
ABSTRACT
HSP110 is induced by different stresses and, through its anti-apoptotic and chaperoning properties, helps the cells to survive these adverse situations. In colon cancers, HSP110 is abnormally abundant. We have recently showed that colorectal cancer (CRC) patients with microsatellite instability (MSI) had an improved response to chemotherapy because they harbor an HSP110 inactivating mutation (HSP110DE9). In this work, we have used patients' biopsies and human CRC cells grown in vitro and in vivo (xenografts) to demonstrate that (1) HSP110 is secreted by CRC cells and that the amount of this extracellular HSP110 is strongly decreased by the expression of the mutant HSP110DE9, (2) Supernatants from CRC cells overexpressing HSP110 or purified recombinant human HSP110 (LPS-free) affect macrophage differentiation/polarization by favoring a pro-tumor, anti-inflammatory profile, (3) Conversely, inhibition of HSP110 (expression of siRNA, HSP110DE9 or immunodepletion) induced the formation of macrophages with a cytotoxic, pro-inflammatory profile. (4) Finally, this effect of extracellular HSP110 on macrophages seems to implicate TLR4. These results together with the fact that colorectal tumor biopsies with HSP110 high were infiltrated with macrophages with a pro-tumoral profile while those with HSP110 low were infiltrated with macrophages with a cytotoxic profile, suggest that the effect of extracellular HSP110 function on macrophages may also contribute to the poor outcomes associated with HSP110 expression.
Abbreviations
| CCL24 | = | Chemokine (C-C motif) Ligand 24 |
| CRC | = | Colorectal Cancer |
| DAMPs | = | Damage Associated Molecular Patterns |
| HSP | = | Heat Shock Protein |
| IL-1β | = | Interleukin-1β |
| M-CSF | = | Macrophage Colony-Stimulating Factor |
| MSI | = | Microsatellite Instability |
| MSS | = | Microsatellite Stability |
| NO | = | Nitric Oxide |
| SR-A | = | Scavenger Receptor-A |
| TLR | = | Toll Like Receptor |
| TNF-α | = | Tumor Necrosis Factor-α |
Introduction
Colorectal cancer (CRC) is a molecularly heterogeneous disease that can be subdivided into several molecular subtypes.Citation1 Approximately 15 to 20% of CRC harbor widespread microsatellite instability (MSI) at DNA repeats (MSI) due to mismatch repair deficiency, in contrast to the majority of CRC tumors, which show microsatellite stability (MSS).Citation2,3 MSI CRCs display particular morphologic features, including greater predilection for the right colon, mucinous histology, low metastatic power and poorer differentiation. They have been consistently reported to show an improved prognosis and a different response to chemotherapeutic agents. The molecular mechanisms of the peculiar MSI CRC pathophysiology have started to be uncovered recently. Notably, we have recently highlighted the important role of heat shock protein-110 (HSP110).Citation4-6 HSPs are a set of highly conserved proteins whose expression is induced in response to a wide variety of physiological and environmental stresses.Citation7,8 They are often overexpressed in cancer cells and contribute to cancer resistance and apoptosis.Citation9,10
HSP110 is a high molecular weight chaperone that accumulates abnormally in CRC cells and whose expression correlates with metastasis and a poor prognosis.Citation11 We have previously shown that a T17 mononucleotide repeat located in intron 8 of HSP110 was systematically mutated in MSI CRC cell lines and primary tumors.Citation6 The shortening of this repeat in tumor DNA correlated with the increased synthesis of an aberrant HSP110 transcript due to exon 9 skipping (HSP110DE9) to the detriment of wild-type HSP110 mRNA. MSI patients with large T17 deletions (low HSP110/high HSP110DE9) have significantly longer relapse-free survival (RFS) than those with small T17 deletions (high HSP110/low HSP110DE9).Citation4 Furthermore, RFS in MSI CRC patients with small T17 deletions (high HSP110 expression/low HSP110DE9) does not seem to differ significantly from that in MSS CRC patients, suggesting a strong dependence of CRC cells on HSP110. In accordance with these clinical data, our in vitro studies have demonstrated that HSP110DE9 acts as a dominant negative mutant that binds to HSP110 wild type, thus impairing its cellular localization and its ability to interact with other chaperones.Citation4,6 HSP110DE9 completely abrogates HSP110 chaperone activity and its cytoprotective function. In vitro, HSP110DE9 expression sensitized colon cancer cells, in a dose-dependent manner, to anticancer agents such as oxaliplatin and 5-fluorouracil.
Immune control of tumors is a well-established mechanism involved in the progression of various cancers, including CRC, where tumor-infiltrating lymphocytes correlate inversely with tumor stage.Citation12,13 Infiltration of activated CD8+ lymphocytes within and around the tumor stroma contributes to a better prognosis. In particular, the number of tumor-infiltrating lymphocytes is higher in MSI CRC than in MSS CRC and, besides HSP110, this may also explain the better prognosis in MSI CRC patients than in MSS CRC patients.Citation14 Tumor-associated macrophages are also abundant tumor-infiltrating cells. In general, tumor-associated macrophages can be found within or surrounding various tumors, where they either promote tumor progression, angiogenesis, migration of tumor cells and T helper 2 responses (so-called M2 or alternative macrophages), or, conversely, they promote resistance to tumors, inflammatory responses and T helper 1 responses (so-called M1 or classical macrophages).Citation15,16 Although macrophages have been found in colorectal tumor sections,Citation17,18 the correlation of their abundance with the prognosis of MSI neoplasms is not yet clear.
Extracellular HSPs are described as damage-associated molecular pattern proteins (DAMPs) with immunogenic properties. Extracellular HSPs such as HSP27 or HSP70 have been reported to influence immune control of tumors, sometimes through immunosuppressive cells.Citation19-21 HSP110 has been described as being able to inhibit immune activation of dendritic cells through scavenger receptor binding.Citation22 However, hardly anything is known about the immune function of HSP110 in cancer. Here, we have addressed the impact of extracellular HSP110 on macrophage profiles in CRC.
Results
In vivo, in patients and mice, expression of HSP110 in colorectal tumors influences the profile of infiltrating macrophages
We first determined the presence of macrophages within tumor biopsies from 10 MSI CRC patients, which were selected based on HSP110 expression and divided into two groups: HSP110-low (large T17 deletions, good prognosis patients group, n = 5) and HSP110-high (small T17 deletions, poor prognosis group, n = 5)Citation4 (). We observed a strong invasion of CD68+ macrophages in all tumor samples regardless of the expression of HSP110 at the invasive front ( ) and in the tumor stroma ( and Fig. S1B, p = 0.55 (ns), n = 5 per group). However, in HSP110 high tumors, compared to HSP110 low tumors, there was a marked increase in macrophages that expressed CD163 ( p = 0.0025, n = 5 per group), a well-described pro-tumoral (M2) macrophage marker ( show the tumor stroma, sup shows the invasive front).
Figure 1. Pro-tumoral macrophages invade tumor bed in CRC expressing high levels of HSP110 (HSP110high) A, B Expression of HSP110 (A), CD68 and CD163 (B) by IHC in stroma of tumor samples from MSI CRC patients belonging to the HSP110high and HSP110 low groups described by Collura et al (reference 4). One representative image is shown (n = 5) Brown color indicates positive staining (200x magnification for A, scale bars, 50 μm. 100x magnification for B, scale bars, 100 μm). (C) Number of CD163 macrophages in tumors biopsy stroma was determined ( p = 0.0025, n = 5) (value for each patient was determined as the average number of stained cells in three distinct sections. (D) Indicated HSPs were analyzed by Immunoblot in HCT116 and HCT116-C22 cells. HSC70 was used as a loading control. (E) Expression of F4/80, iNOS and Arginase by IHC from tumor sections of mice xenografted with HCT116 or HCT116-C22 (One image representative of six mice in each group, 20x magnification, scale bars 40 μm). (F, G) qPCR analysis of Arginase, iNOS mRNA (F) and TNFa (G) from tumor sections of mice xenografted with HCT116 or HCT116-C22. **p < 0.01, ***p < 0.005.
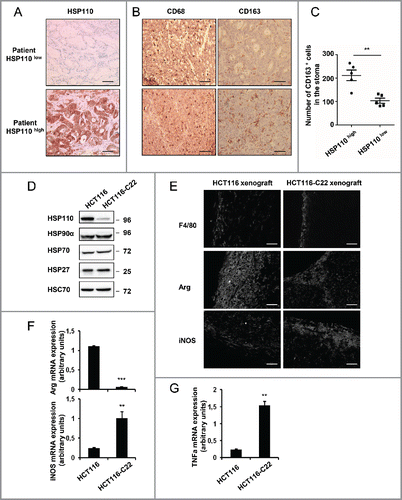
Since a low expression level of HSP110 in MSI CRC cells inversely correlates with the length of the T17 mononucleotide repeat located in intron 8 of HSP110, we next used an HCT116 sub-clone displaying a large HSP110 T17 deletion (HCT116-C22) Citation4 to determine the effect of HSP110 on macrophage phenotypes. We first confirmed the very low expression of HSP110 in this HCT116-C22 clone compared with parental HCT116 (). The expression of no other HSPs other than HSP110 seemed altered in HCT116-C22 (). Furthermore, the low expression of HSP110 in the HCT116-C22 clone did not affect HCT116 spontaneous cell death observed when cultured in vitro (). Tumor xenografts with high or low expression of HSP110 were established by subcutaneously inoculating nude mice with HCT116 or HCT116-C22 cells, respectively. Three weeks after inoculation, xenografts were excised, and the infiltrating macrophages were examined. Immunostaining revealed the presence of F4/80pos macrophages in all tumor slides (). Interestingly, whereas the macrophages from parental HCT116 xenografts expressed the M2 marker Arginase-1 (Arg-1), only the low HSP110-expressing HCT116-C22 xenograft strongly expressed inducible NO synthase (iNOS), which is a marker associated with an M1 cytotoxic phenotype (). Accordingly, mice xenografted with HCT116-C22 expressed more TNFa mRNA (). These data, both in humans and in mice, suggest that levels of HSP110 in MSI tumors may influence the profile of tumor infiltrating macrophages in vivo.
HSP110 is secreted by CRC cells and influences macrophage differentiation
To decipher the mechanism of macrophage polarization observed in the presence or absence of HSP110, we studied the secretome of HCT116 and HCT116-C22. Among the 36 cytokines analyzed, none were significantly modified (data not shown). In contrast, we observed the presence of a soluble HSP110 in the supernatant of HCT116 but not in the HCT166-C22 supernatant (). The presence of extracellular HSP110 in the supernatant was confirmed by ELISA, where the amount of HSP110 secreted by different CRC cells was quantified (). We thus hypothesized that extracellular HSP110 could be involved in the macrophage infiltration and polarization observed herein. To test this, we studied the effect of supernatants of HCT116 and HCT116-C22 cells on primary human monocytes induced to differentiate into macrophages by M-CSF. As shown in and Table S1, supernatants from HCT116-C22, compared to those of HCT116, contributed to the generation of macrophages with stronger expression of HLA-DR, but lower expression of the M2 markers CD163 and CD206. Upon stimulation with LPS, HCT116-C22 supernatants, compared with parental HCT116 supernatants, generated macrophages with higher levels of TNFa mRNA (), secreted higher levels of the pro-inflammatory cytokines TNFa and IL1b, and produced higher level of NO ( and Table S2). Conversely, CCL24, a chemokine associated with the M2 phenotype, was significantly lower. As the ability to stimulate T cell proliferation is a hallmark of pro-inflammatory M1 macrophages, we performed a mixed lymphocyte reaction and observed that more proliferating T cells were generated in the presence of HCT116-C22 supernatant than in the presence of HCT116 supernatant ().
Figure 2. HSP110 is secreted by CRC cell lines and influences macrophage differentiation profile. (A) Immunoblot analysis of HSP110 in the supernatant of HCT116 and HCT1166-C22 cells. Extracellular HSP90 is used here as a loading control. (B) Differentiating macrophages in the presence of HCT116 or HCT116-C22 supernatants were assessed for the expression of HLA-DR (n = 7), CD163 (n = 5) and CD206 (n = 5) by flow cytometry. Results are expressed as mean fluorescence ratio *p < 0.05; ***p < 0.005; Left, data of all experiments; Right, representative data. (C) Monocytes induced to differentiate in the presence of HCT116 or HCT116-C22 supernatants were stimulated for 24 h with LPS. TNFa (n = 3), IL1b (n = 3), CCL24 (n = 4) and NO (n = 4) secreted by macrophages were determined by Milliplex assay *p < 0.05; **p < 0.01. (D) Percentage of proliferating allogeneic T cells after 3 d of culture with macrophages derived from monocytes in the presence of control DMEN medium or supernatants from HCT116 or HCT116-C22 cells (n = 3), *p < 0.05. (E, F) Monocytes were induced to differentiate into macrophages in the presence of supernatant from HCT116 cells either transfected with a HSP110 siRNA or a scrambled control. Expresssion of CD163 (n = 5), and CD206 (n = 5) was determined by flow cytometry (E). (F) TNFa and IL1b from macrophages derived from monocytes in the presence of supernatants as in E, and stimulated for 24 h with LPS (n = 3) *p < 0.05; **p < 0.01.
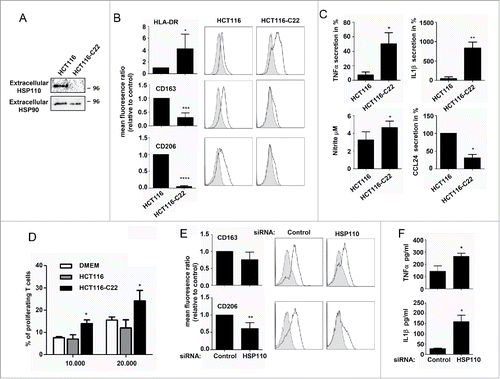
To confirm that this effect on macrophage polarization was HSP110-dependent, we next down-expressed HSP110 using siRNA (). Of note, in our experimental conditions, HSP110 knockdown had no effect on cell viability (). Lower expression of the M2 markers CD163 and CD206 was observed when supernatants from both HSP110-depleted HCT116 ( and Table S3) and SW480 cells were used (Fig. S2C and Table S9), compared to the corresponding controls. Accordingly, upon LPS stimulation, higher levels of TNFa (HCT116, and Table S4; SW480, Fig. S2D and Table S10) and IL1b (HCT116, ) were observed with supernatants of the HSP110-depleted cells.
HSP110DE9 mutant inhibits HSP110 release into the extracellular medium
HSP110DE9 is an HSP110 deletion mutant, which contains only the 1–381 amino acid ATP-domain of HSP110. It is found in all MSI CRC patients with a good prognosis.Citation4 We have previously demonstrated that HSP110DE9 overexpression in CRC cell lines blocks intracellular HSP110 anti-aggregation and anti-apoptotic functions.Citation6 To study whether HSP110DE9 may also affect extracellular HSP110, we analyzed the supernatants from CRC cells overexpressing HSP110DE9 (GFP-tagged). In parallel, supernatants from the same cells overexpressing HSP110 (GFP tagged) were also tested. Interestingly, while as expected HSP110 overexpression led to an increase in its concentration in the supernatant (), HSP110DE9 expression induced a strong decrease in the amount of HSP110 secreted, without altering the intracellular level of HSP110 (), or altering the secretome profile ().
Figure 3. HSP110DE9 hampers HSP110 release (A) Concentration of extracellular HSP110 in the supernatant of Lovo, HCT116 and SW480 transfected with a control GFP plasmid or a plasmid coding HSP110-GFP and measured by ELISA (n = 3) *p < 0.05 ; **p < 0.01. (B) ELISA quantification of HSP110 in the extracellular medium of SW480 transfected with a control GFP or HSP110DE9-GFP plasmid (n = 3) *p < 0.05. (C) Immunoblot analysis of HSP110 in the supernatant of HCT116, transfected with a HSP110-GFP plasmid with or without a plasmid coding HSP110DE9-GFP. (D) Flow cytometry analysis of HLA-DR (n = 6), CD163 (n = 6) and CD206 (n = 6) expression on macrophages derived from monocytes in the presence of supernatant from SW480 cells transfected with a control GFP, HSP110-GFP or a HSP110DE9-GFP plasmid. Left, data of all experiments; Right, representative data *p < 0.05; **p < 0.01; ***p < 0.001. (E) TNFa, and NO (nitrite) secreted by macrophages derived from monocytes in the presence of SW480 transfected as in C, and stimulated for 24 h with LPS (n = 4, *p < 0.05). (F) Percentage of proliferating allogeneic T cells after 3 d of culture with 104 macrophages derived from monocytes in the presence of supernatant from SW480 transfected with a control GFP or a HSP110DE9-GFP (n = 3). *p < 0.05.
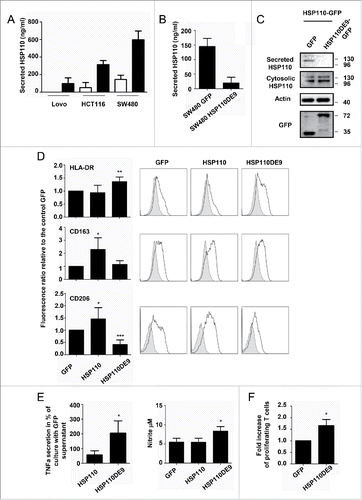
In accordance with these variations in secreted HSP110, stronger HLA-DR expression and a decrease in CD206 expression were observed when HSP110DE9 was expressed ( and Table S5). Conversely, CD163 and CD206 were both significantly increased when HSP110 was overexpressed (). Accordingly, macrophages generated with supernatants from CRC cells that overexpressed HSP110DE9 secreted higher levels of TNFa and had higher levels of NO than those generated with supernatants from control-GFP or from CRC cells overexpressing HSP110 ( and Table S6). Finally, more T cells were induced in the presence of the supernatant of cells expressing HSP110DE9 compared to control ().
Altogether, our results suggest that low secretion of HSP110 (either through siRNA-mediated depletion or by expressing the HSP110DE9 mutant) may favor the generation of macrophages with increased pro-inflammatory and cytotoxic functions.
Immunodepletion of HSP110 modulates the pro-inflammatory phenotype of macrophages
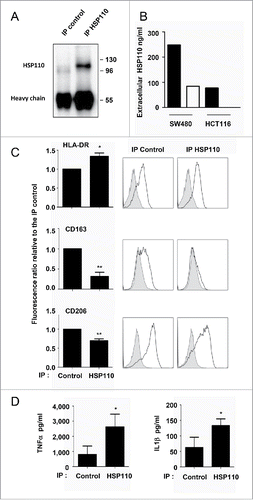
We next studied whether immunodepletion of extracellular HSP110 in the supernatants was enough to switch from the induction of anti-inflammatory macrophages to the induction of macrophages with pro-inflammatory functions. The reduction in extracellular HSP110 (but not other HSPs) after immunodepletion of HSP110 from both HCT116 and SW480 cell supernatants is shown in and Fig. S3A. Macrophages induced to differentiate with the HSP110-immuno-depleted supernatants showed reduced expression of CD163 and CD206, and increased secretion of TNFa and IL1b ( for HCT116, data in tables S7–8, and for SW480 with data in tables S11 and 12). Interestingly, addition of human recombinant HSP110 produced by eukaryotic cells (i.e. LPS free) allows at least a partial recovery of the macrophage phenotypes and TNFα secretion (). We conclude that HSP110 is an essential component in the extracellular medium for the effect observed on macrophage profiles.
Figure 4. Depletion of HSP110 from the supernatants skews the pro-inflammatory phenotype of macrophages. (A) Immunoblot analysis of HSP110 from the immunoprecipitated (IP) fraction of HCT116 supernatant (one image representative of (3). (B) ELISA determination of HSP110 amount in the supernatant of SW480 or HCT116 before (black columns) and after (white columns) HSP110 immunoprecipitation. (C), Expresssion of HLA-DR (n = 5), CD163 (n = 5) and CD206 (n = 4) by flow cytometry on macrophages derived from monocytes in the presence of HSP110-depleted HCT116 supernatant. Left, data of all experiments; Right, representative data *p < 0.05; **p < 0.01. (D) TNFa, and IL1b secreted by macrophages derived from monocytes in the presence of HSP110-depleted HCT116 supernatant, and stimulated for 24 h with LPS (n = 4) *p < 0.05.
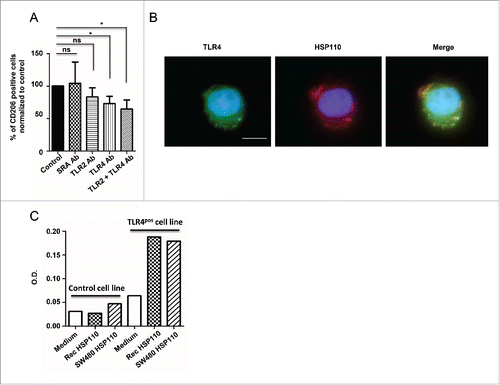
Extracellular HSP110 binds to TLR4 in human monocytes
We previously showed that HSP70 binds to and activates TLR2 on the surface of myeloid-derived suppressive cells.Citation19 To investigate whether the action of extracellular HSP110 on macrophages could also implicate a TLR pathway, we first determined whether its ability to induce the expression of the CD206 macrophage marker was modified in the presence of neutralizing antibodies against TLR2, TLR4 or the scavenger receptor SRA. We found that only neutralizing antibodies against TLR4 significantly reduced the ability of supernatants containing HSP110 to induce CD206 (). Accordingly, when human LPS-free recombinant HSP110 (produced by eukaryotic cells) was added to the cells, co-localization of this extracellular HSP110 with TLR4 was observed (). To confirm the implication of TLR4 in the effect of HSP110 on macrophages, we used TLR4 luciferase reporter cells. We incubated the TLR4-reporter or control cells with either the supernatant of SW480 cells overexpressing HSP110 (where HSP110 was measured by ELISA) or a similar amount of purified HSP110 (600 ng/mL). We found that both sources of HSP110 had a similar effect on TLR4 activation (). Taken together, these results suggest that the effect of extracellular HSP110 in altering the inflammatory profile of macrophages involves TLR4.
Figure 5. Extracellular effect of HSP110 on macrophage profile involves TLR4. (A) The percentage of HSP110-induced CD206-expressing differentiated macrophages in the presence or absence of SRA, TLR2 and/or TLR4 neutralizing antibodies was determined by flow cytometry. Data are expressed as a percentage of the control (no neutralizing Ab added)(n = 6, *p < 0.05.). (B) Fluorescence microscopy analysis of TLR4 and DDK(FLAG)-tagged HSP110 purified from eukaryotic cells (LPS free) on monocytes after 30 min of incubation with HSP110. One representative image is shown. Scale bars 10 μm. (C) TLR4 gene reporter assay (luciferase) using control HSP110-depleted supernatant, supernatant from HSP110-overexpressing HCT116 cells or a similar amount (600 ng/mL) of purified recombinant LPS-free HSP110 (n = 2). *p < 0.05.
Discussion
HSPs are chaperones that are frequently overexpressed in cancer cells. A large body of literature describes how their intracellular function is involved in the increased resistance of tumor cells to cell death notably induced by anticancer drugs or hypoxia.Citation9,10 We recently demonstrated that HSP110 was the main HSP involved in colorectal tumorigenesis. We demonstrated that its expression was directly associated with poor outcomes and that the presence of an HSP110 inactivating mutation was directly associated with a good prognosis of CRC MSI patients.Citation6 Interestingly, in these good-outcome patients, there was also an increase in the immune cells infiltrating the tumor.Citation14,23-26 In this work, we demonstrated that these two events associated with a good CRC prognosis (i.e., HSP110 expression and tumor immunosurveillance) may be linked. Indeed, we showed that HSP110 is secreted by CRC cells and skews the macrophage inflammatory profile. Depletion of extracellular HSP110 by using an antibody or by overexpressing the HSP110DE9 mutant induces macrophages with pro-inflammatory/cytotoxic potential. In contrast, overexpression of HSP110 or the addition of HSP110 recombinant protein (produced in eukaryotes to avoid LPS contamination) favors the formation of macrophages with an anti-inflammatory profile. The fact that the recombinant protein did not completely recapitulate the effect observed with colon cancer supernatant suggests that the extracellular HSP110 might act together with unidentified secreted molecules, and/or that the recombinant protein lacks some specific features of the native protein (e.g., percentage of the molecule containing ADP versus ATP and therefore with an open vs. closed conformation). We are at present performing structural and proteomic approaches to study in deep this question and determine HSP110 extracellular partners.
Although the way through which HSPs are released into the extracellular medium is still a matter of debate (active versus passive secretion), it is well known that some HSPs are abundant extracellular proteins notably in the tumor microenvironment.Citation27 They are believed to act as DAMPs and to have immunogenic properties Citation28 and the term chaperokines has been suggested.Citation29 Our results indicating that HSP110 is released by CRC cells is in agreement with Colgan et al., who reported that soluble HSP110 is a luminal component of the non-malignant human and mouse gastrointestinal tract. Immunohistochemistry showed expression of HSP110 in epithelium from the small and large intestine.Citation30 As in vitro measurements showed low extracellular HSP110 concentrations in MSI-type CRC cell lines, it would be interesting to monitor variations in extracellular HSP110 amounts within body fluids as a new biomarker of disease characterization, or progression.
The anti-inflammatory role of the secreted form of HSP110 in CRC reported here is in agreement with the overall literature about other extracellular HSPs. Indeed, several studies using the immunization of animals with mammalian or mycobacterial HSPs in a context of autoimmune or inflammatory diseases (Arthritis, diabetes…) have shown improvements in animal health, suggesting that HSPs play an immunosuppressive role.Citation31 This effect is probably mediated by the direct modulation/inhibition of antigen-presenting cell activation. In this way, recombinant HSP27 has been shown to directly inhibit dendritic cell differentiation and skew toward a macrophage phenotype.Citation21 Furthermore, these macrophages acquire tolerogenic properties in vitro and in breast cancer patients.Citation20 Similar immunosuppression was observed in higher molecular weight HSPs, like recombinant HSP70, as Ferat-Osio et al showed that highly purified HSP70 inhibits TNFa production by monocytes.Citation32 It is worth noting that the absence of minute amounts of contaminating endotoxin is mandatory to reveal the inherent suppressive functions of these HSPs, as nicely demonstrated by Stocki.Citation33,34 This very low level of contamination could account for the maturation effect of recombinant HSP110 on dendritic cells observed by Manjili et al. Citation35. In our setting, we eliminated this caveat as we used an HSP110 naturally present in the supernatant of CRC cell lines, and when we used purified HSP110, it was produced in a eukaryotic setting with no traces of LPS.
In the absence of LPS, extracellular HSPs have been shown to bind to and activate different TLRs. In particular, we have previously shown that HSP70, expressed at the surface of tumor-derived exosomes, activated myeloid-derived suppressive cells through its binding to TLR2.Citation19 Concerning circulating HSP27, we and others have shown that it inhibits macrophage and dendritic cell differentiation through TLR4 Citation21 and it favors angiogenesis through TLR3.Citation36 We show here that the anti-inflammatory effect of soluble HSP110 on macrophages involves TLR4. Kuang et al. have shown that TLR4 signaling on monocytes could be deleterious for their adequate activation and differentiation.Citation37 They demonstrated that hyaluronan secreted from tumors binds TLR4 on monocytes, which then become refractory to subsequent stimulation. In addition, depending on the context, NFkB signaling induced upon exposure to TLR2/4 ligand can transmit an anti-inflammatory message in macrophages.Citation38-40 Finally, IRAK-M, a negative regulator of TLR signaling that can be upregulated in tumor-infiltrating macrophages in a TLR4-dependent manner, could interfere with adequate polarization.Citation41 Taken together, these mechanisms may provide a rationale for the effect observed with HSP110. It is therefore possible that the early presence of HSP110 during the monocyte differentiation process could hamper the subsequent pro-inflammatory function of macrophages, as a signal of a refractory state.
Other HSP-binding receptors have been described over the last few years. Among them, scavenger receptor SRA/CD204 has been described as a receptor for high molecular weight HSPs such as HSP110.Citation22,42 Although in our experimental setting this receptor was not involved, other authors have shown that it mediates an inhibitory signal in dendritic cells upon HSP110 stimulation, thus supporting our hypothesis that extracellular HSP110 is deleterious to optimal immune responses. Thus, as suggested by W. Van Eden for other HSP family members, HSP110 should therefore not be considered a DAMP (accordingly to the concept of “danger” introduced by P. Matzinger) but rather as a “DAMPer” of the immune system.Citation43
Several groups have investigated the nature of the immune infiltrate within tumors of CRC patients. Though all groups agree on the higher Th1 cell colonization, they provide puzzling observations regarding myeloid cells. The presence of CD163+ macrophages in the tumor microenvironment has been described in MSS and MSI CRC biopsies.Citation18,44 Surprisingly, concomitant increases in M1 and M2 subsets were associated with a better prognosis.Citation17 However, a high expression of PD-L1 (B7-H1), an immune-inhibitory ligand, at the surface of myeloid cells was found at the invasive front and in the stroma of MSI CRC biopsies, suggesting that these cells provide a negative signal to T cells, thus dampening the immune response.Citation44,45 This paradoxal high expression of PD-L1 could be explained by a negative feedback induced by a strong Th1 cell response and mediated by IFNγ.Citation46 This mechanism is intended to avoid tissue and organ damages in non-tumoral conditions, but could be a “double-edge sword” in tumor immunity. Macrophages, therefore, become a cellular target through PD-1/PD-L1 blockage. Such a strategy is currently being investigated in various tumors with mismatch-repair deficiency, including CRC, with encouraging results.Citation45 Several other immune inhibitory checkpoints (B7-H4, PD-1, CTLA-4, IDO and LAG-3) have been described in MSI CRC biopsies and all together play a major role in inhibiting tumor immune-surveillance.Citation44,47 Given that HSP110 low expression level favors a pro-inflammatory environment through macrophage polarization, the existence of a negative feedback involving these immune inhibitory checkpoints expression is worth investigating. We have just started a multicenter study including more than 3,000 CRC biopsies already characterized for HSP110 expressionCitation4 to answer this question.
Studies on the tumor immune microenvironment have distinguished between CRC patients only on the basis of MSS (i.e. MSS vs. MSI). However, we have recently shown that HSP110 expression is a strong and reliable marker to distinguish between MSI CRC patients with good and poor prognosis. In terms of outcomes, MSI CRC patients expressing high levels of HSP110 are probably no different from MSS CRC patients. Here, we showed that these patients with high HSP110 have a greater invasion of CD163+ macrophages than is the case in MSI patients with low HSP110 expression. Based on these results, we have started a clinical study to determine whether a correlation exists between CD163+ macrophages, HSP110 and survival. It also remains to be established whether an association exists between CD163+ cells and lymphocyte infiltration, as this is a well-established predictor of overall survival and relapse in CRC.Citation12
In recent years, HSP110 has become a new point of interest in the field of HSP and cancer, mainly thanks to its excellent chaperoning capacity of peptide antigens, which makes it a powerful tool for vaccine development.Citation48,49 Until our recently published works demonstrating the essential role of HSP110 in CRC, its role in tumor development remained almost unexplored. This study brings new information to the emerging role of extracellular HSP110 in the inhibition of the immune system in the context of tumor microenvironment. It confirms the necessity to target extracellular as well as intracellular HSP110 in CRC and should foster the development of specific inhibitors, which are urgently needed for this HSP.
Materials and methods
Cell culture and macrophage differentiation
CRC cell lines (HCT116 and SW480) were purchased from ATCC (Molsheim, France). The C22 subclone derived from the HCT116 cell line has previously been described (reference 4). All cell lines were cultivated in DMEM supplemented with 10% FBS (Lonza, Amboise, France). Monocytes from human peripheral blood were obtained from healthy donors with informed consent and purified using CD14 microbeads labeling and magnetic cell sorting (Miltenyi Biotec, Paris, France) following the manufacturer's instructions. 0.5 × 106 monocytes were incubated with 500 µL of CRC cell line supernatant for 72 h in a 24-well plate in the presence of macrophage colony-stimulating factor (M-CSF, 100 ng/mL, Miltenyi Biotec) and then characterized. To analyze secreted cytokines, macrophages were incubated for an additional 24 h in the presence of LPS (10 ng/mL). In some experiments, recombinant HSP110 produced in HEK293 cells (OriGene Technologies, Rockville, MD) was added to the culture at 600 ng/mL. For receptor blocking experiments, isolated CD14+ monocytes (5.105) were preincubated for 30 min with antibodies directed against TLR2 (5 µg; MAB2616, R&D systems, Minneapolis, USA), TLR4 (10 µg; AF1478, R&D systems) or SR-A (10 µg; AF2708, R&D systems).
CRC Supernatant preparation
2.5 × 105 HCT116 or HCT116-C22 were cultured in a 12-well plate for 72 h in DMEM supplemented with 10% FBS. The supernatant was then harvested and centrifuged at 450 g for 5 min. For immunoblot experiments, the culture medium was replaced after 72 h of culture by DMEM w/o serum for an additional 8 h and then concentrated using Amicon Ultra centrifugal filters (UFC501096, Merck Millipore, Molsheim, France) according to the manufacturer's instructions.
siRNA and cell transfection
For plasmid transfection, 1.2 × 105 SW480 or 2.5 × 105 HCT116 were implanted and cultured in a 12-well plate for 24 h. Cells were then transfected with 1 µg of plasmid encoding GFP, GFP-HSP110 or GFP-HSP110DE9 using HP Xtreme gene DNA transfection reagent (Roche, Boulogne-Billancourt, France) according to the manufacturer's instructions. 48 h hours later, the supernatant was collected and centrifuged at 450 g for 5 min. For immunoblotting, the culture media were replaced after 48 h of transfection with DMEM w/o serum for an additional 8 h and then concentrated using Amicon Ultra centrifugal filters (UFC501096, Merck Millipore) according to the manufacturer's instructions.
For siRNA transfection, 2.5 × 105 SW480 or 0.5 × 106 HCT116 cells were seeded into a 6-well plate and cultivated for 24 h in DMEM 10% SVF. Cells were then transfected with 50 pmol of control siRNA (ON-TARGETplus Non-targeting Pool, D-001810-10-05) or HSP110 siRNA (ON-TARGETplus HSPH1 siRNA, L-004972-00-0005) from Dharmacon (GE Heathcare, Velizy, France), with Lipofectamine RNAiMAX Reagent (Invitrogen, Cergy Pontoise, France) according to the manufacturer's instructions. The media were replaced after 6 h of incubation with transfection reagent and 24 h after transfection. Media were then collected 72 h after transfection and centrifuged at 450 g for 5 min.
Xenograft model
Nude mice were purchased from Charles River Laboratories (Wilmington, USA) and housed in specific pathogen-free conditions. 10 × 106 HCT116 or HCT116-C22 were injected s.c. into the right flank of each mouse. Tumor growth was then followed every other day for three weeks. Mice were sacrificed when tumors reached 800 mm3. The tumors were collected and included in Tissue-Tek® O.C.T. Compound (Sakura Finetek, Torrance, CA). The mice were treated according to the guidelines of the Ministère de la Recherche et de la Technologie, France.
Flow cytometry analysis
Macrophages were harvested, washed once in PBS and incubated for 20 min with antibody directed against HLA-DR (561224, BD Horizon), CD163 (556018, BD Bioscience) and CD206 (550889, BD Biosciences) at 4°C. Cells were then washed twice in PBS and analyzed using a LSRII flow cytometer (Becton Dickinson, Franklin Lakes, USA). For apoptosis determination, adherent and non-adherent cells were harvested and stained with Annexin V-FITC and 7-AAD (BD PharMingen, Franklin Lakes, USA) according to the manufacturer's recommendations.
Immunodepletion
1.2 × 105 SW480 or 2.5 × 105 HCT116 were seeded and cultivated for 24 h in a 12-well plate. Cells were washed, and 400 µL of media without serum was then added for 8 h. Supernatants were then harvested and centrifuged at 450 g for 5 min. 1 mL of supernatant was then incubated overnight at 4°C under rotation with 2 µg of control antibody (sc-2027, Santa Cruz Biotechnology, Dallas, TX, USA) or directed against HSP110 (sc-6241, Santa Cruz Biotechnology). 25 µL of Protein A-agarose beads (Millipore, Billerica, MA) were added for 1 h 30 min under rotation at 4°C. Supernatants were collected and stored at −20°C. Prior to incubation with monocytes, the immunodepleted supernatant was supplemented with FBS (to a final concentration of 10%). For immunoblot experiments, beads bound to HSP110 were washed three times with PBS and heated for 5 min at 95°C in laemmli buffer.
T lymphocyte proliferation assay
T Lymphocytes from peripheral blood of healthy donors were purified using the pan T-Cells isolation kit (Miltenyi Biotec). T lymphocytes were then stained by using Cell Trace Violet (Invitrogen), according to the manufacturer's procedure. 105 T cells were then incubated for 3 d with 10 × 103 or 20 × 103 macrophages. T-cell division was detected by flow cytometry with an LSRII cytometer (BD Biosciences) and analyzed using ModFit software.
Cytokine and NO quantification
Cytokine secretion by macrophages upon LPS stimulation was determined using the MILLIPLEX MAP Kit “Human high sensitivity T Cell Magnetic Bead Panel” (HSTCMAG-28SK, Millipore, Billerica, MA) according to the manufacturer's instructions. Cytokines secreted by CRC cell lines were analyzed using the Human Cytokine Array Panel A according to the manufacturer's instructions (ARY005, R&D Systems). NO production was evaluated through nitrite measurement using The Griess Reagent System (Promega, Madison, WI).
Cell lysis and immunoblotting
Cells were harvested, washed in PBS and then lysed on ice in lysis buffer (150 mM NaCl, 50 mM Tris pH 6,8, 10 mM NaF, 1mM DTT, 1% Triton X-100) in the presence of protease inhibitors (Roche, Boulogne-Billancourt, France). Cell lysates or concentrated culture media were mixed with laemmli buffer. Proteins were separated and transferred following standard protocols before analysis with a chemiluminescence detection kit (Santa Cruz Biotechnology). Primary antibodies used for immunoblotting were from Santa Cruz biotechnologies directed against HSP110 (sc-6241), HSC70 (sc-7298), from Sigma (Lyon, France) for anti-actin (A1978-200UL), from abcam (Paris, France) for anti-HSP90α (ab59459) and from Enzo Life Sciences (Lyon, France) for anti-HSP70 (ADI-SPA-810) and anti-HSP27 (ADI-SPA-803).
Immunofluorescence
Tumor sections from the xenograft were fixed for 10 min in cold acetone. Slides were dried and rehydrated in PBS for 5 min twice. Slides were then saturated for 20 min in 3% BSA and 2% goat serum in PBS. Slides were incubated overnight at 4°C with (1:100) primary antibody directed against Arginase-1 (sc-20150, Santa Cruz Biotechnology) or NOS2 (sc-651, Santa Cruz Biotechnology), and F4/80 (MCA4971, AbD Serotec, Colmar, France). Slides were washed three times for 5 min in PBS and incubated for 10 min with a blocking reagent (R37107, molecular probes, Saint Aubin, France). (1:1,000) Secondary antibodies (Invitrogen) were added for 45 min at room temperature. Slides were washed three times in PBS for 5 min and mounted with ProLong (Thermo Fisher Scientific, Waltham, MA, USA).
Isolated CD14+ monocytes were incubated for 30 min in 10% RPMI with HSP110-Flag recombinant protein (500 ng/5.105 monocytes). Cells were washed once and incubated for 30 min at room temperature with (1:100) primary antibodies directed against TLR4 (sc-30002, Santa Cruz Biotechnology) and Flag (F1804-1MG, Sigma). Cells were washed once in PBS and fixed for 10 min in 4% PFA PBS. Cells were washed and (1:1,000) secondary antibodies (Invitrogen) were added for 30 min at room temperature. Monocytes were deposited on poly-lysine coated coverslips for 20 min at RT. Coverslips were washed twice in PBS and mounted with ProLong (Sigma).
The TLR4 luciferase reporter gene assay was performed by InvivoGen (Toulouse, France).
Immunohistochemistry
MSI CRC patients were selected as previously described.Citation4 Tumor sections from patients were deparaffinized in xylene and rehydrated in a graded series of alcohol solutions. Antigens were then unmasked in pH 6.0 citrate buffer (30 min, 95°C), cooled for 30 min, washed twice in PBS for 3 min and treated with 3% H202-PBS for 15 min in order to inhibit endogenous peroxidases. After washes in PBS, the slides were saturated for 25 min in 3% BSA PBS. Sections were then incubated with CD68 (1/100, M0814, Dako, Les Ulis, France), CD163 (1/100, NCL-CD163; Leica, Nanterre, France) or HSP110 (1/1200, clone 5812, Leica Biosystems) primary antibody overnight at 4°C (CD68, and CD163) or 1 h at room temperature. After washing in PBS, secondary antibody was added for one hour at room temperature. Slides were washed twice for 5 min in PBS and revealed using Novared kit (Vector, Burlingame, USA). Slides were washed twice in water for 5 min and counterstained with 10% Meyer's hematoxylin. After one wash in water, slides were dehydrated in 100% ethanol and in xylene for 30 s each. The slides were then observed using the Cell Observer station (Zeiss, Germany). Positive cells were counted in three distinct stroma areas of 645 µM by 482 µM for each tumor in a blinded manner.
Quantitative real-time PCR
Total RNA was isolated with Trizol (Invitrogen), reverse transcribed by Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI) with random hexamers (Promega). Primers for real-time PCR were from Bio-Rad (Hercules, CA, USA): Human TNFa (qHsaCED0037461), murine TNFa (qMmuCED0004141), murine Arginase-1 (qMmuCID0022400) and murine iNOS (qMmuCID0023087). HPRT was used as the invariant control (qMmuCED0045738 or qHsaCID0016375).
ELISA
HSP110 concentration was determined by an ELISA. Briefly, a 96-well plate (MaxiSorp Plate; Nunc, Sigma Aldrich, Australia) was coated with 0.2 M sodium carbonate/bicarbonate buffer, pH 9.4, overnight at 4°C with 3 μg/mL of rabbit anti-HSP110 Ab (sc-6241, Santa Cruz Biotechnology). The plates were washed and then blocked with 2% BSA in PBS for 1 h at room temperature. Supernatants were diluted and added to the plates along with recombinant HSP110 produced in HEK293 cells (OriGene Technologies, Rockville, MD) to establish a standard concentration curve. The plates were then incubated 2 h at room temperature, washed three times in PBS 0.5% Tween 20, and incubated for 2 h with a mouse anti-HSP110 Ab (1:200)(NCL-HSP105, Leica Biosystems). After three washes as before, the plates were incubated for 1 h with a Goat anti-mouse IgG conjugated to alkaline phosphatase (SouthernBiotech, Birmingham, Alabama). After three final washes, the ELISA was developed by adding a TMB substrate reagent (OptEIA, BD Biosciences, San Jose, CA). The reaction was stopped after 30 min by adding 2M sulfuric acid. ODs were measured at 450 nm.
HSP27 secreted by CRC cell lines was detected using the Immunoset HSP27 high sensitivity (Human) ELISA according to the manufacturer's instructions (ADI-960-076, Enzo life sciences, Farmingdale, NY).
Statistics
Analyses were performed with GraphPad Prism software (GraphPad Software, San Diego, Calif). The Student paired t test was used where appropriate and a two-tailed p value of .05 or greater was considered significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1170264_supplementary_material.zip
Download Zip (775.2 KB)Funding
This work was supported by grants from the Institut National du Cancer, Agence Nationale de la Recherche, Ligue Nationale Contre le Cancer (‘Labeled teams’ to CG and AD), the Association pour la Recherche sur le Cancer (ARC) and the Conseil Regional de Bourgogne. The work was also supported by a French Government grant managed by the French National Research Agency under the program “Investissements d'Avenir” with reference ANR-11-LABX-0021-01-LipSTIC LabEx. We thank the FEDER for their financial support. K.B. and S.C. have a doctoral fellowship from La Ligue Nationale Contre le Cancer and K.B. from La Foundation pour la Recherche Médicale.
References
- Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Järvinen H, Powell SM, Jen J, Hamilton SR. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812-6; PMID:8484121; http://dx.doi.org/10.1126/science.8484121
- Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993; 363:558-61; PMID:8505985; http://dx.doi.org/10.1038/363558a0
- Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science 1993; 260:816-9; PMID:8484122; http://dx.doi.org/10.1126/science.8484122
- Collura A, Lagrange A, Svrcek M, Marisa L, Buhard O, Guilloux A, Wanherdrick K, Dorard C, Taieb A, Saget A et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil-based chemotherapy. Gastroenterology 2014; 146:401-11 e1; PMID:24512910; http://dx.doi.org/10.1053/j.gastro.2013.10.054
- Duval A, Collura A, Berthenet K, Lagrange A, Garrido C. Microsatellite instability in colorectal cancer: time to stop hiding! Oncotarget 2011; 2:826-7; PMID:22084152; http://dx.doi.org/10.18632/oncotarget.353
- Dorard C, de Thonel A, Collura A, Marisa L, Svrcek M, Lagrange A, Jego G, Wanherdrick K, Joly AL, Buhard O et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med 2011; 17:1283-9; PMID:21946539; http://dx.doi.org/10.1038/nm.2457
- Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 2013; 14:630-42; PMID:24026055; http://dx.doi.org/10.1038/nrm3658
- Doyle SM, Genest O, Wickner S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol 2013; 14:617-29; PMID:24061228; http://dx.doi.org/10.1038/nrm3660
- Jego G, Hazoume A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett 2013; 332:275-85; PMID:21078542; http://dx.doi.org/10.1016/j.canlet.2010.10.014
- Ciocca DR, Arrigo AP, Calderwood SK. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol 2013; 87:19-48; PMID:22885793; http://dx.doi.org/10.1007/s00204-012-0918-z
- Slaby O, Sobkova K, Svoboda M, Garajova I, Fabian P, Hrstka R, Nenutil R, Sachlova M, Kocakova I, Michalek J et al. Significant overexpression of Hsp110 gene during colorectal cancer progression. Oncol Rep 2009; 21:1235-41; PMID:19360299; http://dx.doi.org/10.3892/or_00000346
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960-4; PMID:17008531; http://dx.doi.org/10.1126/science.1129139
- Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pagès F et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 2011; 71:1263-71; PMID:21303976; http://dx.doi.org/10.1158/0008-5472.CAN-10-2907
- Boissiere-Michot F, Lazennec G, Frugier H, Jarlier M, Roca L, Duffour J, Du Paty E, Laune D, Blanchard F, Le Pessot F et al. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. Oncoimmunology 2014; 3:e29256; PMID:25101223; http://dx.doi.org/10.4161/onci.29256
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41:14-20; PMID:25035950; http://dx.doi.org/10.1016/j.immuni.2014.06.008
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity 2014; 41:49-61; PMID:25035953; http://dx.doi.org/10.1016/j.immuni.2014.06.010
- Edin S, Wikberg ML, Dahlin AM, Rutegard J, Oberg A, Oldenborg PA, Palmqvist R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS One 2012; 7:e47045; PMID:23077543; http://dx.doi.org/10.1371/journal.pone.0047045
- Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res 2007; 13:1472-9; PMID:17332291; http://dx.doi.org/10.1158/1078-0432.CCR-06-2073
- Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 2010; 120:457-71; PMID:20093776; http://dx.doi.org/10.1172/JCI40483
- Banerjee S, Lin CF, Skinner KA, Schiffhauer LM, Peacock J, Hicks DG, Redmond EM, Morrow D, Huston A, Shayne M et al. Heat shock protein 27 differentiates tolerogenic macrophages that may support human breast cancer progression. Cancer Res 2011; 71:318-27; PMID:21224361; http://dx.doi.org/10.1158/0008-5472.CAN-10-1778
- Laudanski K, De A, Miller-Graziano C. Exogenous heat shock protein 27 uniquely blocks differentiation of monocytes to dendritic cells. Eur J Immunol 2007; 37:2812-24; PMID:17823891; http://dx.doi.org/10.1002/eji.200636993
- Qian J, Yi H, Guo C, Yu X, Zuo D, Chen X, Kane JM 3rd, Repasky EA, Subjeck JR, Wang XY. CD204 suppresses large heat shock protein-facilitated priming of tumor antigen gp100-specific T cells and chaperone vaccine activity against mouse melanoma. J Immunol 2011; 187:2905-14; PMID:21832164; http://dx.doi.org/10.4049/jimmunol.1100703
- Deschoolmeester V, Baay M, Van Marck E, Weyler J, Vermeulen P, Lardon F, Vermorken JB. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol 2010; 11:19; PMID:20385003; http://dx.doi.org/10.1186/1471-2172-11-19
- Banerjea A, Ahmed S, Hands RE, Huang F, Han X, Shaw PM, Feakins R, Bustin SA, Dorudi S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol Cancer 2004; 3:21; PMID:15298707; http://dx.doi.org/10.1186/1476-4598-3-21
- Phillips SM, Banerjea A, Feakins R, Li SR, Bustin SA, Dorudi S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br J Surg 2004; 91:469-75; PMID:15048750; http://dx.doi.org/10.1002/bjs.4472
- Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, Angell HK, Fredriksen T, Elie N, Fauquembergue E et al. Correlation between Density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res 2015; 75:3446-55; PMID:26060019; http://dx.doi.org/10.1158/0008-5472.CAN-14-3051
- Sherman M, Multhoff G. Heat shock proteins in cancer. Ann N Y Acad Sci 2007; 1113:192-201; PMID:17978282; http://dx.doi.org/10.1196/annals.1391.030
- Joly AL, Wettstein G, Mignot G, Ghiringhelli F, Garrido C. Dual role of heat shock proteins as regulators of apoptosis and innate immunity. J Innate Immun 2010; 2:238-47; PMID:20375559; http://dx.doi.org/10.1159/000296508
- Asea A. Chaperokine-induced signal transduction pathways. Exerc Immunol Rev 2003; 9:25-33; PMID:14686091
- Colgan SP, Pitman RS, Nagaishi T, Mizoguchi A, Mizoguchi E, Mayer LF, Shao L, Sartor RB, Subjeck JR, Blumberg RS. Intestinal heat shock protein 110 regulates expression of CD1d on intestinal epithelial cells. J Clin Invest 2003; 112:745-54; PMID:12952923; http://dx.doi.org/10.1172/JCI200317241
- van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol 2005; 5:318-30; PMID:15803151; http://dx.doi.org/10.1038/nri1593
- Ferat-Osorio E, Sanchez-Anaya A, Gutierrez-Mendoza M, Bosco-Garate I, Wong-Baeza I, Pastelin-Palacios R, Pedraza-Alva G, Bonifaz LC, Cortés-Reynosa P, Pérez-Salazar E et al. Heat shock protein 70 down-regulates the production of toll-like receptor-induced pro-inflammatory cytokines by a heat shock factor-1/constitutive heat shock element-binding factor-dependent mechanism. J Inflamm (Lond) 2014; 11:19; PMID:25053922; http://dx.doi.org/10.1186/1476-9255-11-19
- Stocki P, Dickinson AM. The immunosuppressive activity of heat shock protein 70. Autoimmune Dis 2012; 2012:617213; PMID:23326648; http://dx.doi.org/10.1155/2012/617213
- Stocki P, Wang XN, Dickinson AM. Inducible heat shock protein 70 reduces T cell responses and stimulatory capacity of monocyte-derived dendritic cells. J Biol Chem 2012; 287:12387-94; PMID:22334699; http://dx.doi.org/10.1074/jbc.M111.307579
- Manjili MH, Park J, Facciponte JG, Subjeck JR. HSP110 induces “danger signals” upon interaction with antigen presenting cells and mouse mammary carcinoma. Immunobiology 2005; 210:295-303; PMID:16164037; http://dx.doi.org/10.1016/j.imbio.2005.04.002
- Thuringer D, Jego G, Wettstein G, Terrier O, Cronier L, Yousfi N, Hébrard S, Bouchot A, Hazoumé A, Joly AL et al. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. Faseb J 2013; 27:4169-83; PMID:23804239; http://dx.doi.org/10.1096/fj.12-226977
- Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007; 110:587-95; PMID:17395778; http://dx.doi.org/10.1182/blood-2007-01-068031
- Mancino A, Lawrence T. Nuclear factor-kappaB and tumor-associated macrophages. Clin Cancer Res 2010; 16:784-9; PMID:20103670; http://dx.doi.org/10.1158/1078-0432.CCR-09-1015
- Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, Karin M, Lawrence T. An antiinflammatory role for IKKbeta through the inhibition of “classical” macrophage activation. J Exp Med 2008; 205:1269-76; PMID:18490491; http://dx.doi.org/10.1084/jem.20080124
- Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Göktuna SI, Neuenhahn M, Fierer J, Paxian S et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007; 130:918-31; PMID:17803913; http://dx.doi.org/10.1016/j.cell.2007.07.009
- del Fresno C, Otero K, Gomez-Garcia L, Gonzalez-Leon MC, Soler-Ranger L, Fuentes-Prior P, Escoll P, Baos R, Caveda L, García F et al. Tumor cells deactivate human monocytes by up-regulating IL-1 receptor associated kinase-M expression via CD44 and TLR4. J Immunol 2005; 174:3032-40; PMID:15728517; http://dx.doi.org/10.4049/jimmunol.174.5.3032
- Facciponte JG, Wang XY, Subjeck JR. Hsp110 and Grp170, members of the Hsp70 superfamily, bind to scavenger receptor-A and scavenger receptor expressed by endothelial cells-I. Eur J Immunol 2007; 37:2268-79; PMID:17615582; http://dx.doi.org/10.1002/eji.200737127
- Broere F, van der Zee R, van Eden W. Heat shock proteins are no DAMPs, rather ‘DAMPERs’. Nat Rev Immunol 2011; 11:565; author reply; PMID:21785457; http://dx.doi.org/10.1038/nri2873-c1
- Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015; 5:43-51; PMID:25358689; http://dx.doi.org/10.1158/2159-8290.CD-14-0863
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509-20; PMID:26028255; http://dx.doi.org/10.1056/NEJMoa1500596
- Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 2008; 8:467-77; PMID:18500231; http://dx.doi.org/10.1038/nri2326
- Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, Brumlik M, Cheng P, Curiel T, Myers L et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med 2006; 203:871-81; PMID:16606666; http://dx.doi.org/10.1084/jem.20050930
- Wang XY, Subjeck JR. High molecular weight stress proteins: Identification, cloning and utilisation in cancer immunotherapy. Int J Hyperthermia 2013; 29:364-75; PMID:23829534; http://dx.doi.org/10.3109/02656736.2013.803607
- Mattoo RU, Sharma SK, Priya S, Finka A, Goloubinoff P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J Biol Chem 2013; 288:21399-411; PMID:23737532; http://dx.doi.org/10.1074/jbc.M113.479253