is less clear how steric effects on antibody conformation following binding to the tumor antigen may influence activity T-cell engagement. It is possible that the distribution of GD2 on the cell surface may allow for preferential binding at one angle over another, or that the remaining huOKT3-scFv can bind to T-cells more efficiently when hu3F8-scFv binds to GD2 than when 5F11-scFv binds to GD2. While our current methods cannot rule out the above from contributing to hu3F8-scBA's large increase in potency over 5F11-scBA, we do believe that the difference in affinity is the major contributing factor.
The recent approval of Unituxin™ (anti-GD2 antibody) by FDA for neuroblastoma treatment was the first ever, human chimeric IgG1 approved against a ganglioside tumor target, and the first approved antibody drug selective for pediatric neuroblastoma. The murine 3F8 antibody has also shown promising results among patients with high risk stage 4 disease with follow up of more than 20 y.Citation14 While an IgG can only use FcR(+) effectors cells, hu3F8-BsAb and hu3F8-scBA could harness the underutilized CTLs for more effective tumoricidal activity. To date, at least nine scBAs, retargeting T cells to tumors, are under clinical investigation, with blinatumomab becoming the first in class to receive FDA approvalCitation25 suggesting that this format has strong clinical potential.
In conclusion, we discovered that relatively modest increases in the affinity of an scFv can greatly increase cytotoxicity potency of scBAs, despite reductions in thermal stability. While thermal stability may play an important role in evaluating the developability and manufacturability of therapeutic proteins, our findings indicate that when seeking to improve functional activity for an scFv in bispecific therapeutics, improvements in affinity can have a significantly higher impact on tumoricidal activity.
Materials and methods
Cell lines
LAN-1 (neuroblastoma) and M14 (melanoma) were obtained from University of California, Los Angeles; NMB-7 (neuroblastoma) was obtained from Dr SK Liao of McMaster University, Hamilton, Ontario, CA. SKNBE(2)C (neuroblastoma) was developed at Memorial Sloan Kettering Cancer Center. The following cell lines were obtained from American Type Culture Collection (ATCC), Manassas, VA, USA; neuroblastoma: IMR-32 (CCL-127); melanoma: SKMEL-28 (HTB-72); osteosarcoma: U2OS (HTB-96) and MG-63 (CRL1427), ovarian carcinoma: SK-OV-3 (HTB-77).
Cell transfection, expression and antibody purification
The hu3F8-scBA construct was constructed by ligating hu3F8-scFv to huOKT3-scFvCitation2 with a 15-residue linker (G4S)3 linker in between. The DNA subsequently was cloned into a Glutamine Synthetase (GS) mammalian expression vector and transfected into CHO-S cells (Invitrogen) by nucleofection (using nucleofection solution V and nucleofector electroporation system (Amaxa)). Stable cell lines expressing hu3F8-scBA were selected using 500 µg/mL hygromycin. The highest expressers were picked by single clone selection, and expanded in large volume culture by orbital shaking at 150 rpm in 37°C (8% CO2). Cultures were harvested when they reached desired antibody yield or when viability dropped to < 40%. Hu3F8-scBA proteins secreted into the culture supernatant were purified by Ni2+ sepharose (GE Healthcare Bio-Sciences) and eluted with 300 mM imidazole. Antibody purity was characterized by size-exclusion high-performance liquid chromatography (SEC-HPLC). 180 picomoles of protein was injected into a TSK gel G3000SWXL 7.8 mm × 30 cm, 5 µm column (TOSOH Bioscience) with 0.4 M NaClO4, 0.05 M NaH2PO4, pH 6.0 buffer at flow rate of 0.5 mL/min, and UV detection at 280 nM. A molecular weight standard (Bio-Rad) was run immediately after to allow for estimation of protein size.
ELISA
GD2 ELISAs were performed as previously described.Citation8 Briefly, GD2 (1 µg/mL in 90% ethanol, 20 µl per well) was coated on vinyl 96-well plates overnight at room temperature (RT). The plate was washed five times with PBS in between all steps. The next day, plates were blocked for 1 h with 0.5% BSA in PBS. Subsequently, a titration of each scBA as added and incubated at RT for 2 h. Bound scBA was detected with a two-step method. First, a murine anti-HIS tag antibody (AbD Serotec) (1:1,000 dilutions) was added and incubated for an hour. Next, a goat anti-mouse antibody conjugated to horseradish peroxidase (HRP) (1:3,000 dilutions) (Jackson ImmunoResearch) was added incubated at RT for 1 h. Finally, the plate was developed colorimetrically by using the o-phenylenediamine (OPD) (Sigma). The optical density (OD) of each well was measured at 490 nm using an ELISA plate reader (Dynex Technologies).
Antigen GD2 binding kinetics by surface plasmon resonance
Detailed methods for measuring 5F11-scBA and hu3F8-scBA binding to GD2 have been previously described.Citation3 Briefly, seven different concentrations of scBAs (0–2,000 nM) were diluted in HBS-EP buffer (GE) and injected over the sensor surface at a flow rate of 5 µL/min over 2 min. Following completion of the association phase, dissociation was monitored in HBS-EP buffer for 3 min at the same flow rate. The data were analyzed using the Biacore T-100 evaluation software, and the apparent association on rate constant (kon), dissociation off rate constant (koff) and equilibrium dissociation constant (KD = koff/kon) were calculated, using a 1:1 fitting algorithm.
Thermal stability measurements
The thermal stabilities of hu3F8-scBA were measured by DSF using the Protein Thermal Shift assay (Life Technologies). Hu3F8-scBA (4 µM) was mixed with Protein Thermal Shift dye and fluorescence was monitored using a StepOnePlus quantitative PCR machine (Applied Biosystems) with a 1% thermal gradient from 25 to 99°C. Data were analyzed using Protein Thermal Shift Software (Applied Biosystems) to calculate the Tm. A single domain hu3F8-scFv construct was analyzed and used to appropriately assign the correct peaks for the hu3F8-scFv and huOKT3-scFv in the scBA constructs.
Calcium flux assays
Glass chamber slides (Invitrogen) were coated with lipid bilayers containing biotin as described.Citation1 Briefly, lipid coated slides were incubated with streptavidin, followed by biotinylated GD2 (0.02 µg/mL) and biotinylated ICAM-1 (1 µg/mL). Biointylated-GD2 was kindly provided by the Consortium for Functional Glycomics. Human T cells were loaded with Fura-2 AM (Invitrogen) and then incubated with a given concentration of scBA (1–10 nM) for 30 min at 37°C. Cells were then directly added to a chamber of the coated slide, and imaged as previously described.Citation28 Each chamber was imaged at intervals of 30 s for 30 min (60 images) using a 20x objective lens (Olympus). For each chamber between six and eight imaging fields (∼100 cells/field) were acquired. Calcium induced fluorescence was measured at 340 and 380 nm wavelengths. T-cell activation was quantified using the ratio of 340–380 nm intensities during the plateau phase of the experiment (15–30 min after acquisition). Data was analyzed in Graphpad Prism; un-paired T-tests were used to evaluate statistical significance.
T-cell dependent cytotoxicity assays (51chromium release assay)
Melanoma cells (M14, SKMEL-28), and neuroblastoma cells (LAN-1, NMB-7, IMR-32), were cultured in RPMI1640 (Cellgro) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Life Technologies) at 37°C in a 5% CO2 humidified incubator. Cells were harvested with 2 mM EDTA. T cells were purified from fresh human PBMC using Pan T-cell isolation kit (Miltenyi Biotec). Anti-CD3/CD28 dynabeads (Invitrogen) were then used to stimulate and expand T cells according to manufacturer's protocol. Expanded T cells were cultured and maintained in RPMI supplemented with 10% FBS and 30 U/mL IL-2. T-cell subpopulations were acquired on a FACS Calibur with anti-CD3-PerCP-cy5.5, anti-CD4+-FITC, anti-CD8+-APC and anti-CD56-PE antibodies (BD Biosciences). Analysis was done on FlowJo version 10.
Target cancer cell lines were labeled with sodium chromate (51Cr) (Amersham, Arlington Height, IL) at 100 µCi/106 cells at 37°C for 30 min. After the cells were washed, 5,000 target cells were added per well to a 96-well polystyrene round-bottom plate along with 50,000 effector cells (E:T = 10:1) in the presence of 10-fold serial dilutions of each scBA, in a final volume of 250 µl/well, and incubated at 37°C. After 4 h, the supernatants were harvested after centrifugation for 10 min at 800g and the released 51Cr was counted using a γ-counter (Packed Instrument, Downers Grove, IL). The percentage of specific lysis was calculated from the counts per minute (CPM) using the formula (experimental cpm−background cpm)/(max cpm−background cpm) where cpm represented CPM of 51Cr released. Max cpm was assessed by lysis with 10% SDS (Sigma, St Louis, Mo), background cpm was measured in the absence of effector cells. All reported values were corrected for nonspecific T-cell lysis by subtraction of the % lysis from a control condition with no scBA. Graphs were analyzed by Graphpad Prism 6, using a nonlinear fitting algorithm (log(agonist) vs response-Variable slope (four parameters)) to determine EC50. GD2 expression was evaluated by FACS, with a standard curve of counting beads (BD QuantiBRITE)
Cytokine release assay
Fresh PBMCs were isolated from healthy donor blood by centrifugation using lymphocyte separation medium (Mediatech Inc.). PBMCs were cultured (200,000/well) with or without M14 cells (10,000/well) in the presence of 6.7 nM scBA at 37°C in a 96-well plate. Supernatants were harvested after 24 h. The concentration of four different cytokines (IL-2, IL-10, IFNγ and TNF-α) were assessed using an ELISA based cytokine assay (OptEIA™ human cytokine set, BD Biosciences) according to the manufacturer's instructions.
Xenograft mouse model
The immune deficient mouse strain BALB-Rag2-/-IL-2R-γc-KO (DKO) was kindly provided by Dr Mamoru Ito, Central Institute for Experimental Animals, Kawasaki, Japan and maintained at Memorial Sloan Kettering Cancer Center under sterile conditions. Animals were provided with Sulfatrim food. All procedures were performed in accordance with the protocols approved by our Institutional Animal Care and Use Committee (IACUC) and institutional guidelines for the proper and human use of animals in research. In vivo experiments were performed in 6–10 week old mice. PBMCs of healthy donors were isolated from human blood (New York Blood Center). Erythrocytes were depleted by incubation for 15 min with erythrocyte lysis buffer (Lonza), and thrombocytes removed after differential centrifugation at 100g for 10 min. All fresh PBMC samples used had similar percentages (30–50% CD3(+)) of T-cell subpopulations. In the sc tumor model, purified PBMCs were mixed with M14 or IMR-32 cancer cell lines (at 1:1 ratio) and implanted in DKO mice sc. Treatment with scBA was initiated on day 5 for a total of 12 d. Tumor size was measured using calipers twice a week, and tumor volumes were calculated using the approximated formula V = 0.5 (length × width × width). When tumor volumes reached 2 cm3 or greater, mice were sacrificed. For the intravenous tumor model, 0.5 million luciferase-expressing IMR-32 cells were injected iv into each mouse, where treatment with iv scBA was initiated on day 6, given daily for 13 d. PBMCs were injected iv on days 6 and 12. Tumor growth was assessed by luminescence once a week starting on day 2.
Molecular modeling
The homology model of hu3F8 scFv was generated from the crystal structure of murine 3F8 (PDB code 3VFG) using Discovery Studio (Dassault Systemes, San Diego CA). Images were rendered using PyMol (Schrodinger LLC, New York NY).
Disclosure of potential conflicts of interest
NK Cheung and M Ahmed have a financial interest in hu3F8, which was licensed by MSKCC to Ymabs Inc. The other authors declare they have no competing interests as defined by the Journal, or other interests that might be perceived to influence the results and discussion reported in this paper.
KONI_A_1168557_s02.pptx
Download MS Power Point (827.7 KB)Acknowledgments
We thank Dr Mamoru Ito of Central Institute for Experimental Animals, Kawasaki, Japan, for providing the DKO mice, Dr Gloria Koo, Hospital for Special Surgery, New York, NY, for her advice in handling DKO mice, and Dr Irene Cheung for reviewing the manuscript.
Funding
This study was supported in part by grants from the following: Isabella Foundation Band of Parents, Kids Walk for Kids with Cancer NYC, Cookies for Kids' Cancer, Robert Steel Foundation, Geoffrey Beene Cancer Research Center of Memorial Sloan Kettering Cancer Center (MSK), Technology Development Fund of MSKCC, Technical service provided by the MSK Small-Animal Imaging Core Facility and Molecular Cytology Core Facility were supported in part by the NIH Cancer Center Core Support Grant P30 CA008748-48.
References
- Abeyweera TP, Merino E, Huse M. Inhibitory signaling blocks activating receptor clustering and induces cytoskeletal retraction in natural killer cells. J Cell Biol 2011; 192:675-90; PMID:21339333; http://dx.doi.org/10.1083/jcb.201009135
- Adair JR, Athwal DS, Bodmer MW, Bright SM, Collins AM, Pulito VL, Rao PE, Reedman R, Rothermel AL, Xu D et al. Humanization of the murine anti-human CD3 monoclonal antibody OKT3. Hum Antibodies Hybridomas 1994; 5:41-7; PMID:7858182; http://dx.doi.org/10.3233/HAB-1994-51-206
- Ahmed M, Cheng M, Cheung I, Cheung NK. Human derived dimerization tag enhances tumor killing potency of a T-cell engaging bispecific antibody. OncoImmunology 2015; 4:e989776; PMID:26137406; http://dx.doi.org/10.4161/2162402X.2014
- Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett 2014; 588:288-97; PMID:24295643; http://dx.doi.org/10.1016/j.febslet.2013.11.030
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974-7; PMID:18703743; http://dx.doi.org/10.1126/science.1158545
- Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother 2010; 59:1197-209; PMID:20309546; http://dx.doi.org/10.1007/s00262-010-0844-y
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 2009; 15:5323-37; PMID:19723653; http://dx.doi.org/10.1158/1078-0432.CCR-09-0737
- Cheng M, Ahmed M, Xu H, Cheung NK. Potency and structural design of disialoganglioside and CD3-bispecific antibodies for redirecting T cells for tumor therapy. Int J Cancer 2014; 136:476-486; PMID:24895182; http://dx.doi.org/10.1002/ijc.29007
- Cheng M, Ahmed M, Xu H, Cheung NK. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer 2015; 136:476-86; PMID:24895182; http://dx.doi.org/10.1002/ijc.29007
- Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, Modak S. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-Cis-Retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol 2012a; 30:3264-70; PMID:22869886; http://dx.doi.org/10.1200/JCO.2011.41.3807
- Cheung NK, Guo H, Hu J, Tassev DV, Cheung IY. Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. OncoImmunology 2012b; 1:477-86; PMID:22754766; http://dx.doi.org/10.4161/onci.19864
- Choi BD, Kuan CT, Cai M, Archer GE, Mitchell DA, Gedeon PC, Sanchez-Perez L, Pastan I, Bigner DD, Sampson JH. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc Natl Acad Sci USA 2013; 110:270-5; PMID:23248284; http://dx.doi.org/10.1073/pnas.1219817110
- Cioffi M, Dorado J, Baeuerle PA, Heeschen C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin Cancer Res 2012; 18:465-74; PMID:22096026; http://dx.doi.org/10.1158/1078-0432.CCR-11-1270
- Dobrenkov K, Cheung NK. GD2-targeted immunotherapy and radioimmunotherapy. Semin Oncol 2014; 41:589-612; PMID:25440605; http://dx.doi.org/10.1053/j.seminoncol.2014.07.003
- Ewert S, Honegger A, Pluckthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods 2004; 34:184-99; PMID:15312672; http://dx.doi.org/10.1016/j.ymeth.2004.04.007
- Hagihara Y, Saerens D. Engineering disulfide bonds within an antibody. Biochim Biophys Acta 2014; 1844:2016-23; PMID:25038323; http://dx.doi.org/10.1016/j.bbapap.2014.07.005
- Kipriyanov SM, Moldenhauer G, Schuhmacher J, Cochlovius B, Von der Lieth CW, Matys ER, Little M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J Mol Biol 1999; 293:41-56; PMID:10512714; http://dx.doi.org/10.1006/jmbi.1999.3156
- Le Gall F, Reusch U, Little M, Kipriyanov SM. Effect of linker sequences between the antibody variable domains on the formation, stability and biological activity of a bispecific tandem diabody. Protein Eng Des Sel 2004; 17:357-66; PMID:15126676; http://dx.doi.org/10.1093/protein/gzh039
- Miller BR, Demarest SJ, Lugovskoy A, Huang F, Wu X, Snyder WB, Croner LJ, Wang N, Amatucci A, Michaelson JS et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng Des Sel 2010; 23:549-57; PMID:20457695; http://dx.doi.org/10.1093/protein/gzq028
- Orcutt KD, Slusarczyk AL, Cieslewicz M, Ruiz-Yi B, Bhushan KR, Frangioni JV, Wittrup KD. Engineering an antibody with picomolar affinity to DOTA chelates of multiple radionuclides for pretargeted radioimmunotherapy and imaging. Nucl Med Biol 2011; 38:223-33; PMID:21315278; http://dx.doi.org/10.1016/j.nucmedbio.2010.08.013
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012; 12:278-87; PMID:22437872; http://dx.doi.org/10.1038/nrc3236
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67:95-106; PMID:25637431; http://dx.doi.org/10.1016/j.molimm.2015.01.003
- Suzuki M, Cheung NK. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert Opin Ther Targets 2015; 19:349-62; PMID:25604432; http://dx.doi.org/10.1517/14728222.20.986459
- Thakkar S, Nanaware-Kharade N, Celikel R, Peterson EC, Varughese KI. Affinity improvement of a therapeutic antibody to methamphetamine and amphetamine through structure-based antibody engineering. Sci Rep 2014; 4:3673; PMID:24419156; http://dx.doi.org/10.1038/srep03673
- Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 2015; 16:57-66; PMID:25524800; http://dx.doi.org/10.1016/S1470-2045(14)71170-2
- Torisu-Itakura H, Schoellhammer HF, Sim MS, Irie RF, Hausmann S, Raum T, Baeuerle PA, Morton DL. Redirected lysis of human melanoma cells by a MCSP/CD3-bispecific BiTE antibody that engages patient-derived T cells. J Immunother 2011; 34:597-605; PMID:21904216; http://dx.doi.org/10.1097/CJI.0b013e3182307fd8
- Wiedermann U, Davis AB, Zielinski CC. Vaccination for the prevention and treatment of breast cancer with special focus on Her-2/neu peptide vaccines. Breast Cancer Res Treat 2013; 138:1-12; PMID:23340862; http://dx.doi.org/10.1007/s10549-013-2410-8
- Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T Cells to GD2 Pentasaccharide on Human Tumors Using Bispecific Humanized Antibody. Cancer Immunol Res 2015; 3:266-77; PMID:25542634; http://dx.doi.org/10.1158/2326-6066.CIR-14-0230-T
- Yankelevich M, Kondadasula SV, Thakur A, Buck S, Cheung NK, Lum LG. Anti-CD3 x anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr Blood Cancer 2012; 59:1198-205; PMID:22707078; http://dx.doi.org/10.1002/pbc.24237
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010; 363:1324-34; PMID:20879881; http://dx.doi.org/10.1056/NEJMoa0911123
- Zhao Q, Ahmed M, Guo HF, Cheung IY, Cheung NK. Alteration of electrostatic surface potential enhances affinity and tumor killing properties of anti-ganglioside GD2 monoclonal antibody hu3F8. J Biol Chem 2015; 290:13017-27; PMID:25851904; http://dx.doi.org/10.1074/jbc.M115.650903