
ABSTRACT
We used a murine model to monitor changes to myeloid cell subsets, i.e., myeloid-derived suppressor cells (MDSCs), M1 macrophages that secrete pro-inflammatory cytokines and express CD40 and CD80 and suppressive M2 macrophages that secrete anti-inflammatory cytokines and express CD206 and CX3CR1, during mesothelioma progression and during chemotherapy or immunotherapy-induced tumor regression. In vitro studies showed that mesothelioma-conditioned media generated CD206−CX3CR1+MCP-1+TGF-β+ macrophages that induced T cell proliferation but prevented T cell IFNγ production. In vivo studies showed that co-inoculation of macrophages with mesothelioma cells led to faster tumor growth, and depleting macrophages using anti-F4/80 antibody induced tumor regression. Flow cytometry revealed increasing levels of different suppressive myeloid cells in lymphoid organs: MDSCs dominated bone marrow (BM) and spleens, M2 macrophages dominated tumor-draining lymph nodes (DLN) and a mixed IL-10+TNF-α+CD206−CX3CR1+ M1/M2 (M3) macrophage subset dominated the mesothelioma microenvironment. Ki67 staining and cell cycle analysis showed that tumor-associated M1 and M3, but not M2, macrophages were proliferating in situ, with M1 cells arrested in the G1 phase while M3 cells progressed to mitosis. Immunohistochemistry showed that M1 and M3 cells were co-located supporting the hypothesis that M1 cells transition to M3 cells during proliferation. Gemcitabine reduced tumor-associated M3 and MDSCs, but not M2 macrophages, the latter likely contributing to the tumor outgrowth seen following treatment cessation. In contrast, IL-2/agonist anti-CD40 antibody therapy reduced M3 cells and polarized macrophages into M1 cells coinciding with tumor regression. These data show that myeloid cells, particularly M3 cells, represent a therapeutic target for the generation of antitumor immunity.
Introduction
Malignant Mesothelioma, a devastating tumor usually located in the mesothelial lining of the pleural cavity, is resistant to conventional chemotherapy; however, weCitation1-4 and othersCitation5,6 have shown that it is susceptible to immunotherapy. This may be because mesothelioma tumors induce an endogenous T cell response that is insufficient to prevent disease progressionCitation7,8 without exogenous help, often in the form of cytokines.Citation5,9 We have previously shown that intra-tumoral, but not systemic, IL-2 induces regression of small mesothelioma tumors.Citation1 Larger tumors did not respond to IL-2 despite the promotion of effector rather than regulatory T cells.Citation10 These data imply the development of potent immune suppression with increasing tumor burden.Citation10 We have also shown that while regulatory T cells play an important suppressive role during the early stages of mesothelioma tumor evolution they are not key suppressor cells in larger tumors in this model. Furthermore, we have shown that stromal tissue within murine mesothelioma tumors contains a significant number of tumor-associated macrophages.Citation11 Others have shown similar data in human mesothelioma.Citation12 These results prompted us to hypothesize that myeloid cells and macrophages in particular are a major regulatory cell in larger tumor burdens. Yet, to the best of our knowledge, until now, macrophage subsets had not been systematically examined in mesothelioma during tumor progression and following chemotherapy or immunotherapy.
Macrophages can be broadly classified into two subsets, M1 and M2 cells. Classically-activated M1 macrophages secrete pro-inflammatory cytokines, such as interleukin-12 (IL-12), generate toxic intermediates, and are potent effector cells with antitumoricidal activity.Citation13 Alternatively-activated M2 macrophages secrete anti-inflammatory cytokines such as IL-10 and transforming growth factor-β (TGF-β) which promote angiogenesis and tissue repair.Citation14,15 Tumor-associated macrophages are reported to be predominately suppressive M2 macrophages.Citation16-18 Myeloid-derived suppressor cells (MDSCs) are highly suppressive macrophage-like immature cells that are often increased in cancer and have similarities to M2 macrophages.Citation19-21
Importantly, macrophages can polarize from the M1 to the M2 phenotype.Citation14,22 Thus, targeting macrophage subsets within mesothelioma tumors represents a potential therapeutic strategy.Citation23,24 We have also previously shown that while injecting an agonist anti-CD40 antibody (Ab) i.v. was ineffective, direct intra-tumoral injection induced rejection of small, but not large, tumors.Citation2 This approach did not improve tumor antigen presentation with CD11c+ dendritic cell (DC) numbers diminishing in tumors and DLNs during treatment. The anti-CD40 Ab monotherapy did not increase tumor-specific CTL activity and a significant number of mice experienced anti-CD40 Ab-driven survival in the absence of CD8+ T cells. These data suggest that the DC/T cell axis is not as critical as expected.
Others have shown that use of an agonist anti-CD40 Ab in human and murine pancreatic cancer activated tumoricidal M1 macrophages leading to inhibition of tumor growth.Citation25 When we intra-tumorally injected a combination of IL-2 with agonist anti-CD40 Ab large tumors regressed.Citation3 Systemic injection of IL-2/CD40 was unacceptably toxic, even in young adult mice, and significantly less effective than intra-tumoral administration. IL-2/CD40-induced tumor regression involved a collaborative effort between highly activated T cells and neutrophils.Citation3 The role of macrophages was not examined in those studies. However, we now have evidence that targeting and repolarizing tumor-associated macrophages using IL-2/CD40 may contribute to tumor regression.Citation26,27 Others have shown that targeting and depleting tumor-associated macrophages inhibits mesothelioma tumor development.Citation23 In these studies, we compared the effects of the IL-2/anti-CD40 Ab combination immunotherapy with a chemotherapy used for mesothelioma patients (gemcitabine) on macrophages. Gemcitabine is reported to eliminate MDSCs and inhibit tumor growth.Citation28 Therefore, this study aimed to monitor changes to myeloid cells including the M1 and M2 macrophage subsets, as well as MDSCs in tumors and secondary lymphoid organs, during tumor growth and following treatment with gemcitabine or local IL-2/anti-CD40 Ab immunotherapy. Secondary lymphoid organs were examined as they might reveal local suppression which could thwart the induction and/or maintenance of antitumor immunity.
Results
Macrophages contribute to mesothelioma tumor growth
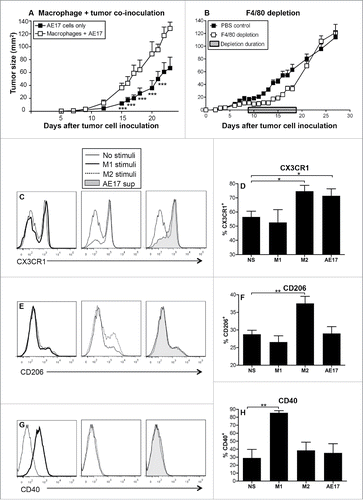
There is evidence that tumor-associated macrophages play an important role in tumor progression.Citation17,29,30 However, the role of macrophages in mesothelioma tumor growth is not yet clear. Therefore, we first aimed to assess the contribution of macrophages to mesothelioma tumor growth in vivo using a murine model. BM-derived macrophages were co-inoculated with AE17 mesothelioma tumor cells and tumor growth monitored. Macrophage/tumor co-inoculation led to significantly faster tumor growth rate compared to AE17 tumor only controls (). We next assessed the role of tumor-associated macrophages in small established mesothelioma tumors (15–20 mm2) using the F4/80 antibody as it has been reported to deplete macrophages in vivo.Citation31 To ensure that tumor-associated macrophages were targeted the anti-F4/80 Ab was injected on alternate days into the peritoneal cavity then the tumor for 9 d. Tumors regressed after the first anti-F4/80 Ab injection, then grew slowly until the anti-F4/80 Ab injections were stopped at day 18 (). However, after ceasing anti-F4/80 Ab injections tumors rapidly regrew, likely mediated by macrophage re-population (). These data imply that macrophages contribute to mesothelioma tumor growth.
Figure 1. Macrophages contribute to mesothelioma tumor growth. C57BL/6J mice were inoculated s.c. with AE17 mesothelioma tumor cells (3 × 105 cells) or co-inoculated with BM-derived macrophages (1 × 105 cells) and AE17 cells (3 × 105 cells, ratio 1 macrophage:3 tumor cells) and tumor growth monitored (). In a separate experiment, mice were inoculated with 5 × 105 AE17 tumor cells s.c and tumors left to grow to 25–30 mm2 before anti-F4/80 Ab treatment commenced (). Daily anti-F4/80 Ab injections (100 μg/mL in 100 µL) were alternately given i.p. then i.t. for 9 d. Control mice were given the PBS diluent. Pooled data is shown as mean ± SEM; n = 9 mice/group. Peritoneal macrophages from C57BL/6J mice were cultured overnight with IFNγ/LPS (M1 stimuli), IL-4/IL-13 (M2 stimuli), 50% AE17 mesothelioma-derived supernatant or left untouched (no stimuli, NS) and analyzed by flow cytometry for surface expression of CX3CR1 (), CD206 (), and CD40 (G and H). Pooled data from four experiments is shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Mesothelioma tumor-derived factors induce M2-like macrophages
The next experiments assessed if mesothelioma tumor cell-derived factors polarize macrophages. Murine peritoneal macrophages cultured with AE17 mesothelioma-conditioned media (AE17 supernatant), IL-4/IL-13 (M2 stimulus control) or IFNγ/LPS (M1 stimulus control) were stained for expression of CD40 and CD80 (M1 macrophages are CD40hi CD80hi), as well as CX3CR1 and CD206, (M2 macrophages are CD206hi CX3CR1hi)Citation14,32 and analyzed by flow cytometry. Similar to IL-4/IL-13-stimulated M2 macrophages, macrophages exposed to mesothelioma tumor supernatant upregulated CX3CR1 (). However, tumor-exposed macrophages did not upregulate the M2 macrophage marker CD206 (). Expression of M1 macrophage markers CD40 () and CD80 (data not shown) did not change after exposure to mesothelioma-derived conditioned media. These data suggest that mesothelioma tumor-derived factors partially polarize macrophages into M2 cells.
Supernatants from the macrophage subsets described above were assessed for cytokine secretion as this is indicative of macrophage pro- or anti-inflammatory function. Similar to M2 macrophages, mesothelioma-exposed macrophages did not secrete the pro-inflammatory M1 cytokines, IFNγ and TNF-α (data not shown). However, mesothelioma-exposed macrophages produced significantly more anti-inflammatory TGF-β () and significantly more monocyte chemoattractant protein-1 (MCP-1; ) than any other stimulus used. MCP-1 is a chemokine that recruits monocytes to inflammatory sites,Citation29 suggesting that mesothelioma tumor cells induce macrophages to secrete MCP-1 to recruit more macrophages into the tumor microenvironment. Overall, these data suggest that mesothelioma-derived factors drive incomplete polarization into an M1/M2-like phenotype, termed as M3 cells.Citation22,33
Figure 2. Mesothelioma tumor-derived factors polarize macrophages into M2-like cells. Peritoneal macrophages from C57BL/6J mice were cultured overnight with IFNγ/LPS (M1 stimuli), IL-4 (M2 stimuli), 50% AE17 mesothelioma-derived supernatant or left untouched (no stimuli, NS). Supernatants were analyzed for TGF-β (2A) and MCP-1 () by CBA. AE17 mesothelioma supernatant contains TGF-β (438 ± 194 pg/mL) and MCP-1 (4658 ± 136 pg/mL), therefore the data shown for AE17-exposed macrophages in were calculated by subtracting AE17 supernatant only. CFSE-labeled Balb/c non-adherent splenocytes were added to macrophages at varying ratios. After 5 d, cells were stained for CD8+ T cells and analyzed by flow cytometry. The percentage of CD8+ T cell proliferation was calculated based on loss of CFSE staining intensity of the parent peak (example histograms and graphed data from four experiments in ). Supernatants from the macrophage:T cell ratio of 1:2 MLR assay for IFNγ by CBA (), the data shown was calculated by subtracting the IFNγ levels seen in the M1 controls (i.e., 54.3 ± 16.1 pg/mL) where appropriate. Pooled data from four experiments is shown as mean ± SEM. *p < 0.05, **p < 0.01.
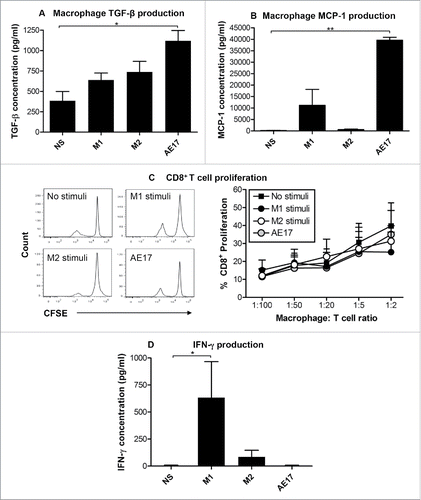
We next assessed whether tumor-exposed macrophages could induce T cell proliferation and T cell IFNγ secretion. Peritoneal macrophages from C57BL/6J mice were cultured with AE17 mesothelioma supernatant, M1 or M2 stimuli, as described above. CFSE-labeled, allogeneic Balb/c non-adherent splenic cells consisting of 40% T cells were then added to the macrophages at varying ratios. After 5 d, cells were stained with anti-CD4+ and anti-CD8+ antibodies and CD4+ and CD8+ T cell proliferative levels calculated based on loss of CFSE staining intensity of the parent peak. Neither M1 or M2 stimuli nor AE17 supernatant increased CD4+ (data not shown) or CD8+ () T cell proliferation relative to the no stimuli controls. Supernatants from the 1:2 macrophage:T cell ratio were used to measure IFNγ production by T cells. M1 and M2-stimulated macrophages, but not mesothelioma-exposed macrophages induced T cells to secrete IFNγ (). These data show that mesothelioma-exposed macrophages can stimulate T cell proliferation; however, they cannot induce T cells to secrete IFNγ. These data imply that mesothelioma modulates macrophages such that they compromise T cell function.
MDSCs dominate the spleen and bone marrow of mesothelioma tumor-bearing mice
The data described above suggested that mesothelioma tumor cells promote suppressive CX3CR1+CD206− “M2-like” rather than CX3CR1+CD206+ M2 macrophages in vivo. Therefore, we characterized macrophage subsets as well as MDSCs in tumors and lymphoid organs during disease progression. Lymphoid organs and tumors were collected from small (15.4 ± 2.8 mm2) and large (113.4 ± 8.4 mm2) AE17 mesothelioma tumor-bearing mice and stained for M1 macrophages (CD11b+ F4/80+ Ly6Chi CX3CR1lo), M2 macrophages (CD11b+ F4/80+ Ly6Clo CX3CR1hi) or MDSCs (CD11b+ F4/80lo Ly6C+ Ly6G+) and analyzed by flow cytometry (gating strategy shown in ).
Figure 3. MDSCs dominate the spleens and bone marrow of mesothelioma-bearing mice. C57BL/6J mice were inoculated s.c. with 5 × 105 AE17 tumor cells. Spleens and BM collected from mice with small (15.4 ± 2.8 mm2) or large (113.4 ± 8.4 mm2) tumors were stained for macrophage subsets and MDSCs. After gating on CD11b+F4/80+ macrophages () CD11b+F4/80+Ly6ChiCX3CR1lo M1 cells and CD11b+F4/80+Ly6CloCX3CR1hi M2 cell were identified. Gating on CD11b+F4/80lo cells was used to identify spleen and BM MDSCs (CD11b+F4/80loLy6C+Ly6G+; ). Data from healthy control mice (n = 10); small tumor-bearing (n = 20); and large tumor-bearing mice (n = 12) shows CD11b+F4/80+ macrophages in spleens () and BM (), splenic CD11b+F4/80+ macrophage intracellular IL-10 production (, n = 5–7/group), as well as splenic and BM macrophage subsets (, respectively). Data is shown with each point representing an individual mouse, the line shows the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
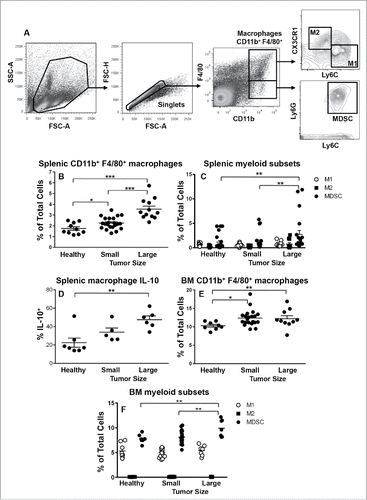
The proportion of total tumor-DLN cells that were CD11b+F4/80+ macrophages (Fig. S1A) did not change with disease progression relative to healthy controls. However, as DLNs increase in size in tumor-bearing mice this means that total CD11b+F4/80+ macrophage numbers did increase (data not shown). Similarly, the percent of total cells that were M1 and M2 macrophage in DLNs did not change with disease progression (Fig. S1B). MDSCs in lymphoid organs have been identified as CD11b+ and F4/80− cellsCitation34 and there was no difference between MDSCs in DLNs from healthy or mesothelioma-bearing mice (Fig. S1B).
Splenic CD11b+F4/80+ macrophage proportions significantly increased as tumors progressed (). As spleens increase in size in tumor-bearing mice total CD11b+F4/80+ macrophage numbers will have increased as well. Similarly, the percentage of MDSCs significantly increased in spleens with tumor progression with MDSCs being the dominant splenic subset in mice with large tumors (). While the percentage of M1 and M2 macrophages did not change (), there was a significant increase in percentage of splenic macrophages that were positive for IL-10 production with tumor progression ().
The percentage of CD11b+F4/80+ macrophages within BM slightly but significantly increased with tumor progression (). The percentage of BM M1 and M2 macrophages remained unchanged throughout tumor growth with M2 macrophages representing a very low proportion (). MDSCs significantly increased in the BMs of mice-bearing large tumors () and were the dominating BM macrophage-like subset during tumor progression. No change in macrophage IL-10 levels was observed in the BM (data not shown).
M3 macrophages emerge in mesothelioma tumors
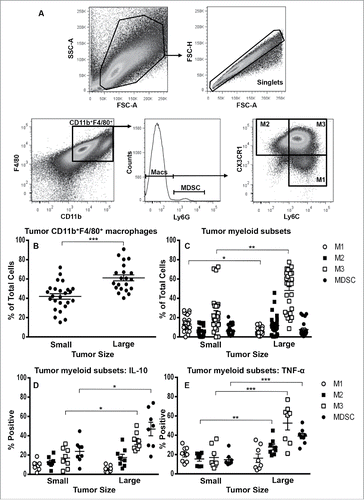
The proportion of total cells that were CD11b+F4/80+ tumor-associated macrophages increased significantly with disease progression (; gating strategy in 4A). In contrast to other studies (reviewed by,Citation17 MDSCs and M2 macrophages represented a low percentage of total cells in small and large tumors (). Interestingly, M1 macrophages significantly decreased with tumor progression () and a macrophage subset expressing a mixed M1 and M2 phenotype (CD11b+ F4/80+ Ly6Chi CX3CR1hi), i.e., M3 macrophagesCitation22,33 represented the dominant subset in small and large tumors (). M3 cells were not seen in significant numbers in DLNs, spleens or BMs during tumor progression (Fig. S1C). All tumor-associated macrophage subsets and MDSCs secreted IL-10 () and TNF-α (); with increasing proportions of M3 and MDSCs secreting IL-10, and M2, M3 and MDSCs secreting TNF-α with increasing tumor burden.
Figure 4. M3 macrophages dominate the mesothelioma tumor microenvironment. Small or large tumors were stained for macrophage subsets and MDSCs to identify M1 cells, M2 cells and M3 macrophages (CD11b+F4/80+Ly6ChiCX3CR1hi) and MDSCs. The gating strategy () demonstrates gating on CD11bhiF4/80hi macrophages as there were no F4/80low macrophages or F4/80loCD11b+Ly6G+MDSCs. Data from small tumor-bearing mice (n = 25) and large tumor-bearing mice (n = 21) for CD11b+F4/80+ macrophages (), macrophage subsets () and tumor-associated macrophage subset IL-10 (, n = 8/group) and TNF-α (, n = 8/group). Data is shown with each point representing an individual mouse, line shows the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
M3 macrophages proliferate in tumors
Large tumors were focused on in the next series of experiments as the data above showed they contained more total macrophages and more M3 cells and MDSCs. Ki67 staining, which detects most phases of the cell cycle, i.e., Gap (G)1, DNA synthesis (S), G2 and mitosis (M), but not G0 in resting cells showed that M1 macrophages (70.4 ± 7.6%), M3 macrophages (67.7 ± 1.4%), and MDSCs (55 ± 6%) were proliferating in situ (); very low proportions of M2 macrophages (8 ± 1.4%) were proliferating. Cell cycle analysis using DAPI revealed that most M1 and M2 macrophages and MDCSs were in the G0/G1 phase; however, the Ki67 data implies that M2 macrophages were in the G0 resting phase while M1 cells and MDSCs were in the G1 phase. In contrast, the majority of M3 macrophages were in the G2/M phase (); similar data was seen in small tumors (data not shown). These data imply that while M1, M3 and MDSCs proliferate in situ, M1 cells and MDSCs are mostly arrested in the G1 phase and only M3 macrophages progress further into the cell cycle to reach mitosis and cell division. Therefore, M3 macrophages may be induced and expanded in tumors. The data reveal the possibility that as M1 cells progress through the cell cycle from the G1 phase to the G2/M phase they may also be transitioning into an M3 phenotype.
Figure 5. M3 cells expand in tumors during disease progression. Tumor-associated cells were first stained for M1, M2, M3 and MDSCs as described for and then stained with Ki67 or DAPI for cell cycle analysis (, respectively). Data from AE17-tumor-bearing mice (n = 5–6/group) show the proportions of M1, M2, M3 cells and MDSCs that were in G2 mitosis phase (). Data is shown with each point representing an individual mouse, the line shows the mean ± SEM; ***p < 0.001, ****p < 0.0001. Immunofluorescence on large frozen AE17 mesothelioma tumor sections (n = 3) stained with F4/80 (red), CX3CR1 (green) and Ly6C (blue) revealed micro-niches of M1 and M3 cells (). Exposure times were determined by comparing each fluorescence stain to the relevant isotype control; a representative photograph is shown, magnification 200×.
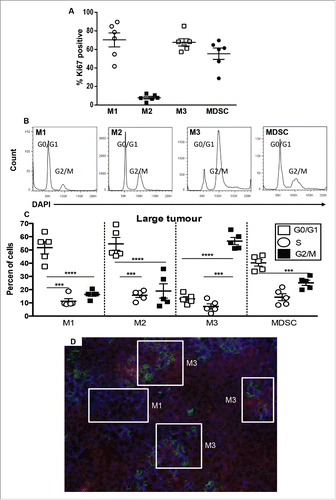
Macrophage subsets form niches in the mesothelioma tumor microenvironment
As described above, different macrophage subsets were observed in mesothelioma tumors by flow cytometry, therefore immunohistochemistry was used to identify their location. Triple label immunofluorescence with F4/80, Ly6C (M1 marker) and CX3CR1 (M2 marker) revealed microenvironmental niches of M1 (F4/80+ Ly6C+ CX3CR1lo) and M3 macrophages (F4/80+ Ly6C+ CX3CR1hi) in mesothelioma tumors (). In some areas M1 and M3 macrophages were juxtaposed, which supports the idea that the two cells are positioned such that they interact.
Chemotherapy does not eliminate all macrophage subsets in vivo
Our data show that M1 and M3 macrophages and MDSCs, as well as a few M2 macrophages, proliferate in tumors rendering them susceptible to destruction by chemotherapy that targets cell cycle. Gemcitabine is an anti-metabolite that prevents cells from making or repairing DNA thereby inducing apoptosis. Gemcitabine treatment has been shown to eliminate MDSCs in several tumor models including mesotheliomaCitation28; this response may contribute to the slowed tumor growth seen with gemcitabine treatment via improved T cell activity.Citation19 However, it is possible that macrophage subpopulations may also be affected.
We first examined the in vitro effect of gemcitabine on murine peritoneal macrophages. MTT assays showed that high concentrations of gemcitabine (3 µg/mL) induced macrophage cell death (Fig. S2A). We then performed in vivo studies in mesothelioma tumor-bearing mice and confirmed that gemcitabine-retarded tumor growth (). To identify which myeloid subpopulations were affected by gemcitabine lymphoid organs and tumors were collected mid-way through gemcitabine treatment when tumors were responding and analyzed as described above.
Figure 6. Gemcitabine does not affect suppressive M2 cells in spleens and tumors. C57BL/6J mice were inoculated with 5 × 105 AE17 tumor cells s.c. and tumors left to grow to 15–20 mm2 before gemcitabine treatment commenced. Treatment consisted of three i.p. injections (120 µg/g of body weight) every 3 d (). Pooled data is shown as mean ± SEM, n = 11 mice/group. In a separate experiment, AE17-bearing mice were given three injections of PBS (100 µL/dose) or gemcitabine every 3 d. Tumors (), spleens () and DLNs () were collected 4 d after the last dose, stained for CD11b+F4/80+ macrophages, M1, M2 or M3 subsets and MDSCs and analyzed by flow cytometry. Data is shown with each point representing an individual mouse (n = 5 mice/group), the line shows the mean ± SEM. *p < 0.05, **p < 0.01.
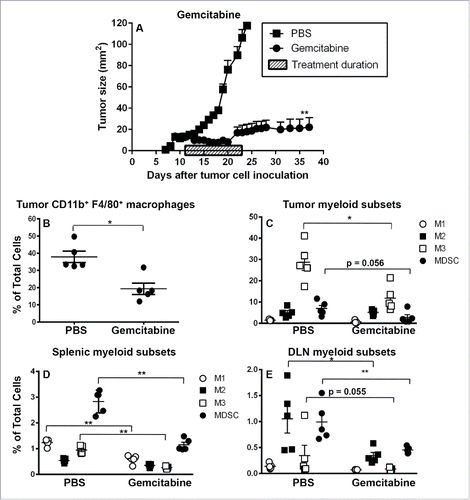
The percentage of CD11b+F4/80+ macrophages in tumors (), spleens and DLNs (data not shown) decreased significantly with gemcitabine treatment. Examination of tumor-associated myeloid subsets showed that gemcitabine significantly decreased M3 macrophages, likely due to their active proliferation (they were Ki67+ and in the G2/M phase), while M1 and M2 macrophage (that were mostly in the G0/G1 phase) proportions remained constant (). MDSC proportions trended downwards (p = 0.056). Nonetheless, M3 macrophages were still the dominant intra-tumoral macrophage subset and suppressive M2 macrophages and MDSCs were preserved. The preservation of M3, M2 and MDSC cells in tumors, and M2 and MDSC cells in spleen and DLNs may contribute to the tumor outgrowth seen following gemcitabine cessation ().
Examination of lymphoid organs showed that gemcitabine treatment was associated with: (1) significantly decreased MDSCs, M1 and M3 macrophages, but not M2 cells in spleens (); and (2) significantly decreased M2 macrophages and MDSCs, a decreasing trend for M3 cells (p = 0.055), while M1 macrophages remained unchanged in DLNs (). Nonetheless, MDSC and M2 cells remained the dominating DLN myeloid subset () and MDSCs were the dominating splenic myeloid subset ().
IL-2/agonist anti-CD40 antibody immunotherapy is not toxic to macrophages
The next experiments examined whether tumor-associated macrophages could be modulated by targeted immunotherapy with IL-2/anti-CD40 Ab. Both agents have been shown to induce an M1 phenotype.Citation35-37 MTT assays confirmed that IL-2 and/or anti-CD40 Ab were not toxic to peritoneal macrophages, instead they induced a proliferative response in macrophages relative to untreated controls (Fig. S2B–C).
IL-2/anti-CD40 Ab immunotherapy reduces M3 macrophages in tumors
In vivo studies were performed to assess whether i.t. IL-2/anti-CD40 Ab could polarize macrophages. In agreement with our previous studies,Citation3,38 IL-2/anti-CD40 Ab-inhibited tumor growth compared to PBS controls (). A group of tumor-bearing mice were depleted of macrophages using the F4/80 Ab. There was no difference in tumor growth between the IL-2/anti-CD40 Ab only and the IL-2/anti-CD40 Ab with anti-F4/80 Ab depletion during treatment groups () as both groups demonstrated tumor regression compared to PBS controls. However, upon IL-2/anti-CD40 Ab treatment cessation tumors in the macrophage-depleted group re-emerged, while the immunologically intact group demonstrated persisting inhibition of tumor growth.
Figure 7. IL-2/anti-CD40 Ab promotes M1 cells in draining lymph nodes. C57BL/6J mice were inoculated with 5 × 105 AE17 tumor cells s.c. and tumors left to grow to 15–20 mm2 before anti-F4/80 Ab injections commenced. IL-2/anti-CD40 Ab treatment consisted of three i.t. injections (20 µg IL-2 and 40 µg anti-CD40 Ab, 100 µL/dose) given every 2 d (). Daily anti-F4/80 Ab injections were given i.p. and i.t. alternately for 9 d. Pooled data shown as mean ± SEM, n = 7–10 mice/group. In a separate experiment, AE17 tumor-bearing mice were given three doses of either PBS (100 µL/dose) or IL-2/anti-CD40 Ab i.t. every 2 d and samples collected 3 d after the final dose. Tumors (), spleens (, and DLNs () were stained for CD11b+F4/80+ macrophages, M1, M2 or M3 cells and MDSCs for analysis by flow cytometry. Data is shown with each point representing an individual mouse (n = 5 mice/group), the line shows the mean ± SEM. *p < 0.05, **p < 0.01.
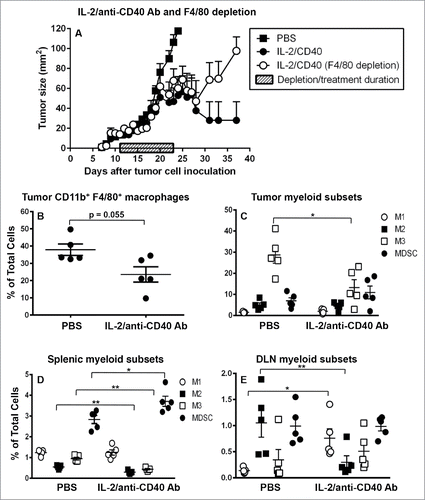
Tumors and lymphoid organs were again collected mid-way through treatment when tumors were responding and analyzed as described above. The percentage of tumor-associated macrophages decreased with IL-2/anti-CD40 Ab treatment () which may reflect a dilution effect due to massive infiltration of B cells, T cells and neutrophils Citation3; this was associated with a significant decrease in the proportion of M3 macrophages in IL-2/anti-CD40 Ab-treated tumors ().
Splenic CD11b+F4/80+ macrophage proportions in IL-2/anti-CD40 Ab-treated mice decreased relative to PBS controls (Fig. S3A). This was associated with significantly decreased proportions of splenic M2 and M3 macrophages yet MDSCs increased ().
IL-2/anti-CD40 Ab immunotherapy promotes M1 macrophages in DLNs
IL-2/anti-CD40 Ab did not alter the percentage of macrophages in DLNs (Fig. S3B). Interestingly, there was a decrease in DLN M2 macrophages with a concomitant increase in M1 macrophages with IL-2/anti-CD40 Ab treatment relative to PBS controls (). IL-2/anti-CD40 Ab had no impact on MDSC proportions in DLNs, nonetheless the significant increase in M1 cells and decrease in M2 cells should reduce the suppressive milieu. Moreover, it is likely that M2 macrophages were polarized into M1 macrophages to promote the effective anti-mesothelioma T cell immunity we have previously described.Citation3,38
Discussion
M2 macrophages and MDSCs are associated with cancer progression, yet until now, their role in progressing mesothelioma had not been extensively studied. Therefore, this study aimed to investigate the role of myeloid cell subsets in murine mesothelioma progression. In vivo studies showed that co-inoculation of AE17 mesothelioma tumor cells with macrophages led to faster tumor growth, and F4/80-antibody depletion of macrophages led to tumor regression. Note that use of a rat IgG2a control confirmed F4/80-antigen specificity and that all tumor-associated macrophages were F4/80high meaning they were likely to be targeted by the F4/80 antibody, although it is possible that F4/80lo monocytes and F4/80− splenic and subcapsular sinus lymph node macrophagesCitation39,40 may not have been affected. These novel data clearly show that macrophages play an important role in promoting mesothelioma tumor growth in vivo. This has been shown in other models of cancers such as lung cancer and teratocarcinomas, where depletion of macrophages and monocytes using clodronate-encapsulated liposomes inhibited tumor growth.Citation41-45 However, a significant M1 population was seen in mesothelioma tumors early during tumor growth. These pro-inflammatory M1 macrophages may be tissue resident macrophages that are rapidly alerted upon tumor cell injection, as could be occurring in our studies, or upon local inflammatory conditions induced by asbestos fibers that lead to mesothelial cell transformation,Citation46 as in human mesothelioma. M1 macrophages may contribute to the long latency period observed in this modelCitation1 and in human mesotheliomaCitation47 via secretion of tumoricidal pro-inflammatory cytokines (IL-1β, IL-12 and TNF-α; reviewed byCitation17,48). However, they may eventually contribute to tumor growth as M1 macrophages in chronic inflammation can promote tumor developmentCitation49,50; this might be further exacerbated by TNF-α secretion by all myeloid subsets including MDSCs as we have shown here. MDSC and macrophage-derived IL-10, among other mechanisms, may then function to suppress T cell function.
Our data also highlight the complexity of the effect of a tumor (and anticancer therapy) on local and distal myeloid cells as organ-specific, immune-suppressive myeloid patterns developed in mice-bearing progressing mesotheliomas. The presence of a mesothelioma tumor led to increased total splenic and BM macrophage proportions, plus higher numbers of MDSCs, as well as the emergence of an interesting macrophage subset expressing IL-10+TNF-α+CD206−CX3CR1+ in tumors; populations expressing a mixed M1/M2 phenotype have been described by others as suppressive M3 macrophages that promote tumor growth.Citation33 In contrast, total myeloid proportions did not change in DLN which were dominated by M2 macrophages.
The effects in the BM could reflect an increasing demand for myeloid precursors in spleens and progressing tumors. BM MDSCs may have responded to tumor-derived signals to proliferate and emigrate into spleens and tumors where our Ki67 and DAPI data show they continued to proliferate and expand. Mesothelioma tumors secrete prostaglandins,Citation51 M-CSFCitation52 and vascular endothelial growth factor (VEGF),Citation53 all of which block the differentiation of myeloid precursors in the BM leading to accumulation of MDSCs (reviewed byCitation54).
MDSCs in spleens and tumors are likely to mediate potent immune suppression by: oxidative stress induced by ROS and reactive nitrogen species production; starving T cells of amino acids; inducing T cell exhaustion or apoptosis via expression of inhibitory B7 family members such as PD-L1; preventing T cell homing to LN by reducing specific selectins; inhibiting T cell activation; as well as by recruiting/inducing immunosuppressive T regulatory cells (Tregs)Citation55,56 and M2 macrophages (reviewed byCitation57,58). The latter may be occurring in our mesothelioma tumors with M1 cells responding to MDSC and tumor-derived factors and beginning to transition into M2 cells but only reaching an intermediate M3 cell. MDSCs can also directly promote and expand cancer stem cells thereby affecting cancer stemnessCitation59 that is likely to reduce the efficacy of chemotherapy. The combination of lymphoid and tumor MDSCs, M2 macrophages in DLNs and M3 macrophages in tumors represents potent local and systemic suppression as well as the opportunity to develop cancer stem cells and remodel tissues for tumor dispersal and establishment of metastatic deposits.
Tumor M1 and M3 cells and MDSCs, but not M2 macrophages were proliferating in situ suggesting self-renewal. However, our data also suggest that M1 cells were mostly in the G1 phase while M3 cells were in the G2/M phase. These data suggest that during proliferation tumor M1 cells respond to factors from MDSCs and tumor cells and transition into more suppressive M3 cells that increase in numbers in tumors with increasing disease burden. Thus, polarization of M1 to M2 tumor-associated macrophages appears to be initiated in the tumor site, but is incomplete, likely due to concurrent exposure to pro- and anti-inflammatory cytokines. This was further supported by our in vitro studies showing that macrophages exposed to mesothelioma-derived supernatant adopted an incomplete M2-like phenotype as they expressed CX3CR1 and not CD206. It is also feasible that migration/re-distribution could contribute to M1-to-M3, and/or to M3-to-M1, transition. In this case it may be that F4/80lo monocytes migrate into tumors to an adopt an M1 or an M3 phenotype depending on the factors they encounter, and then as they move through tumors in response to chemotactic signals they may further respond to highly localized factors that initiate their transition to M3 or M1 cells, respectively.
Mesothelioma-exposed macrophages secreted increased levels of pro-inflammatory (MCP-1) and anti-inflammatory cytokines (TGF-β), and were able to induce T cells to proliferate but, unlike M1 stimulated macrophages, could not induce T cells to secrete IFNγ. Other studies have shown that tumor-associated M3 macrophages exert immunosuppressive functions and promote tumor growthCitation33; our studies imply a suppressive role for M3-derived IL-10. Thus, the emergence of CX3CR1+CD206− M3 macrophages is likely to contribute to mesothelioma tumor progression, as CX3CR1+ macrophages have been shown to promote angiogenesis and metastasis.Citation60 Expression of CX3CR1 provides further evidence of in situ self-renewal as the CX3CR1-CX3CL1 axis has been shown to promote survival and self-renewal in tumors.Citation60 Our data imply that M3 cells and MDSCs drive disease progression directly by affecting cancer cells and remodeling local and distal tissue and indirectly via potent immune suppression, as discussed above.
Tumor-induced local and systemic immune suppression by macrophages and MDSCs represents a potential therapeutic target. Gemcitabine exerted direct cytotoxic activity to macrophages in vitro and likely in vivo as gemcitabine targets proliferating cells, and M3 macrophages and to a lesser extent MDSCs, were proliferating within tumors and were therefore depleted, while non-proliferating M2 macrophages were not targeted. As a result, gemcitabine reduced the proportion of total macrophages in tumors. Gemcitabine also significantly reduced MDSCs in spleens. We did not examine whether splenic myeloid cells were cycling, however, these data suggest that splenic MDSC could have been proliferating explaining their susceptibility to gemcitabine. Reduced tumor M3 cells and splenic MDSCs may contribute to the tumor regression seen during gemcitabine treatment by releasing immune suppression mediated by both cell types, and by depleting cycling tumor cells. Systemic elimination of MDSCs by gemcitabine has been observed in mesothelioma and other cancer models leading to tumor regression.Citation28,61 However, several studies including this one have shown that gemcitabine-induced tumor regression is transient, and tumor outgrowth occurs when gemcitabine is ceased.Citation62,63 This may be because M3 macrophages remained the dominant subset within tumors, and M2 macrophages were preserved in all tissues, particularly the DLNs. The latter may contribute to tumor relapse by impairing tumor-specific cytotoxic T cells in DLNs. Thus, while chemotherapy removes some suppressive myeloid subsets from the tumor microenvironment and lymphoid organs, preservation of suppressive M2 macrophages implies that antitumor immunity can still be thwarted. Moreover, cancer stem cells previously promoted by MDSCs that are resistant to chemotherapy,Citation59 in association with re-populating suppressive MDSCs and M3 cells (likely driven by MDSC and tumor-derived factorsCitation19) upon gemcitabine cessation may also account for the re-emergence of tumors. This is supported by recent observations that MDSC cell death in the periphery directly contributes to enhanced expansion of MDSCs in the BM.Citation64
In contrast to gemcitabine treatment, IL-2/anti-CD40 Ab-polarized macrophages from the M2 toward the M1 phenotype in DLNs but not in tumors. It is not clear why macrophages in tumors did not respond IL-2/anti-CD40 Ab. It is possible is that the M3 cells in tumors are “overwhelmed” by tumor-derived factors as well as by direct signals from tumor cells and cannot respond to activating signaling molecules including IL-2 or CD40. Alternatively, the IL-2/anti-CD40 Ab may be diluted out by tumor-derived factors. It is also possible that macrophages in the tumor that may be rapidly transitioning from M1 to M3 cells cannot be re-polarized back to M1 cells. Another possibility is that even though we inject IL-2/anti-CD40 Ab directly into tumors we do not get full tumor penetration and there is likely to be leakiness to DLNs. Therefore, macrophages in DLNs that are not exposed to such high levels of tumor-derived factors or to cell-to-cell contact with tumors cells can be polarized into M1 cells. Thus, the DLNs are likely to be a critical site for IL-2/anti-CD40 Ab-induced antitumor immunity. We have previously shown that IL-2/anti-CD40 Ab leads to improved antitumor T cell responses, memory development and long-term tumor regression.Citation3,38 These studies suggest that IL-2/anti-CD40 Ab-driven DLN M1 macrophages secrete pro-inflammatory cytokines that help stimulate the proliferation and activation of tumor-specific T cells.Citation25,29 This, in turn could promote the activation and trafficking of effector and memory T cells into tumors as we have reported with IL-2/anti-CD40 Ab.Citation38 IL-2/anti-CD40 Ab also reduced M3 macrophages in the mesothelioma tumor microenvironment which could be a dilution effect due to the recruitment of massive numbers of B cells, cytotoxic T cells and neutrophils into the tumor, as we have previously shown.Citation3 Reduced M3 macrophage proportions could also reflect a less suppressive microenvironment.
Large tumor burdens are resistant to most mono-therapies, including conventional chemotherapy and novel therapeutic approaches, and their combination with therapeutics that target macrophage subsets may lead to improved anti-mesothelioma immunity. Future studies are required to determine if gemcitabine concurrently or sequentially combined with IL-2/anti-CD40 Ab improves antitumor immunity.
In summary, this study showed that mesothelioma tumors exerted distinct local and systemic effects on myeloid subsets. Tumor progression was associated with the local induction and expansion of suppressive M3 macrophages within the tumor microenvironment, and accumulation of MDSCs in tumors, spleens and BM, demonstrating systemic disabling of antitumor immunity. While gemcitabine significantly reduced cycling, tumor-associated M3 macrophages and splenic MDSCs, M3 cells were still the dominant tumor-associated subset and M2 macrophages were preserved in all lymphoid organs, implying global immune suppression that would contribute to eventual therapeutic failure. In contrast, IL-2/anti-CD40 Ab diluted M3 macrophages and MDSCs within mesothelioma tumors, and importantly, polarized DLN macrophages toward pro-inflammatory M1 macrophages. Our data shows that, myeloid cells represent a potential therapeutic target for the successful generation of antitumor immunity and improved therapies for mesothelioma patients.
Materials and methods
Mice
Female C57BL/6J mice aged 6 to 8 weeks were obtained from the Animal Resource Center. Mice were maintained under Specific Pathogen Free conditions in the Curtin University animal holding facility. All experiments were performed according to the Australian Code of Practice for the care and use of animals for scientific purposes as per Curtin University Animal Ethics Committee (AEC) approval number AEC-2011-16. The definition of small (< 50 mm2) or large tumor (> 100 mm2) is relative to the maximum allowable size, 140 mm2.
The AE17 cell line, tumor growth and treatment
AE17 is a malignant mesothelioma cell line derived from the peritoneal cavity of C57BL/6J mice injected with asbestos fibers.Citation1 Mice were inoculated subcutaneously (s.c.) with 5 × 105 AE17 mesothelioma cells. Tissues and tumors were collected during normal tumor growth or mid-way through treatment when tumors were responding. Tumor sizes were determined by multiplying two perpendicular axes that are at right angles to each other (measured using microcallipers) to get an area calculation. Volumes were not calculated as the tumors do not reliably develop into spherical, ellipsoid or hemi-ellipsoid shapes that have defined formulae. Furthermore, tumor shape often changes in response to therapy introducing large errors if volume measurements were to be used.
For chemotherapy, AE17 tumor-bearing mice (39.8 mm2 ± 7.3) were intra-peritoneally (i.p.) treated every 3 d for 9 d with PBS (100 µL/dose; Invitrogen) or gemcitabine (120 µg/g bodyweight in PBS) as previously described.Citation62 For immunotherapy, AE17 tumor-bearing mice (100.6mm2 ± 17.11) were intra-tumorally (i.t.) treated every 2 d for 6 d with PBS (100 µL/dose) or IL-2 (20 µg/dose; Cetus Corporation) and agonist rat anti-mouse CD40 Ab (FGK45, IgG2a isotype, 40 µg/dose; Absolutions) as previously described.Citation3
Macrophage depletion and treatment
Macrophages were depleted in vivo when tumor sizes reached 4–9 mm2. Mice were injected i.p. and i.t. on alternate days with 100 μg of rat anti-mouse F4/80 IgG2a Ab (clone BM8, Absolutions) in 100 µL PBS for 9 d. This approach was used to ensure tumor-associated macrophages were targeted. Control mice received i.p. and i.t. injections of the PBS diluent. Note that we have used the control rat isotype IgG2a antibody (Absolutions) to determine if IgG2a antibodies such the anti-CD40 Ab (FGK45) and the anti-F4/80 Ab, activate non-antigen specific antibody dependent cellular cytoxicity or antibody dependent cellular phagocytosis.Citation2 The endotoxin levels in these antibodies are less than 0.1 EU/mL (measured by the supplier using an endotoxin detection kit, documentation can be supplied). The IgG2a isotype control did not affect tumor growth and therefore did not activate complement, neutrophils, natural killer cells or macrophages with effector antitumor function, therefore any tumor response seen must be specific and mediated via the antigen binding site
Bone-marrow macrophage preparation and co-inoculation
Tibia and femur were collected from C57BL/6J mice and BM cells isolated by flushing with media (RPMI/10%FCS/gentamicin/2-ME, all from Invitrogen) using a 25g needle. Cells were cultured for 7 d in media supplemented with 30 ng/mL macrophage colony stimulating factor (M-CSF, Shenandoah Biotechnology), replacing with fresh media (containing 30 ng/mL M-CSF) at days 3 and 6. Macrophage purity (based on F4/80+CD11b+ cells using flow cytometry) was > 95% (data not shown). Cells were removed with trypsin-EDTA, washed twice in PBS, and co-inoculated subcutaneously with AE17 tumor cells into C57BL/6J mice at a ratio of 1:3 (1 × 105 macrophages:3 × 105 tumor cells). Tumor growth was monitored as described above.
Peritoneal macrophage preparation
Cells obtained from the peritoneal cavity by washing with ice cold PBS were incubated for 2–4 h at 37°C, after which non-adherent cells were removed and the remaining adherent population was > 95% F4/80+ macrophages (data not shown). Macrophages were cultured overnight with 50% AE17 tumor cell-derived conditioned media, M2 stimuli (20 ng/mL murine IL-4, 20 ng/mL murine IL-13; Shenandoah Biotechnology) or M1 stimuli (20 ng/mL interferon-gamma, IFNγ; Shenandoah Biotechnology; and 1 µg/mL lipopolysaccharide, LPS; Sigma-Aldrich).
MTT assay
Treatments were added in triplicate to 1 × 105 peritoneal macrophages in vitro. Gemcitabine was used at 7.5 × 10−6 µg/mL (low), 0.015 µg/mL (medium) or 3 µg/mL (high) concentrations. IL-2 and/or anti-CD40 Ab were used at 0.001 µg/mL (low), 0.01 µg/mL (medium) or 0.1 µg/mL (high) concentrations. Plates were incubated for 24 h at 37°C, and in the last 4 h treated with 2 mg/mL 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich). Plates were then centrifuged, supernatants removed and dimethyl sulfoxide (Sigma-Aldrich) added for 30 min. Absorbance was measured at 600 nm using a VICTOR X Multilabel Plate Reader (Perkin Elmer).
Flow cytometry
Peritoneal macrophages treated with various stimuli were removed with trypsin-EDTA for flow cytometric staining. Tissue samples were disaggregated into single-cell suspensions using frosted glass slides and stained for flow cytometric analysis. Combinations of the following anti-mouse primary antibodies (diluted in PBS/2% FCS) were incubated for 1 h at 4°C in the dark:anti-CD11b-PE-Cy7 (Biolegend), anti-F4/80-APC-Cy7 (Biolegend), anti-Ly6C-biotin (Biolegend), anti-CD40-PE (BD), anti-Ly6G-PE (Biolegend) and rabbit anti-CX3CR1 (Abcam, Massachusetts, USA). Following three washes in PBS/2% FCS, secondary Ab streptavidin-PerCP-Cy5.5 (Biolegend) and Alexafluor® 488-conjugated anti-rabbit Ab (Biolegend) were incubated for 30 min at 4°C in the dark. Samples for intracellular staining or cell cycle analysis were fixed in 1% paraformaldehyde on ice for 15 min, followed by permeabilization with PBS/2% FCS solution containing 0.1% saponin on ice. Samples were then stained with either DAPI (Sigma), anti-CD206-Alexafluor® 647 (Biolegend), anti-Ki67-Alexafluor® 647 (Santa Cruz Biotechnology), anti-TNF-α Alexafluor® 647 (Biolegend) or anti-IL-10-Brilliant Violet 421 (Biolegend). Cells were washed twice and re-suspended in PBS/2% FCS for analysis on a FACSCanto II using FACSDiva (BD). PMT voltages were set using unstained control cells and compensation was calculated using single stained samples. Representative data sets demonstrating the gating strategies (cross checked with fluorescence minus one controls) are shown in .
Cell culture supernatants from polarized macrophages were analyzed using Mouse Th1/Th2 Cytometric Bead Array (CBA; BD), Mouse Inflammation CBA (BD) and TGF-β CBA (BD). The assays were optimized and carried out according to manufacturers' instructions. Data acquisition was performed on a FACSCanto II with FACSDiva software (BD) and analyzed using FlowJo software (TreeStar).
Allogeneic mixed lymphocyte reaction (MLR)
Spleens from allogeneic BALB/c mice were disaggregated into single-cell suspensions by gentle dispersion between two frosted glass slides. Splenocytes were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) a fluorescent dye that binds permanently to cell membranes.Citation26,27 CFSE-labeled splenocytes were incubated for 2–4 h (37x°C, 5% CO2) to remove adherent macrophages and DCs and collect non-adherent cells (consisting of 40% T cells). CFSE-labeled splenocytes (2 × 105) were co-cultured with varying ratios (1:2, 1:5, 1:10, 1:20, 1:50 and 1:100) of polarized macrophages for 5 d at 37°C, 5% CO2. A positive control, consisting of splenocytes cultured with 5 μg/mL Concanavalin A (Sigma-Aldrich) was included. On day 5, cells were stained for 30 min on ice with APC-Cy7-conjugated rat anti-mouse CD4 (BD) and PerCP-Cy5.5-conjugated rat anti-mouse CD8a (BD) antibodies to identify CD4+ and CD8+ T cells using a FACS Canto II flow cytometer (BD). As T cells proliferate, CFSE segregates equally between each daughter population. In flow cytometric analysis, each round of proliferation is seen as sequential halving of CFSE intensity. The percentage of T cell proliferation was calculated based on the loss of staining intensity of parental peak. Supernatants collected from the macrophage:T cell ratio of 1:2 MLR assay were assayed for IFNγ by cytokine bead array.
Immunofluorescence
Frozen sections (10 μm) of OCT-embedded tumors were fixed in ice-cold acetone (Sigma-Aldrich) for 10 min, air dried and the following steps performed at room temperature for 1 h. Sections were blocked with PBS/2% FCS and incubated with anti-F4/80-PE (BD), anti-Ly6C-biotin and rabbit anti-CX3CR1 Ab. After 3 PBS washes, secondary Ab streptavidin-AMCA (Jackson Immunoresearch, Pennsylvania, USA) and Alexafluor® 488-conjugated anti-rabbit Ab were added. Slides were washed and mounted in Citifluor™ anti-fadent (ProScitech) for visualization on an Olympus IX51 microscope (Olympus) with DP71 controller/manager software.
Statistical analysis
Statistical analyses were performed using GraphPad PRISM version 4 (GraphPad Software Inc.). Mann–Whitney U-test determined differences between two populations. One-way analysis of variance determined differences between more than two populations.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1173299_s02.pdf
Download PDF (258.5 KB)Acknowledgments
The authors acknowledge the facilities, and the scientific and technical assistance of the Curtin Health Innovation Research Institute core facility, Curtin University.
Funding
This work was funded by the Dust Disease Board of NSW, Australia, Cancer Council WA and the School of Biomedical Sciences, Curtin University.
References
- Jackaman C, Bundell CS, Kinnear BF, Smith AM, Filion P, van Hagen D, Robinson BW, Nelson DJ. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol 2003; 171:5051-63; PMID:14607902; http://dx.doi.org/10.4049/jimmunol.171.10.5051
- Jackaman C, Cornwall S, Graham PT, Nelson DJ. CD40-activated B cells contribute to mesothelioma tumor regression. Immunol Cell Biol 2011; 89:255-67; PMID:20628372; http://dx.doi.org/10.1038/icb.2010.88
- Jackaman C, Lew AM, Zhan Y, Allan JE, Koloska B, Graham PT, Robinson BW, Nelson DJ. Deliberately provoking local inflammation drives tumors to become their own protective vaccine site. Int Immunol 2008; 20:1467-79; PMID:18824504; http://dx.doi.org/10.1093/intimm/dxn104
- Jackaman C, Nelson DJ. Cytokine-armed vaccinia virus infects the mesothelioma tumor microenvironment to overcome immune tolerance and mediate tumor resolution. Cancer Gene Ther 2010; 17:429-40; PMID:20150930; http://dx.doi.org/10.1038/cgt.2009.85
- Caminschi I, Venetsanakos E, Leong CC, Garlepp MJ, Scott B, Robinson BW. Interleukin-12 induces an effective antitumor response in malignant mesothelioma. Am J Respir Cell Mol Biol 1998; 19:738-46; PMID:9806738; http://dx.doi.org/10.1165/ajrcmb.19.5.3257m
- Thomas A, Hassan R. Immunotherapies for non-small-cell lung cancer and mesothelioma. Lancet Oncol 2012; 13:e301-10; PMID:22748269; http://dx.doi.org/10.1016/S1470-2045(12)70126-2
- Marzo AL, Lake RA, Robinson BW, Scott B. T-cell receptor transgenic analysis of tumor-specific CD8 and CD4 responses in the eradication of solid tumors. Cancer Res 1999; 59:1071-9; PMID:10070965
- Robinson BW, Scott BM, Lake RA, Stumbles PA, Nelson DJ, Fisher S, Marzo AL. Lack of ignorance to tumor antigens: evaluation using nominal antigen transfection and T-cell receptor transgenic lymphocytes in Lyons-Parish analysis–implications for tumor tolerance. Clin Cancer Res 2001; 7:811s-7s; PMID:11300477
- Vachani A, Sterman DH, Albelda SM. Cytokine gene therapy for malignant pleural mesothelioma. J Thorac Oncol 2007; 2:265-7; PMID:17409795; http://dx.doi.org/10.1097/01.JTO.0000263706.23579.35
- Jackaman C, Cornwall S, Lew AM, Zhan Y, Robinson BW, Nelson DJ. Local effector failure in mesothelioma is not mediated by CD4+ CD25+ T-regulator cells. Eur Respir J 2009; 34:162-75; http://dx.doi.org/10.1183/09031936.00101008
- Jackaman C, Cornwall S, Lew AM, Zhan Y, Robinson BW, Nelson DJ. Local effector failure in mesothelioma is not mediated by CD4+ CD25+ T-regulator cells. Eur Respir J 2009; 34:162-75; PMID:19251786; http://dx.doi.org/10.1183/09031936.00101008
- Burt BM, Rodig SJ, Tilleman TR, Elbardissi AW, Bueno R, Sugarbaker DJ. Circulating and tumor-infiltrating myeloid cells predict survival in human pleural mesothelioma. Cancer 2011; 117:5234-44; PMID:21523763; http://dx.doi.org/10.1002/cncr.26143
- Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci 2008; 13:453-61; PMID:17981560; http://dx.doi.org/10.2741/2692
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 2007; 204:1057-69; PMID:17485518; http://dx.doi.org/10.1084/jem.20070075
- Deonarine K, Panelli MC, Stashower ME, Jin P, Smith K, Slade HB, Norwood C, Wang E, Marincola FM, Stroncek DF. Gene expression profiling of cutaneous wound healing. J Transl Med 2007; 5:11; PMID:17313672; http://dx.doi.org/10.1186/1479-5876-5-11
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood 2006; 107:2112-22; PMID:16269622; http://dx.doi.org/10.1182/blood-2005-01-0428
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002; 23:549-55; PMID:12401408; http://dx.doi.org/10.1016/S1471-4906(02)02302-5
- Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, Bottazzi B, Colombo MP, Mantovani A, Sica A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res 2006; 66:11432-40; PMID:17145890; http://dx.doi.org/10.1158/0008-5472.CAN-06-1867
- Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 2007; 179:977-83; PMID:17617589; http://dx.doi.org/10.4049/jimmunol.179.2.977
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004; 6:409-21; PMID:15488763; http://dx.doi.org/10.1016/j.ccr.2004.08.031
- Pelegrin P, Surprenant A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J 2009; 28:2114-27; PMID:19536133; http://dx.doi.org/10.1038/emboj.2009.163
- Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther 2008; 7:788-99; PMID:18375821; http://dx.doi.org/10.1158/1535-7163.MCT-07-0579
- Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, Hegmans JP. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010; 10:464; PMID:20804550; http://dx.doi.org/10.1186/1471-2407-10-464
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011; 331:1612-6; PMID:21436454; http://dx.doi.org/10.1126/science.1198443
- Jackaman C, Radley-Crabb HG, Soffe Z, Shavlakadze T, Grounds MD, Nelson DJ. Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell 2013; 12:345-57; PMID:23442123; http://dx.doi.org/10.1111/acel.12062
- Jackaman C, Dye DE, Nelson DJ. IL-2/CD40-activated macrophages rescue age and tumor-induced T cell dysfunction in elderly mice. Age (Dordr) 2014; 36:9655; PMID:24744051; http://dx.doi.org/10.1007/s11357-014-9655-y
- Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 2005; 11:6713-21; PMID:16166452; http://dx.doi.org/10.1158/1078-0432.CCR-05-0883
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol 2008; 66:1-9; PMID:17913510; http://dx.doi.org/10.1016/j.critrevonc.2007.07.004
- Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, Charles K, Gordon S, Balkwill FR. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol 2006; 176:5023-32; PMID:16585599; http://dx.doi.org/10.4049/jimmunol.176.8.5023
- Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, Closset R, Dewals B, Thielen C, Gustin P et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest 2009; 119:3723-38; PMID:19907079; http://dx.doi.org/10.1172/JCI39717
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 2010; 70:5728-39; PMID:20570887; http://dx.doi.org/10.1158/0008-5472.CAN-09-4672
- Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ, Lee CC, McBride WH, Chiang CS, Hong JH. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys 2007; 68:499-507; PMID:17398016; http://dx.doi.org/10.1016/j.ijrobp.2007.01.041
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141:39-51; PMID:20371344; http://dx.doi.org/10.1016/j.cell.2010.03.014
- Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J Immunol 2005; 174:6013-22; PMID:15879094; http://dx.doi.org/10.4049/jimmunol.174.10.6013
- Economou JS, McBride WH, Essner R, Rhoades K, Golub S, Holmes EC, Morton DL. Tumour necrosis factor production by IL-2-activated macrophages in vitro and in vivo. Immunology 1989; 67:514-9; PMID:2788610
- Puddu P, Carollo M, Pietraforte I, Spadaro F, Tombesi M, Ramoni C, Belardelli F, Gessani S. IL-2 induces expression and secretion of IFN-gamma in murine peritoneal macrophages. J Leukoc Biol 2005; 78:686-95; PMID:15951352; http://dx.doi.org/10.1189/jlb.0105035
- Jackaman C, Nelson DJ. Intratumoral interleukin-2/agonist CD40 antibody drives CD4(+)-independent resolution of treated-tumors and CD4 (+)-dependent systemic and memory responses. Cancer Immunol Immunother 2011; 61(4):549-60; PMID:22002241; http://dx.doi.org/10.1007/s00262-011-1120-5
- Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 1981; 11:805-15; PMID:7308288; http://dx.doi.org/10.1002/eji.1830111013
- Gordon S, Hamann J, Lin H-H, Stacey M. F4/80 and the related adhesion-GPCRs. Eur J Immunol 2011; 41:2472-6; PMID:21952799; http://dx.doi.org/10.1002/eji.201141715
- Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012; 12:35; PMID:22273460; http://dx.doi.org/10.1186/1471-2407-12-35
- Zaynagetdinov R, Sherrill TP, Polosukhin VV, Han W, Ausborn JA, McLoed AG, McMahon FB, Gleaves LA, Degryse AL, Stathopoulos GT et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J Immunol 2011; 187:5703-11; PMID:22048774; http://dx.doi.org/10.4049/jimmunol.1100558
- Kim SW, Kim JS, Papadopoulos J, Choi HJ, He J, Maya M, Langley RR, Fan D, Fidler IJ, Kim SJ. Consistent interactions between tumor cell IL-6 and macrophage TNF-α enhance the growth of human prostate cancer cells in the bone of nude mouse. Int Immunopharmacol 2011; 11:862-72; PMID:21251905; http://dx.doi.org/10.1016/j.intimp.2011.01.004
- Fritz JM, Tennis MA, Orlicky DJ, Lin H, Ju C, Redente EF, Choo KS, Staab TA, Bouchard RJ, Merrick DT et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol 2014; 5:587; PMID:25505466; http://dx.doi.org/10.3389/fimmu.2014.00587
- Kruse J, von Bernstorff W, Evert K, Albers N, Hadlich S, Hagemann S, Günther C, van Rooijen N, Heidecke CD, Partecke LI. Macrophages promote tumour growth and liver metastasis in an orthotopic syngeneic mouse model of colon cancer. Int J Colorectal Dis 2013; 28:1337-49; PMID:23657400; http://dx.doi.org/10.1007/s00384-013-1703-z
- Yang H, Rivera Z, Jube S, Nasu M, Bertino P, Goparaju C, Franzoso G, Lotze MT, Krausz T, Pass HI et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc Natl Acad Sci U S A 2010; 107:12611-6; PMID:20616036; http://dx.doi.org/10.1073/pnas.1006542107
- Bianchi C, Giarelli L, Grandi G, Brollo A, Ramani L, Zuch C. Latency periods in asbestos-related mesothelioma of the pleura. Eur J Cancer Prev 1997; 6:162-6; PMID:9237066
- Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 2008; 205:1261-8; PMID:18490490; http://dx.doi.org/10.1084/jem.20080108
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005; 7:211-7; PMID:15766659; http://dx.doi.org/10.1016/j.ccr.2005.02.013
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860-7; PMID:12490959; http://dx.doi.org/10.1038/nature01322
- Edwards JG, Faux SP, Plummer SM, Abrams KR, Walker RA, Waller DA, O'Byrne KJ. Cyclooxygenase-2 expression is a novel prognostic factor in malignant mesothelioma. Clin Cancer Res 2002; 8:1857-62; PMID:12060628
- Demetri GD, Zenzie BW, Rheinwald JG, Griffin JD. Expression of colony-stimulating factor genes by normal human mesothelial cells and human malignant mesothelioma cells lines in vitro. Blood 1989; 74:940-6; PMID:2787682
- Kumar-Singh S, Weyler J, Martin MJ, Vermeulen PB, Van Marck E. Angiogenic cytokines in mesothelioma: a study of VEGF, FGF-1 and -2, and TGF β expression. J Pathol 1999; 189:72-8; PMID:10451491; http://dx.doi.org/10.1002/(SICI)1096-9896(199909)189:1%3c72::AID-PATH401%3e3.0.CO;2-0
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499-506; PMID:19342621; http://dx.doi.org/10.4049/jimmunol.0802740
- Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 2006; 66:1123-31; PMID:16424049; http://dx.doi.org/10.1158/0008-5472.CAN-05-1299
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 2007; 13:828-35; PMID:17603493; http://dx.doi.org/10.1038/nm1609
- Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv Cancer Res 2015; 128:95-139; PMID:26216631; http://dx.doi.org/10.1016/bs.acr.2015.04.002
- Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 2015; 125:3356-64; PMID:26168215; http://dx.doi.org/10.1172/JCI80005
- Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y, Thomas DG et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013; 39:611-21; PMID:24012420; http://dx.doi.org/10.1016/j.immuni.2013.08.025
- Zheng J, Yang M, Shao J, Miao Y, Han J, Du J. Chemokine receptor CX3CR1 contributes to macrophage survival in tumor metastasis. Mol Cancer 2013; 12:141; PMID:24245985; http://dx.doi.org/10.1186/1476-4598-12-141
- Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol 2009; 9:900-9; PMID:19336265; http://dx.doi.org/10.1016/j.intimp.2009.03.015
- Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res 2003; 63:4490-6; PMID:12907622
- Broomfield S, Currie A, van der Most RG, Brown M, van Bruggen I, Robinson BW, Lake RA. Partial, but not complete, tumor-debulking surgery promotes protective antitumor memory when combined with chemotherapy and adjuvant immunotherapy. Cancer Res 2005; 65:7580-4; PMID:16140921; http://dx.doi.org/10.1158/0008-5472.CAN-05-0328
- Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, Tracey KJ, Ostrand-Rosenberg S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res 2014; 74:5723-33; PMID:25164013; http://dx.doi.org/10.1158/0008-5472.CAN-13-2347