
ABSTRACT
Pediatric cancers, including Ewing sarcoma (ES), are only weakly immunogenic and the tumor-patients' immune system often is devoid of effector T cells for tumor elimination. Based on expression profiling technology, targetable tumor-associated antigens (TAA) are identified and exploited for engineered T-cell therapy. Here, the specific recognition and lytic potential of transgenic allo-restricted CD8+ T cells, directed against the ES-associated antigen 6-transmembrane epithelial antigen of the prostate 1 (STEAP1), was examined. Following repetitive STEAP1130 peptide-driven stimulations with HLA-A*02:01+ dendritic cells (DC), allo-restricted HLA-A*02:01− CD8+ T cells were sorted with HLA-A*02:01/peptide multimers and expanded by limiting dilution. After functional analysis of suitable T cell clones via ELISpot, flow cytometry and xCELLigence assay, T cell receptors' (TCR) α- and β-chains were identified, cloned into retroviral vectors, codon optimized, transfected into HLA-A*02:01− primary T cell populations and tested again for specificity and lytic capacity in vitro and in a Rag2−/−γc−/− mouse model. Initially generated transgenic T cells specifically recognized STEAP1130-pulsed or transfected cells in the context of HLA-A*02:01 with minimal cross-reactivity as determined by specific interferon-γ (IFNγ) release, lysed cells and inhibited growth of HLA-A*02:01+ ES lines more effectively than HLA-A*02:01− ES lines. In vivo tumor growth was inhibited more effectively with transgenic STEAP1130-specific T cells than with unspecific T cells. Our results identify TCRs capable of recognizing and inhibiting growth of STEAP1-expressing HLA-A*02:01+ ES cells in vitro and in vivo in a highly restricted manner. As STEAP1 is overexpressed in a wide variety of cancers, we anticipate these STEAP1-specific TCRs to be potentially useful for immunotherapy of other STEAP1-expressing tumors.
Introduction
Ewing Sarcoma (ES) is an aggressive bone and soft tissue tumor with a peak incidence in adolescents and young adults. Although prognosis for patients with localized ES has improved during the past years, metastatic and recurrent disease still represents a therapeutic problem with an overall 5-y survival of less than 30%.Citation1,2 Especially, multifocal bone disease and early relapse are associated with poor prognosis.Citation3 Bone marrow involvement is a catastrophic event with little chance of cure, even with high-dose therapies including allogeneic stem-cell transplantation (SCT). Furthermore, current treatment regimens are associated with high toxicity, prompting the search for new therapeutic treatment modalities.
Allogeneic SCT is an established treatment for leukemia and is explored as a treatment for a variety of other hematologic and non-hematologic malignancies,Citation4 but seems less effective in solid cancers.Citation5 In ES patients, allogeneic SCT represents a therapy option,Citation6-8 but is limited by the extraordinary toxicity of allogenic SCTCitation5,9,Citation10 and is not efficacious in patients with bone marrow involvement (Uwe Thiel, personal communication).
During the past 15 y, methods emerged to identify, isolate and expand tumor-peptide-specific allo-restricted T cells ex vivo.Citation11-14 Further, characterization, cloning and expression of tumor-specific T cell receptors (TCRs) derived from such T cells and their subsequent expression in T cells for adoptive transferCitation15 is now an established procedure. Landmark clinical trials with a TCR specific for NY-ESO-1 already demonstrated the great potential of this approach.Citation14,16,Citation17
Allo-restricted TCRs have the additional advantage that they can be used to target tumor-associated antigens (TAA), overcoming the problem of negative selection of high-affinity TCRs within the thymus during development.Citation5,12,Citation13
We previously identified a number of genes that are highly upregulated in ESCitation18 with the 6-transmembrane epithelial antigen of the prostate 1 (STEAP1) being one of them. In addition to ES, where STEAP1 influences the invasive behavior and oxidative stress phenotype,Citation19 STEAP1 is also overexpressed in many cancers including prostate and bladder carcinoma,Citation20 but almost absent in normal tissues, except for urothelium and prostate.
In this study, we selected allo-restricted tumor-antigen-specific T-cell lines from an allo-reactive T cell pool, based on an HLA-A*02:01-multimer approach using peptides derived from STEAP1. We identified the specific TCRs from these T-cell lines, engineered transgenic, allo-restricted cytotoxic T cells directed against STEAP1 and demonstrate their in vitro and in vivo efficacy, rendering them a personalized treatment option for patients with STEAP1-expressing tumors.
Results
STEAP1130 is a suitable target peptide for adoptive cellular therapy (ACT)
We previously identified STEAP1 being highly overexpressed in primary ES, influencing proliferation and invasiveness of this tumor via alteration of intracellular reactive oxygen species (ROS) levels.Citation19 Apart from minor expression in prostate and urothelium, STEAP1 is only weakly expressed in normal tissues (Figs. S1 and S2A). To determine a suitable STEAP1 peptide that could be targeted by cytotoxic T cells, in silico prediction of HLA-A*02:01 binding and proteasomal cleavage was performed using BIMAS, NetCTL and SYFPEITHI web tools. Scores of various peptides calculated from three algorithms are shown in Table S1. Subsequently, we performed binding assays, wherein TAP transporter-deficient HLA-A*02:01+ T2 cells were loaded with varying concentrations of the relevant peptide and analyzed by flow cytometry. STEAP1130 (YLPGVIAAI) manifested to be the best HLA-A*02:01 binder with affinities comparable to the well-described influenza (GILGFVFTL) peptide (Fig. S3) and was used for subsequent in vitro priming of CD8+ T cells.
STEAP1130 T cell line specifically recognizes target structures
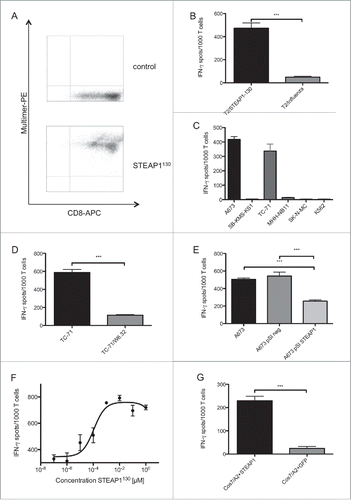
For the generation of allo-restricted STEAP1130-specific cytotoxic T cells, HLA-A*02:01− CD8+ T cells were primed with peptide-loaded HLA-A*02:01+ mature dendritic cells (DC). After 14 d of co-cultivation, cells were specifically stained by HLA-A*02:01/STEAP1130 multimer and anti-CD8 mAb. Double positive cells were FACS sorted and expanded via limiting dilution (Fig. S4A). Several lines of STEAP1130-multimer+ CD8+ T cells with specific recognition of STEAP1130 peptide-loaded T2 cells and HLA-A*02:01+ ES (Figs. S4B and S4C) were further expanded. One line (P2A5) was subsequently characterized in detail. This line stained positive for the HLA-A*02:01/STEAP1130 multimer () and was able to specifically recognize STEAP1130 peptide-loaded T2 cells () as well as STEAP1+HLA-A*02:01+ ES cell lines in interferon-γ (IFNγ) ELISpot assays, whereas HLA-A*02:01− or STEAP1-negative cells were not recognized (). The HLA-restricted detection of ES cells was reduced after blocking target cells with MHC-I-specific antibody W6.32 (). The quantity of released IFNγ corresponded to the quantity of presented peptide, since less IFNγ was secreted after specific siRNA mediated knock down of STEAP1 in A673 ES cells ( and Figs. S2B, C). Additionally, decreasing amounts of IFNγ release were observed after down-titration of STEAP1130 peptide onto T2 cells (). To confirm processing and transport of the predicted STEAP1130 nonamer to the surface of target cells, Cos7 cells were double-transfected with HLA-A*02:01 and STEAP1 cDNA or GFP, respectively. T cells released markedly more IFNγ upon co-incubation with STEAP1-transduced cells than upon incubation with GFP controls (), verifying processing and presentation as well as specific recognition of the target nonamer.
Figure 1. ES specificity of STEAP1130-specific T cell line P2A5. (A) Multimer staining of STEAP1130-P2A5 with CD8-APC and specific HLA-A*02:01/STEAP1130 multimer (bottom) or irrelevant multimer as control (top) (B–D), IFNγ release of STEAP1130-P2A5 during co-culture with STEAP1130 and influenza-pulsed T2 cells, respectively. (B) HLA-A*02:01+ (A673, TC71) and HLA-A*02:01− (SB-KMS-KS1, SK-N-MC, K562) tumor cells expressing STEAP1 or lacking STEAP1 expression (MHH-NB11). (C) TC-71 cells with and without MHC-I specific blocking mAB W6.32. (D) A673 cells with and without STEAP1 knock down. (E) T2 cells pulsed with titrated amounts of STEAP1130 peptide. (F) Cos 7 cells transfected with HLA-A*02:01 and either STEAP1 or GFP. (G) All analyzed in triplicates via IFNγ ELISpot. Error bars indicate SEM. p values < 0.05 were considered as statistically significant (*p < 0.05; **p < 0.005; ***p < 0.0005).
STEAP1130 T cell line specifically inhibits growth of target cells
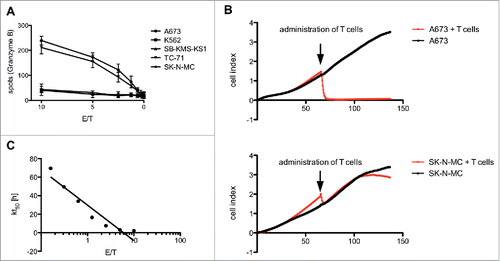
To show the ability of the STEAP1130-specific T cell line P2A5 to lyse target cells, we examined the release of granzyme B (GB) after co-incubation with HLA-A*02:01+ STEAP1+ double positive target cells A673 and TC-71 and HLA-A*02:01− cells SK-N-MC, SB-KMS-KS1 and K562 as controls. T cells released GB upon incubation with HLA-A*02:01+ STEAP1+ cells in a dose-dependent manner. Only baseline recognition was observed after stimulation with negative controls (). To further demonstrate the direct inhibition of ES cell growth, an impedance-based xCELLigence assay was performed, where a rapid lysis of A673 cells was observed after administration of T cell line P2A5, but almost no killing of HLA-A*02:01− ES cell line SK-N-MC (). The rate of killing was cell concentration-dependent as shown in .
Figure 2. Antitumor reactivity of STEAP1130-specific T cell clone P2A5. (A) Effector:target ratio- (E/T) dependent granzyme B release of STEAP1130-P2A5 after co-culture with various tumor cell lines. (B) Target-specific tumor cell lysis of A673 and SK-N-MC (E/T: 10) by STEAP1130-P2A5, detected via xCELLigence assay. (C) E/T-dependent timeframe needed for killing of 50% of tumor cells (kt50) by STEAP1130-P2A5.
Cloning and expression of STEAP1130-specific TCRs
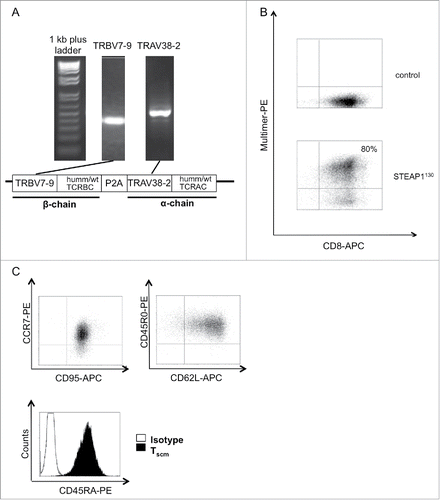
Clonality of TCR line P2A5 and the TCR repertoire of additional STEAP1130-specific, allo-restricted cytotoxic T-cells lines was determined using degenerated primers for the amplification and subsequent sequencing of TCR α- (TRAV) and TCR β-chains (TRBV). Sequence analysis revealed the TCR of line P2A5 to be dominant and this TCR was expressed in all of the three characterized STEAP1130-specific T cell lines, subsequently referred as STEAP1P2A5 T-cell clone. The identified TCR comprised TRAV38-2 and TRBV7-9 based on the International Immunogenetics nomenclature (). For subsequent analysis, both TCR chains were either codon optimized and minimally murinized (humm)Citation21 or cloned as wild-type (wt) sequence, linked with a self-cleaving P2A element into a pMP-71 retroviral backbone. Sequences for both constructs are documented in Table S2. For expression of STEAP1P2A5 in primary human T cells, RD114-pseudotyped retroviruses were used. We analyzed transduction of the TCRs into naive T cells and PBMCs, respectively, with transduction rates of up to 80% as analyzed by HLA-multimer staining in flow cytometry (). Comparable transduction rates were obtained for wt and humm STEAP1P2A5 TCRs (data not shown). Results obtained with wt STEAP1P2A5 TCRs are shown below.
Figure 3. Generation of STEAP1130 TCR-transgenic TSCM. (A) Identification of TCR α- (TRAV) and β- (TRBV) chains via PCR. Additionally, a scheme of the TCR construct, containing either minimal murinized and codon optimized (humm) or wild type (wt) TCR α- and β-chains linked with a self-cleaving P2A element and integrated into the pMP-71 vector is shown. (B) Multimer staining of STEAP1130 TCR-transgenic T cells with CD8-APC and HLA-A*02:01/STEAP1130 multimer (bottom) or irrelevant multimer as control (top). (C) Dot plots of T cells, co-stained with CCR7-PE/CD95-APC, CD45R0-PE/CD62L-APC and histogram plot of CD45RA-PE stained CD8+ T cells.
STEAP1P2A5 TCR-transgenic T cells revealed similar recognition patterns as T-cell clone P2A5
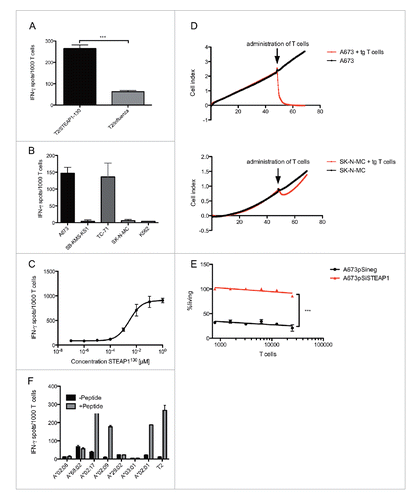
We then used the protocol of Cieri and colleaguesCitation22 for the infection and generation of STEAP1P2A5 TCRs transgenic stem-cell-memory-like T cells (TSCM). Accordingly, we stimulated naive T cells with anti-CD3/CD28 magnetic beads and moderate amounts of rhIL-7 and rhIL-15 (5 ng/mL) during infection. The transgenic CD8+ T cells stained positive with the STEAP1130-specific HLA-multimer and revealed a stem-cell-memory-like phenotype (CCR7+CD62L+CD45RA+CD45R0+CD95+, ). In addition, the STEAP1P2A5 TCR-transgenic T cells were able to release IFNγ upon stimulation with STEAP1130 peptide-pulsed T2 cells as well as HLA-A*02:01+ ES cell lines, whereas negative controls were not recognized (). The STEAP1130 peptide avidity of STEAP1P2A5 TCR-transgenic T cells was diminished in comparison to the initial T cell clone P2A5, but killing of target cells occurred with comparable efficacy (). Furthermore, lysis of target cells was antigen dependent, as shown in a cytotoxicity assay with A673 cells (A673pSineg) expressing wt amounts of STEAP1 or A673 cells with a STEAP1 knock-down (A673pSiSTEAP1) ().Citation19 The recognition of STEAP1130 in the context of HLA-A*02 was not restricted to HLA-A*02:01, as STEAP1P2A5 TCR-transgenic T cells released IFNγ upon co-incubation with STEAP1130 peptide-pulsed HLA-A*02:09+ and HLA-A*02:17+ LCL cell lines ().
Figure 4. Antigen specificity of STEAP1130 TCR-transgenic CD8+ T cells. (A–C) IFNγ release of STEAP1P2A5 TCR-transgenic T cells during co-culture with STEAP1130- and influenza-pulsed T2 cells, respectively. (A) HLA-A*02:01+ (A673, TC71) and HLA-A*02:01− (SB-KMS-KS1, SK-N-MC, K562) tumor cells expressing STEAP1. (B) T2 cells pulsed with titrated amounts of STEAP1130 peptide. (C) All analyzed in triplicates via IFNγ ELISpot. Error bars indicate SEM. p values < 0.05 were considered as statistically significant (***p < 0.0005). (D) Target-specific tumor cell lysis of A673 and SK-N-MC (E/T: 10) by STEAP1130 TCR-transgenic T cells, detected via xCELLigence assay. (E) T cell dose-dependent lysis of A673 cells after STEAP1 knock down (A673pSiSTEAP1, see supplementary information) in comparison to control transfected A673 cells (A673pSineg), 2 h after T cell inoculation. (F) IFNγ release of STEAP1P2A5 TCR-transgenic T cells upon co-culture with STEAP1130 peptide-loaded or unloaded, respectively, LCL cell lines in the context of various HLA-A subtypes.
Transgenic STEAP1130-specific TSCM inhibit tumor growth in a Rag2−/−γc−/− mouse model
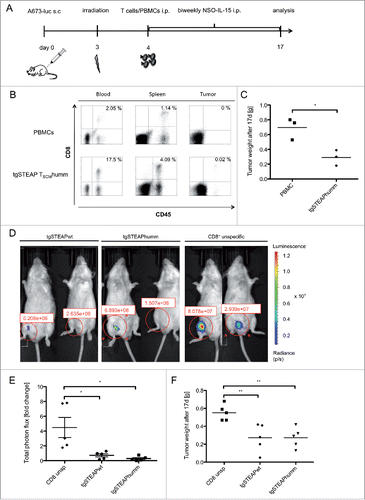
To examine in vivo efficacy of stem-cell-like STEAP1P2A5 TCR-transgenic T cells, immune-deficient Rag2−/−γc−/− mice were inoculated subcutaneously (s.c.) with 2 × 106 A673 cells and in a first experiment, 4 d later, i.p. injected with 3 × 106 STEAP1P2A5 TCR-transgenic CD8+ TSCM cells together with 5 × 106 CD8+-depleted, autologous PBMCs or with unspecific PBMCs as a control (), respectively. Tumor size and CD8+ T-cell infiltration into spleen, blood and tumor were analyzed after 17 d. Human CD8+ T cell engraftment could be detected in mice of both treatment groups, but only tumors of animals treated with specific T cells showed CD8+ T cell infiltration (). Furthermore, tumors of animals treated with STEAP1P2A5 TCR-transgenic TSCM were markedly reduced, compared to animals treated with unspecific PBMCs (). In a second experiment, we increased the T cell dose and the mice received either 5 × 106 wt or humm STEAP1P2A5 TCR-transgenic CD8+ TSCM cells or unspecific CD8+ T cells together with 5 × 106 CD8+-depleted autologous PBMCs. Tumor size and total photon flux of luciferase-transduced A673 tumors were analyzed. Tumor growth was markedly delayed in animals treated with STEAP1P2A5 TCR-transgenic T cells, compared to mice receiving unspecific CD8+ T cells (, Fig. S5A). Finally, tumor size in animals treated with STEAP1P2A5 TCR-transgenic CD8+ T cells was significantly reduced, compared to the group of mice receiving unspecific CD8+ T cells (). We observed no difference between animals receiving wt or humm STEAP1P2A5 TCR-transgenic TSCM. Manual counting of CD3+ cells in histological sections demonstrated a stronger tumor infiltration by administered T cells in animals receiving STEAP1P2A5 TCR-transgenic CD8+ TSCM cells, compared to animals receiving unspecific T cells (Figs. S5A and S5B).
Figure 5. In vivo anti ES reactivity of STEAP1130TCR-transgenic TSCM. 2 × 106 A673 cells were injected into the groin of Rag2−/−γc−/− mice. Animals were irradiated on day 3 (3.5 Gy) and received 5 × 106 T cells the day after. 1.5 × 107 human IL-15 producing cells were injected twice a week i.p. (A) Scheme of the experimental set-up for in vivo experiments. (B–C) Animals treated with either human codon optimized/minimal murinized (humm) STEAP1P2A5 TCR-transgenic (tg) T cells or unspecific PBMCs were analyzed 17 d after inoculation with A673 cells. (B) FACS analysis of blood, spleen and tumor of animals. Whole blood was stained with anti-human CD45 mAb and anti-human CD8 mAb. Percentages of double positive cells are given. A representative experiment of three mice per group is shown. (C) Analysis of total tumor weight, each dot corresponds to one animal. (D–F) Animals received 2 × 16 luciferase expressing A673 cells and were irradiated on day 3 (3.5 Gy). One day later, 5 × 106 STEAP1P2A5 TCR-transgenic TSCM were injected i.p. (D) Total photon flux (p/s) of luciferase expressing A673 cells, 3 d after i.p injection of either wild type (wt) or humm STEAP1-transgenic TSCM or unspecific CD8+ T cells, respectively. (E) Fold change of total photon flux 3 d after i.p injection of either wt or humm STEAP1-transgenic TSCM or unspecific CD8+ T cells, respectively. (F) Absolute tumor weight of tgSTEAP1wt- or tgSTEAP1humm-treated animals and unspecific CD8+ T-cell-treated controls, 17 d after initial tumor cell injection. Each dot corresponds to one animal.
Discussion
ES appear weakly immunogenic, presumably due to the activity of tumor-associated macrophages,Citation23 a high proportion of bone marrow T cells with regulatory phenotypeCitation24 and the low mutation rate observed for this tumor.Citation25-28 However, the addition of adjunctive, immune-activating therapies during first remission demonstrated the potential to reduce recurrence rates and exploit immunotherapy approaches for ES patients.Citation29
Balanced chromosomal EWS/ETS translocations that give rise to oncogenic chimeric proteins (EWS-ETS), the most common being EWS-FLI1 as a consequence of the t(11;22)(q24;q12) translocation,Citation30,31 are the characteristic driver event of ES tumorgenesis.Citation32 However, despite an MHC-class-II-restricted peptide derived from the fusion region of EWS-FLI1 that is able to initiate a CD4+ T-cell response,Citation33 no immunogenic ES-specific MHC-class-I-binding peptides derived from this fusion region have been identified,Citation34 prompting the search for further immunogenic epitopes of this disease.
STEAP1 is a part of an ES-specific signature identified previously.Citation18 STEAP proteins are homologs of NADPH-oxidases (NOX), involved in cellular ROS metabolism and frequently overexpressed in cancers. We demonstrated that STEAP1 is induced by EWS-FLI1 and that its expression promotes proliferation, invasiveness, anchorage-independent colony formation, tumorigenicity and metastasis of ES cells,Citation19 as well as impacting on patient survival.Citation35 Apart from minor expression in prostate and urothelium, STEAP1 is only weakly expressed in normal tissues.Citation36
Controversial results have been observed with autologous SCT for patients with high risk or recurrent ES. Whereas some studies reported improved disease-free survival over historical controls,Citation37-39 others observed no long-term benefit compared to conventional therapies.Citation40,41 Further, there was no improvement of survival of ES patients receiving reduced intensity conditioning compared to high-dose conditioning prior to allogeneic SCT with HLA-matched grafts, implicating absence of a clinically relevant graft versus ES effect.Citation42
T-cell recognition of conventional and allogeneic antigens share similarities.Citation43,44 Allo-restricted T cells mainly recognizing peptide in the context of specific MHC, are now easily isolated from an allogeneic T cell population by the use of peptide MHC multimers.Citation13,15 Using this technique, we succeeded in establishing T cell clones directed against the HLA-A*02:01-restricted STEAP1130 peptide (YLPGVIAAI) that turned out to be the best HLA-A*02:01-binding peptide and generated an allo-restricted, cytotoxic CD8+ T-cell response, able to kill ES cells specifically in an HLA-A*02:01-restricted fashion. We choose HLA-A*02:01 due to its dominant expression in the Caucasian population (around 50%) and similar prevalence in ES patients.Citation45 The STEAP1130 peptide (YLPGVIAAI) was not only well recognized by allo-restricted T cells but similarly seemed processed and presented on HLA-A*02:01, based on HLA-A*02:01 and STEAP1 cDNA co-transfection experiments into Cos-7 cells.
The subsequent characterization of the STEAP1130 peptide-specific TCRs suggested the presence of a dominant T-cell response since in all of the three analyzed STEAP1130-specific T-cell clones the same TCR was present. We codon-optimized variable and minimally murinized constant TRAV and TRBV domains to increase expression based on higher affinity of murine sequences to human CD3Citation46 and specific hetero-dimerization. STEAP1P2A5 TCR-transgenic T cells turned out to be similarly effective in comparison to the native T cell clone P2A5, although a decreased avidity of the STEAP1P2A5 TCR-transgenic T cells toward the STEAP1130 peptide was observed, which has previously also been described for other transgenic TCRs.Citation5
The STEAP1130-restricted TCRs, even though isolated from an allogeneic T-cell population, seemed HLA-A2-restricted and peptide-specific, since we did not observe any cross-reactivity against other MHC molecules or STEAP1-negative cell lines. STEAP1130 peptide presentation on other HLA class I molecules generated a T-cell response upon co-incubation of HLA-A*02:09+ and HLA-A*02:17+ LCL cell lines pulsed with STEAP1130 peptide, indicating the ability of this peptide to bind onto and be recognized on HLA-A2-related MHC class I molecules. Furthermore, analysis of in vivo efficacy of STEAP1P2A5 TCR-transgenic T cells did not cause any overt adverse effects and tissue toxicity in immune-deficient Rag2−/−γc−/− mice.Citation47 However, further toxicity testing of this TCR before transfer into the clinic is designated in future experiments.
What type of donor T cell will be most appropriate for TCR gene transfer is under intense investigation. Recent results suggest that virus-specific central memory T cells are good candidates,Citation48 but others also propose that certain characteristics of naive T cells may possess superior traits for adoptive T cell immunotherapy.Citation49 For instance, in a study in mice, it was demonstrated that a single naive T cell can expand in vivo and give rise to effector, central memory and effector memory T cells.Citation50 In vitro human naive T cells can be easily transduced, demonstrated superior tumor cell elimination, and stronger proliferation while retaining longer telomeres,Citation50 all desired properties for future therapeutic T cells. For the generation of STEAP1P2A5 TCR-transgenic T cells, we used the protocol of Cieri and colleagues.Citation22 The transgenic CD8+ T cells stained positive with the STEAP1130-specific HLA-A*02:01-multimer and revealed a stem-cell-memory-like phenotype (CCR7+CD62L+CD45RA+CD45R0+CD95+). They were able to release IFNγ upon contact with their specific target antigen. Tumor growth was markedly delayed in tumor-bearing animals treated with STEAP1P2A5 TCR-transgenic TSCM compared to mice receiving unspecific PBMC or unspecific CD8+ T cells.
Thus, we demonstrated here the generation of highly specific and efficacious STEAP1P2A5 TCR-transgenic T cells, able to recognize and inhibit the growth of STEAP1-expressing HLA-A*02:01+ ES cells in vitro as well as in vivo in a highly restricted fashion. These STEAP1-specific TCRs are potentially useful for immunotherapy of other STEAP1-expressing tumors and may already yet open the avenue for new therapeutic strategies, such as allogeneic stem cell and effector cell transplantation for the treatment of patients with STEAP1-positive tumors.
Materials and methods
Cell lines
ES cell lines TC-71 and SK-N-MC as well as the neuroblastoma cell line MHH-NB11 were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ). A673 (ES cell line) and Cos7 (Simian SV40-transformed fibroblasts) were obtained from ATCC (LGC Standards GmbH). TAP-transporter-deficient HLA-A*02:01+ (HLA-A2) T2 cells were from P Cresswell (Yale University School of Medicine, New Haven, CT, USA). K562 (erythroid leukemia cell line) was a gift from A Knuth and E Jäger (Krankenhaus Nordwest, Frankfurt, Germany). The HLA-A*02:01− ES cell line SB-KMS-KS1 (former SBSR-AKS) was described previously.Citation13 Retroviral packaging cell line RD114 was a gift of Manuel Caruso (Center de Recherche en Cancérologie, Quebec, Canada). Human IL-15 producing NSO cells were a kind gift of S. Riddell (Seattle, Washington, USA). LCL cell lines (kindly provided by A. Krackhardt), expressing various common HLA subclasses were routinely tested for HLA-A*02:01 status. All ES and neuroblastoma cell lines were cultured in RPMI 1640 medium (Invitrogen) containing 10% FBS (Biochrom) and 100 U/mL penicillin and 100 µg/mL streptomycin (Invitrogen). For LCL and T2 cells, RPMI 1640 medium was additionally provided with 1mM Na-pyruvate and non-essential amino acids (Invitrogen). T cell clones as well as transgenic T cells were cultured in AIM-V medium (Invitrogen) containing 5% human AB serum (Lonza) and antibiotics (T cell medium, TCM). DCs were cultivated in X-VIVO 15 medium (Lonza) enriched with 1% human AB serum and no antibiotics. RD114 packaging cells and hIL-15 producing NSO cells were cultured in DMEM containing 10% FBS, 1 mM Na-pyruvate, 1 mM non-essential amino acids and antibiotics. All cell lines were present for more than 6 mo in the lab and routinely tested for purity (e.g. EWS-FLI1 translocation product, surface antigen expression or HLA-phenotype) and Mycoplasma contamination.
In silico prediction of suitable HLA-A*02:01-binding peptides
An in silico analysis of STEAP1 peptide nonamers was executed to find feasible targets for adoptive cell transfer (ACT). We used BIMAS, NetCTL and SYFPEITHI algorithm tools to check for HLA-A*02:01 binding and proteasomal cleavage as well as TAP transport. Peptides that were among the top 10 binders of all the three web tools and at least the top five in one were chosen for further analysis.
HLA-A*02:01-binding assay
To confirm suitable presentation of STEAP1-derived peptides to HLA-A*02:01, predicted by the in silico analysis, a binding assay using the TAP transporter-deficient T2 cell line was performed. 50 and 100 µM as well as titrated amounts of various STEAP1 peptide nonamers were loaded onto T2 cells for 16 h at 37°C and 5% CO2. Subsequently, stabilized HLA-A2 molecules on the cell surface were stained with a FITC-conjugated anti-HLA-A2 antibody (BD Bioscience) and fluorescence intensity compared to a well-established influenza peptide (GILGFVFTL).
Isolation of PBMCs
The PBMCs human peripheral blood samples (obtained with IRB approval and informed consent from the DRK-Blutspendedienst Baden-Wuerttemberg-Hessen, Ulm, Germany) were isolated via density-gradient centrifugation using ficoll paque (GE Healthcare), according to the supplier's instructions.
Generation of dendritic cells (DC)
After isolation of PBMCs out of a healthy HLA-A*02:01+ donor, CD14+ monocytes were isolated using anti-human CD14 magnetic particles (BD Bioscience), according to the supplier's instructions. 100 ng/mL rhIL-4 (R&D) and 800 U/mL rhGM-CSF (ImmunoTools) were added and replaced on day 3 to induce transformation into DCs. Addition of a cytokine cocktail containing 1,000 U/mL rhIL-6 (R&D), 10 ng/mL rhIL-1β (Pan Biotech), 10 ng/mL rhTNF-α (R&D) and 1 µg/mL PGE2 (Cayman Chemicals) on day 5 induced maturation of DCs. On day 9, cells were checked for maturation markers CD83, CD86 and HLA-DR by flow cytometry.
Isolation of CD8+ T-cell subtypes
To obtain an untouched CD8+ T-cell population, the irrelevant cell populations of healthy HLA-A*02:01− donor PBMCs were magnetically labeled using the human CD8+ T-cell isolation kit (Miltenyi) and subsequently depleted on LS columns (Miltenyi), following the manufacturer's instructions. For further isolation of untouched naive CD8+ T cells, cytotoxic T cells were enriched for CCR7+CD45RA+ cells, using the Naive Pan T-cell Isolation Kit (Miltenyi) according to the supplier's protocol.
In vitro priming
Mature DCs were pulsed with 38 µM STEAP1130 peptide, supported by 20 µg/mL β2-microglobulin (Sigma Aldrich), in TCM for 4 h at 37°C and 5% CO2. Subsequently, untouched CD8+ T cells from a HLA-A*02:01− healthy donor were co-cultured with pulsed DCs in TCM, containing 10 ng/mL rhIL-12 (Pan Biotech) and 1,000 U/mL rhIL-6 for 1 week at an effector to target ratio of 1:20. After one week, T cells were re-primed with the same amount of peptide-loaded DCs together with 100 U/mL rhIL-2 (Novartis) and 5 ng/mL rhIL-7 (R&D).
Multimer staining and FACS sorting
For FACS sorting of STEAP1130-specific T cells, cells were pooled after 2 weeks of co-culture and stained with HLA-A*02:01/STEAP1130-specific phycoerythrin (PE) labeled multimers (home-made, as previously describedCitation51) and anti-CD8 monoclonal antibody (mAb, BD Bioscience). Isotype IgG (BD Bioscience) and irrelevant multimer served as controls. Sorting was performed on a FACS Aria (BD Bioscience) at the Institute of Medical Microbiology (TUM).
Expansion of STEAP1130 specific T cell clones
After sorting, STEAP1130-specific CD8+ T cells were expanded via limiting dilution in 96-well round bottom plates. Irradiated LCLs (100 Gy) and PBMCs (30Gy) of five different donors served as feeder cells. Cells were activated using 30 ng/mL anti-CD3 mAb (Okt3), 100 U/mL rhIL-2 and 2 ng/mL rhIL-15 (R&D). After 7 d, the medium and cytokines were refreshed. Expanded cells were subsequently tested in ELISpot and xCELLigence assays.
Functional characterization of T cell clone P2A5 and STEAP1130-transgenic T cells
To test expanded T cell lines and transgenic T cells for their specificity, IFNγ and GB release upon co-incubation with target cells and controls, respectively, was examined using ELISpot assays (Mabtech), according to the supplier's instructions. Effector and target cells were incubated for 20 h at 37°C and 5% CO2 at a ratio of 1:20. To monitor in situ cytotoxicity of T cells, an impedance-based xCELLigence assay (Roche Diagnostics) was performed, enabling continuous tracking of effector cell activity against ES cell lines A673 and SK-N-MC. Growth curves of untreated tumor cells served as controls.
Cell lines with STEAP1 knock down
ES cell lines with stable silencing of STEAP1 expression were previously described.Citation19
Identification of TCR Vα- and Vβ-chain and synthesis of retroviral TCR construct
To identify the TCR Vα- and Vβ-repertoire of specific T cell clones, we used a set of degenerated PCR primers, covering a bulk of published Vα- and Vβ-chains, to amplify specific TCR Vα- and Vβ-chains.Citation12,15 After separating the PCR products in an agarose gel, specific DNA fragments were excised and sequenced. The resulting sequence was analyzed using the IMGT database. Afterward, a fully functional TCR was designed using the GeneART® Gene Synthesis web tool. To this end, Vα- and Vβ-chains of the TCR were complemented with their respective constant chain, linked with a self-cleaving P2A element and integrated into a pMP-71 retroviral backbone.Citation12,15 In addition to the native TCR (wt), a human codon optimized and minimal murinized TCR (humm) was designed. Sequences of both TCRs are given in Table S2.
Retroviral transduction of T cells
The fully functional TCR was transduced into human PBMCs or TSCM, using the retroviral vector pMP-71. Briefly, PBMCs were activated with an anti-CD3 mAb and 100 U/mL IL-2. Two days later, spin transduction was performed using high-titer virus supernatant produced by the packaging cell line RD114. Infection was repeated on the following day and transduction success determined via FACS staining after one week. For the generation of transgenic TSCM, naive CD8+ T cells were activated with αCD3/CD28 beads (Invitrogen), at a bead to cell ratio of 3:1, together with 5 ng/mL rhIL-7 and rhIL-15 followed by an identical transduction procedure.
In vivo activity of transgenic T cells
To investigate the in vivo activity of transgenic T cells in an initial experiment, we injected 2 × 106 A673 cells s.c. into the groin of Rag2−/−γC−/− mice (BALB/c background, obtained from the Central Institute for Experimental Animals, Kawasaki, Japan). Animals were irradiated sub-lethally (3.5 Gy) on day 3 followed by i.p injection of 3 × 106 STEAP1P2A5 TCR-transgenic CD8+ TSCM cells together with 5 × 106 CD8+-depleted, autologous PBMCs or unspecific PBMCs of a HLA-A*02:01− donor, respectively, the day after. Twice a week, 1.5 × 107 irradiated (80 Gy) hIL-15 producing NSO cells were injected i.p. The experiments were stopped at day 17 and tumors were weighed and analyzed for T-cell infiltration using flow cytometry. Subsequently, a second experiment was performed where animals were inoculated s.c. with 2 × 106 luciferase expressing A673 cells and 4 d later i.p. injected with 5 × 106 STEAPP2A5 TCR-transgenic CD8+ TSCM cells together with 5 × 106 CD8+-depleted, autologous PBMCs or unspecific CD8+ T cells, respectively. After 17 d, tumors were weighed and analyzed for immune cell infiltration by immunohistochemistry. Additionally, tumors were evaluated by measuring total photon flux after administration of 150 mg luciferin/kg body weight (Caliper life science).
Statistical analysis
Descriptive statistics is used to determine parameters like mean, standard deviation and standard error of the mean (SEM). Differences were analyzed by unpaired 2-tailed student's t-test as indicated using Excel (Microsoft) or Prism 5 (GraphPad Software); p values < 0.05 were considered statistically significant (*p < 0.05; **p < 0.005; ***p < 0.0005).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1175795_s02.zip
Download Zip (9.3 MB)Acknowledgments
Experimental support by Kristina von Heyking, Henrieke Gerdes, Lynette Henkel and Isabel Storz is gratefully acknowledged.
Funding
This work was supported by grants from the Else Kröner-Fresenius-Stiftung (2013_A49), the Wilhelm Sander-Stiftung (2006.109.2), as well as the SFB824 (TP C10) and the TR-SFB 36 (TP B13) of the Deutsche Forschungsgemeinschaft. It is part of the Translational Sarcoma Research Network (TransSaRNet; 01GM1104B), “Rare Diseases” and Prospective Validation of Biomarkers in Ewing Sarcoma for Personalised Translational Medicine (PROVABES; 01KT1311), Funding Programs of the Federal Ministry of Education and Research (BMBF), Germany. T.G.P.G. is supported by a grant from “Verein zur Förderung von Wissenschaft und Forschung an der Medizinischen Fakultät der LMU München (WiFoMed),” the Daimler and Benz Foundation in cooperation with the Reinhard Frank Foundation, by LMU Munich's Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative, the “Mehr LEBEN für krebskranke Kinder – Bettina-Bräu-Stiftung,” the Fritz Thyssen Foundation (Ref. 40.15.0.030MN), the Walter Schulz foundation and by the German Cancer Aid (DKH-111886).
References
- Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA, Juergens H. Ewing's sarcoma family of tumors: current management. Oncologist 2006; 11:503-19; PMID:16720851; http://dx.doi.org/10.1634/theoncologist.11-5-503
- Rodriguez-Galindo C, Navid F, Liu T, Billups CA, Rao BN, Krasin MJ. Prognostic factors for local and distant control in Ewing sarcoma family of tumors. Ann Oncol 2008; 19:814-20; PMID:17998282; http://dx.doi.org/10.1093/annonc/mdm521
- Burdach S, Jurgens H. High-dose chemoradiotherapy (HDC) in the Ewing family of tumors (EFT). Crit Rev Oncol Hematol 2002; 41:169-89; PMID:11856593; http://dx.doi.org/10.1016/S1040-8428(01)00154-8
- Childs R, Chernoff A, Contentin N, Bahceci E, Schrump D, Leitman S, Read EJ, Tisdale J, Dunbar C, Linehan WM et al. Regression of metastatic renal-cell carcinoma after nonmyeloablative allogeneic peripheral-blood stem-cell transplantation. N Engl J Med 2000; 343:750-8; PMID:10984562; http://dx.doi.org/10.1056/NEJM200009143431101
- Cieri N, Mastaglio S, Oliveira G, Casucci M, Bondanza A, Bonini C. Adoptive immunotherapy with genetically modified lymphocytes in allogeneic stem cell transplantation. Immunol Rev 2014; 257:165-80; PMID:24329796; http://dx.doi.org/10.1111/imr.12130
- Burdach S, Thiel U, Schoniger M, Haase R, Wawer A, Nathrath M, Kabisch H, Urban C, Laws HJ, Dirksen U et al. Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 2010 Mar; 45(3):483-9; PMID:19684633; http://dx.doi.org/doi:10.1038/bmt.2009.184
- Koscielniak E, Gross-Wieltsch U, Treuner J, Winkler P, Klingebiel T, Lang P, Bader P, Niethammer D, Handgretinger R. Graft-versus-Ewing sarcoma effect and long-term remission induced by haploidentical stem-cell transplantation in a patient with relapse of metastatic disease. J Clin Oncol 2005; 23:242-4; PMID:15625381; http://dx.doi.org/10.1200/JCO.2005.05.940
- Lucas KG, Schwartz C, Kaplan J. Allogeneic stem cell transplantation in a patient with relapsed Ewing sarcoma. Pediatr Blood Cancer 2008; 51:142-4; PMID:18266223; http://dx.doi.org/10.1002/pbc.21503
- Storb R, Gyurkocza B, Storer BE, Sorror ML, Blume K, Niederwieser D, Chauncey TR, Pulsipher MA, Petersen FB, Sahebi F et al. Graft-versus-host disease and graft-versus-tumor effects after allogeneic hematopoietic cell transplantation. J Clin Oncol 2013; 31:1530-8; PMID:23478054; http://dx.doi.org/10.1200/JCO.2012.45.0247
- Burdach S, van Kaick B, Laws HJ, Ahrens S, Haase R, Korholz D, Pape H, Dunst J, Kahn T, Willers R et al. Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. Stem-cell transplant programs at Dusseldorf University Medical Center, Germany and St. Anna Kinderspital, Vienna, Austria. Ann Oncol 2000; 11:1451-62; PMID:11142486; http://dx.doi.org/10.1023/A:1026539908115
- Moris A, Teichgraber V, Gauthier L, Buhring HJ, Rammensee HG. Cutting edge: characterization of allorestricted and peptide-selective alloreactive T cells using HLA-tetramer selection. J Immunol 2001; 166:4818-21; PMID:11290755; http://dx.doi.org/10.4049/jimmunol.166.8.4818
- Schuster IG, Busch DH, Eppinger E, Kremmer E, Milosevic S, Hennard C, Kuttler C, Ellwart JW, Frankenberger B, Nössner E et al. Allorestricted T cells with specificity for the FMNL1-derived peptide PP2 have potent antitumor activity against hematologic and other malignancies. Blood 2007; 110:2931-9; PMID:17626842; http://dx.doi.org/10.1182/blood-2006-11-058750
- Thiel U, Pirson S, Muller-Spahn C, Conrad H, Busch DH, Bernhard H, Richter GH. Specific recognition and inhibition of Ewing tumour growth by antigen-specific allo-restricted cytotoxic T cells. Br J Cancer 2011; 104:948-56; PMID:21407224; http://dx.doi.org/10.1038/bjc.2011.54
- June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med 2015 Oct;149(4):1042-52; PMID:25810311; http://dx.doi.org/10.1053/j.gastro.2015.05.055
- Dargel C, Bassani-Sternberg M, Hasreiter J, Zani F, Bockmann JH, Thiele F, Bohne F, Wisskirchen K, Wilde S, Sprinzl MF et al. T cells engineered to express a T-cell receptor specific for Glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology 2015; 149(4):1042-52; PMID:26052074; http://dx.doi.org/10.1053/j.gastro.2015.05.055
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011; 29:917-24; PMID:21282551; http://dx.doi.org/10.1200/JCO.2010.32.2537
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; http://dx.doi.org/10.1126/science.aaa4967
- Staege MS, Hutter C, Neumann I, Foja S, Hattenhorst UE, Hansen G, Afar D, Burdach SE. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res 2004; 64:8213-21; PMID:15548687; http://dx.doi.org/10.1158/0008-5472.CAN-03-4059
- Grunewald TG, Diebold I, Esposito I, Plehm S, Hauer K, Thiel U, da Silva-Buttkus P, Neff F, Unland R, Müller-Tidow C et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res 2012; 10:52-65; PMID:22080479; http://dx.doi.org/10.1158/1541-7786.MCR-11-0524
- Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood 2006; 108:1388-94; PMID:16609065; http://dx.doi.org/10.1182/blood-2006-02-003681
- Sommermeyer D, Uckert W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J Immunol 2010; 184:6223-31; PMID:20483785; http://dx.doi.org/10.4049/jimmunol.0902055
- Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, Bondanza A, Bordignon C, Peccatori J, Ciceri F et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013; 121:573-84; PMID:23160470; http://dx.doi.org/10.1182/blood-2012-05-431718
- Fujiwara T, Fukushi J, Yamamoto S, Matsumoto Y, Setsu N, Oda Y, Yamada H, Okada S, Watari K, Ono M et al. Macrophage infiltration predicts a poor prognosis for human Ewing sarcoma. Am J Pathol 2011; 179:1157-70; PMID:21771572; http://dx.doi.org/10.1016/j.ajpath.2011.05.034
- Brinkrolf P, Landmeier S, Altvater B, Chen C, Pscherer S, Rosemann A, Ranft A, Dirksen U, Juergens H, Rossig C. A high proportion of bone marrow T cells with regulatory phenotype (CD4+CD25hiFoxP3+) in Ewing sarcoma patients is associated with metastatic disease. Int J Cancer 2009; 125:879-86; PMID:19480009; http://dx.doi.org/10.1002/ijc.24461
- Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 2014; 10:e1004475; PMID:25010205; http://dx.doi.org/10.1371/journal.pgen.1004475
- Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 2014; 4:1326-41; PMID:25186949; http://dx.doi.org/10.1158/2159-8290.CD-13-1037
- Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetête-Lalami S, Rusch M et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 2014; 4:1342-53; PMID:25223734; http://dx.doi.org/10.1158/2159-8290.CD-14-0622
- Agelopoulos K, Richter GH, Schmidt E, Dirksen U, von Heyking K, Moser B, Klein HU, Kontny U, Dugas M, Poos K et al. Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin Cancer Res 2015; 21(21):4935-46; PMID:26179511; http://dx.doi.org/10.1158/1078-0432.CCR-14-2744
- Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, Tesso M, Long LM, Grindler D, Merino M et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res 2008; 14:4850-8; PMID:18676758; http://dx.doi.org/10.1158/1078-0432.CCR-07-4065
- Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Ann Rev Path 2012; 7:145-59; PMID:21942527; http://dx.doi.org/10.1146/annurev-pathol-011110-130237
- Mackintosh C, Madoz-Gúrpide J, Ordóñez JL, Osuna D, Herrero-Martín D. The molecular pathogenesis of Ewing's sarcoma. Cancer Biol Therapy 2010; 9:655-67; PMID:20215864; http://dx.doi.org/10.4161/cbt.9.9.11511
- Kovar H. Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert Opin Ther Targets 2014; 18:1315-28; PMID:25162919; http://dx.doi.org/10.1517/14728222.2014.947963
- Meyer-Wentrup F, Richter G, Burdach S. Identification of an immunogenic EWS-FLI1-derived HLA-DR-restricted T helper cell epitope. Pediatr Hematol Oncol 2005; 22:297-308; PMID:16020117; http://dx.doi.org/10.1080/08880010590935194
- Evans CH, Liu F, Porter RM, O'Sullivan RP, Merghoub T, Lunsford EP, Robichaud K, Van Valen F, Lessnick SL, Gebhardt MC et al. EWS-FLI-1-targeted cytotoxic T-cell killing of multiple tumor types belonging to the Ewing sarcoma family of tumors. Clin Cancer Res 2012; 18:5341-51; PMID:22879388; http://dx.doi.org/10.1158/1078-0432.CCR-12-1985
- Grunewald TG, Ranft A, Esposito I, da Silva-Buttkus P, Aichler M, Baumhoer D, Schaefer KL, Ottaviano L, Poremba C, Jundt G et al. High STEAP1 expression is associated with improved outcome of Ewing's sarcoma patients. Ann Oncol 2012; 23:2185-90; PMID:22317770; http://dx.doi.org/10.1093/annonc/mdr605
- Grunewald TG, Bach H, Cossarizza A, Matsumoto I. The STEAP protein family: versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol Cell 2012; 104:641-57; PMID:22804687; http://dx.doi.org/10.1111/boc.201200027
- Burdach S, Meyer-Bahlburg A, Laws HJ, Haase R, van Kaik B, Metzner B, Wawer A, Finke R, Göbel U, Haerting J et al. High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 2003; 21:3072-8; PMID:12915596; http://dx.doi.org/10.1200/JCO.2003.12.039
- Burdach S. Treatment of advanced Ewing tumors by combined radiochemotherapy and engineered cellular transplants. Pediatr Transplant 2004; 8:67-82; PMID:15125708; http://dx.doi.org/10.1111/j.1398-2265.2004.00186.x
- Burdach S, Jurgens H, Peters C, Nurnberger W, Mauz-Korholz C, Korholz D, Paulussen M, Pape H, Dilloo D, Koscielniak E et al. Myeloablative radiochemotherapy and hematopoietic stem-cell rescue in poor-prognosis Ewing's sarcoma. J Clin Oncol 1993; 11:1482-8; PMID:8101562
- Meyers PA, Krailo MD, Ladanyi M, Chan KW, Sailer SL, Dickman PS, Baker DL, Davis JH, Gerbing RB, Grovas A et al. High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 2001; 19:2812-20; PMID:11387352
- Cotterill SJ, Ahrens S, Paulussen M, Jurgens HF, Voute PA, Gadner H, Craft AW. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 2000; 18:3108-14; PMID:10963639
- Thiel U, Wawer A, Wolf P, Badoglio M, Santucci A, Klingebiel T, Basu O, Borkhardt A, Laws HJ, Kodera Y et al. No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 2011; 22:1614-21; PMID:21245159; http://dx.doi.org/10.1093/annonc/mdq703
- Felix NJ, Allen PM. Specificity of T-cell alloreactivity. Nat Rev Immunol 2007; 7:942-53; PMID:18007679; http://dx.doi.org/10.1038/nri2200
- Burdach S, Kolb HJ. The vigor of defense against non-self: potential superiority of allorestricted T cells in immunotherapy of cancer?. Front Oncol 2013; 3:100; PMID:23653891; http://dx.doi.org/10.3389/fonc.2013.00100
- Thiel U, Wolf P, Wawer A, Blaeschke F, Grunewald TG, von Luttichau IT, Klingebiel T, Bader P, Borkhardt A, Laws HJ et al. Human leukocyte antigen distribution in German Caucasians with advanced Ewing's sarcoma. Klin Padiatr 2012; 224:353-8; PMID:22821288; http://dx.doi.org/10.1055/s-0032-1321730
- Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 2006; 66:8878-86; PMID:16951205; http://dx.doi.org/10.1158/0008-5472.CAN-06-1450
- Fast LD, DiLeone G, Cardarelli G, Li J, Goodrich R. Mirasol PRT treatment of donor white blood cells prevents the development of xenogeneic graft-versus-host disease in Rag2-/-gamma c-/- double knockout mice. Transfusion 2006; 46:1553-60; PMID:16965583; http://dx.doi.org/10.1111/j.1537-2995.2006.00939.x
- Turtle CJ, Riddell SR. Genetically retargeting CD8+ lymphocyte subsets for cancer immunotherapy. Curr Opin Immunol 2011; 23:299-305; PMID:21237630; http://dx.doi.org/10.1016/j.coi.2010.12.012
- Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, Muranski P, Kern SJ, Logun C et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci USA 2009; 106:17469-74; PMID:19805141; http://dx.doi.org/10.1073/pnas.0907448106
- Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, Busch DH. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity 2007; 27:985-97; PMID:18082432; http://dx.doi.org/10.1016/j.immuni.2007.10.012
- Busch DH, Pilip IM, Vijh S, Pamer EG. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity 1998; 8:353-62; PMID:9529152; http://dx.doi.org/10.1016/S1074-7613(00)80540-3