
ABSTRACT
Perforin, a pore-forming toxin released from secretory granules of NK cells and CTLs, is essential for their cytotoxic activity against infected or cancerous target cells. Bi-allelic loss-of-function mutations in the perforin gene are invariably associated with a fatal immunoregulatory disorder, familial haemophagocytic lymphohistiocytosis type 2 (FHL2), in infants. More recently, it has also been recognized that partial loss of perforin function can cause disease in later life, including delayed onset FHL2 and haematological malignancies. Herein, we report a family in which a wide range of systemic inflammatory and neoplastic manifestations have occurred across three generations. We found that disease was linked to two missense perforin gene mutations (encoding A91V, R410W) that cause protein misfolding and partial loss of activity. These cases link the partial loss of perforin function with some solid tumors that are known to be controlled by the immune system, as well as haematological cancers. Our findings also demonstrate that perforin gene mutations can contribute to hereditary cancer predisposition.
Introduction
Perforin (PRF, encoded by the PRF1 gene) is a pore-forming toxinCitation1 stored in the secretory granules of cytotoxic lymphocytes. During an immune response, these “killer” cells form an immune synapse with a virus-infected or cancer target cell, and release PRF and granzyme serine proteases into the synaptic cleft.Citation2 PRF forms membrane pores that are essential for cytotoxic lymphocyte pro-apoptotic serine proteases, granzymes, to enter the target cell cytoplasm, where they trigger apoptosis.Citation3
Bi-allelic mutations in PRF1 that completely abrogate function are classically associated with the pediatric immunoregulatory disorder familial haemophagocytic lymphohistiocytosis type 2 (FHL2),Citation4 which accounts for 30–60% of all FHL cases.Citation5 However, it is possible that FHL2 represents one extreme of a spectrum of diseases caused by PRF deficiency.Citation6 Thus, missense mutations causing partial loss of expression or function of PRF are more often associated with later atypical onset FHL2Citation7 and/or haematological malignancy.Citation8-12 It is unknown whether PRF deficiency can predispose to solid tumors, nor has a possible contribution of PRF1 mutations to familial cancer ever been explored.
Herein, we describe a UK family of Italian origin, in which adult onset FHL2, haematological and various solid tumors have affected three generations. We show that these diseases are consistently associated with PRF1 missense mutations, with defective protein folding causing partial loss of PRF expression. These findings highlight a novel hereditary cancer predisposition syndrome associated with PRF1 mutations.
Methods
DNA sequencing
Oligonucleotide primers used for PRF1 sequencing are available on request.
PRF expression
PBMNCs were surface stained using anti-CD56-PE, CD4+-PerCP, CD8+-APC, then fixed, permeabilized, and stained with cytofix/cytoperm and anti-human PRF-FITC or isotype control (BD Biosciences). 1,00,000 lymphocytes were acquired on FACs Calibur and analyzed by sequential gating using CellQuest Pro (BD Biosciences).
Assessment of R410W function
Primary murine Prf1−/− CD8+ T cells transgenic for the OT1 TCR were transiently transfected with either WT or mutant R410W PRF cDNA, as described previously.Citation11,13 Cells were then used in 4-h 51Cr release assays against OVA257 (SIINFEKL peptide) labeled EL-4 thymoma target cells.Citation11,13 Percentage-specific 51Cr release was estimated as described.Citation11
Study approval
All clinical investigation was conducted according to Declaration of Helsinki principles. All participants gave full informed consent for analysis of PRF expression and PRF1 mutation screening, and for the results to be reported in this study.
Results and discussion
A family pedigree showing members in whom PRF1 was analyzed is shown in . Further details, including PRF1 mutational analysis and NK cell PRF expression are summarised in .
Figure 1. Characterization of PRF1 defects within the family. Family pedigree showing the relationship between family members in which PRF1 analysis was performed. An arrow indicates the index case. Each family member's cancer history is also indicated (H = haematological malignancy; S = solid cancer; DF = disease free; U = unknown cancer history).
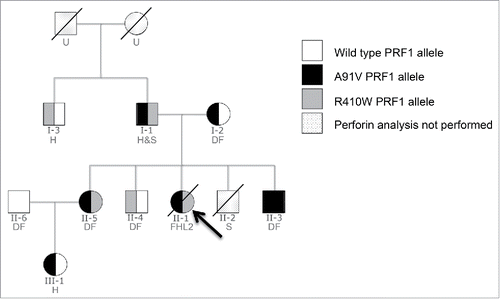
Table 1. Summary of PRF defects and associated disease in family members. Analysis of PRF included PRF1 genotype (assessed by Sanger sequencing) and PRF expression in NK cells (assessed by intracellular FACS). The penetrance of PRF mutations was found to be over 50% with five out of nine carriers affected by disease, including solid tumors.
The index case, a 43 y-old woman (II-1), presented with lethargy, pyrexia, and moderate splenomegaly. She was found to have pancytopenia, hyperferritinaemia, hypertriglyceridaemia, and extensive haemophagocytosis in her bone marrow, leading to a diagnosis of HLH, based on the HLH-2004 diagnostic criteria. Despite extensive investigation, no acquired cause for HLH could be found; rather, the diagnosis of late onset FHL2 was supported by mutations in PRF1 and reduced intracellular PRF expression (). Treatment was according to the HLH-1994 protocol, but the patient developed progressive multi-organ failure and died.
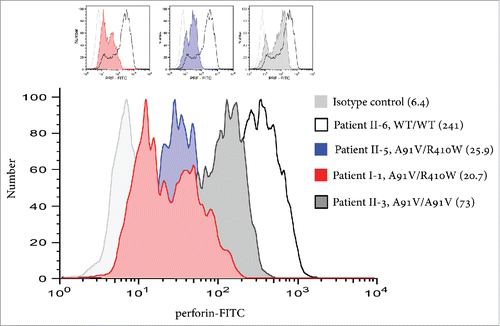
Figure 2. Intracellular PRF expression in selected family members. Intracellular PRF expression in CD56+CD8− NK cells in patients I-1, II-3, II-5, and II-6 (serving as WT perforin control); in parenthesis is the median fluorescence intensity. Of note, patient II-3, who is homozygous for the A91V mutation but has remained healthy, demonstrates reduced, but appreciable PRF levels.
Subsequently, two family members also carrying PRF1 mutations and with reduced intracellular PRF presented with de novo leukemia. The index case's niece (III-1) was diagnosed with acute lymphoblastic leukemia (ALL), and her uncle (I-3) with chronic myelomonocytic leukemia (CMML), which subsequently transformed to acute myeloid leukemia (AML) (). A link between PRF1 mutations and lymphoid malignancies has been reported,Citation8,10,Citation11 but to our knowledge this is the first case of CMML in association with PRF deficiency.
Further investigation of the family's medical history revealed that the father of the index case (I-1), who carried the same mutations as his daughter (II-1), had developed multiple primary malignancies as an adult, including renal cell carcinoma at 54 y of age, but had never been affected by HLH (). No other predisposing environmental or genetic cancer risk had been identified. A brother of the index case (II-2) developed intracranial glioma, which was rapidly fatal despite radiation therapy. Genotyping was not performed, but it could be deduced from his parents' genotype that he would have carried at least one PRF1 mutation, with a 50% probability of two mutations. To our knowledge, this is the first report of an association between human PRF1 mutations and solid tumors, some of which have previously been described to be under the control of the immune system.Citation14,15
The A91V allele identified in this family is by far the commonest hypomorphic PRF1 variant, being found in ∼8% of Caucasians.Citation5 A91V adversely affects PRF folding and this is typically thought to markedly reduce PRF levels and the cytotoxicity of CTL/NK cells.Citation16,17 Although misfolded A91V PRF is detectable by standard methodologies in healthy A91V heterozygotes,Citation17 earlier reports suggested that A91V homozygote patients (or those who co-inherited A91V together with a null mutation) have severely reduced or absent PRF levels.Citation16,18 In analyzing patient II-3, who had bi-allelic A91V mutations but has remained healthy, we demonstrated, for the first time, that A91V homozygosity is indeed compatible with reduced, but still detectable PRF levels (). This discrepancy may be related to PRF levels being assessed at times when a patient is extremely ill.
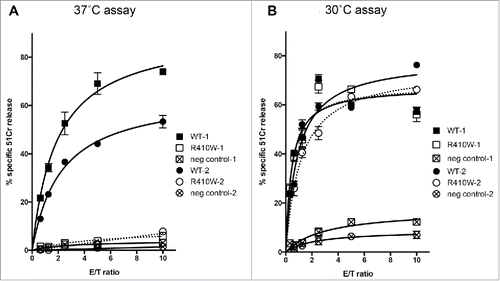
In contrast, the R410W mutation, that also affects this family, has not been previously investigated. We found that R410W causes near-total loss of function, as the activity of Prf1−/− mouse CTLs transfected with the mutant was <5% of the CTLs reconstituted with wild-type PRF (). As with other cancer-associated PRF1 mutations,Citation11 when R410W-transfected T cells were cultured at reduced temperature (30°C), their activity was restored to wild-type level (). The fact that the loss of activity of R410W is temperature-dependent strongly suggests that protein misfolding is responsible for its functional impairment.Citation11
Figure 3. Determining the nature of the R410W mutation. (A) At 37°C, reconstitution of Prf1−/− CTLs with R410W mutant PRF leads to cytolytic activity (assessed by 51Cr release assays) <5% of Prf1−/− CTLs reconstituted with wild-type perforin. (B) At 30°C (a permissive temperature for protein folding),Citation11 Prf1−/− CTLs transfected with R410W mutant PRF have cytolytic activity that is indistinguishable from Prf1−/− CTLs transfected with wild-type PRF. A complete recovery of R410W activity indicates that the only reason for its marginal activity at 37°C is a severe misfolding, and the mutation itself does not affect PRF function.
A91V and R410W both lead to PRF misfolding, which reduces but does not abolish PRF activity. Such in vitro studies on PRF mutants have previously been shown to be strong predictors of their behavior in vivo in carriers of these mutations.Citation11,17 Whereas, the complete loss of PRF function typically presents in early childhood as FHL2, the subtotal loss of PRF activity causes systemic inflammatory disease that is delayed beyond infancy (as in the index case II-1) or may present as a different immunopathology.Citation6 In these instances, consistent with Burnet's hypothesis of tumor immune surveillance,Citation19 reduced cytotoxic lymphocyte activity caused by partial loss of PRF may be expected to predispose to neoplasia, and this is indeed supported by previous studies.Citation9-12 It is not clear, however, why these malignancies thus far reported in humans in association with PRF deficiency are predominantly haematological. It is possible that PRF defects lead to a reactive proliferation of the lymphoid and histiocytic pools, as typically observed in FHL, causing genomic instability and a predisposition to neoplasia in one of these lineages. Alternatively, haematological malignancies may be more susceptible to immune control than solid tumors and may thus arise more frequently in the absence of a fully functional immune system. This report supports the strong link between partial PRF deficiency and haematological malignancy. Critically, it also shows for the first time that multiple solid tumors, some known to be controlled by immune system,Citation14,15 can occur in humans in this context, thus validating earlier investigations in murine models, where many types of solid tumor, including prostate, mammary, and lung carcinoma, were controlled by PRF-dependent cytotoxicity.Citation20,21 Overall, in the current study, the penetrance of PRF1 mutations was over 50% with five out of nine carriers affected by disease (). Excluding patient II-2 (where genetic analysis was not performed), a similar penetrance was observed in both carriers of mono-allelic PRF1 mutations (two out of four carriers affected) and family members with bi-allelic mutations (two out of four carriers affected).
Of particular note, the cases discussed in this report involve many family members and several generations. This is highly suggestive of a hereditary cancer predisposition syndrome,Citation22 and we believe this is the first association of PRF1 mutations with such a disorder. Such findings align closely with a recent report highlighting increased incidence of cancer in relatives of patients with FHL.Citation23
Although hereditary cancer syndromes are typically autosomal dominant with incomplete penetrance, the pattern of inheritance is less clear in this family, with both mono-allelic and bi-allelic carriers of PRF1 mutations developing disease. One explanation for this variable pattern in inheritance is that PRF mutants such as A91V have been previously shown to exhibit dominant negative activity,Citation17,24 and thus may cause pathology when only present on one allele. Furthermore, although PRF1 mutations may be acting alone, it is possible that another undetected mutation may be present in this family. For example, it has recently been shown that patients with FHL may co-inherit mutations in two different genes-regulating lymphocyte cytotoxicity, although this is a rare event leading to FHL mostly at a younger age.Citation25 Besides, in the context of the family described here, co-inheritance of three pathological mutations would be required, with one leading to defective perforin secretion (UNC13D or STX11 or STXBP2). While this is theoretically possible, the probability of such an event is exceedingly small. Alternatively, PRF deficiency may be acting in concert with classical cancer predisposition genes unrelated to cytotoxic lymphocyte function. The latter possibility potentially opens up a new paradigm, which we believe warrants further investigation: that deficiency of an extrinsic tumor suppressor such as PRF may co-operate with an intrinsic genetic defect to predispose to familial cancer syndromes.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
M.S.C. researched the study, collected and analyzed the data, and wrote the manuscript; K.G. performed experiments and contributed to writing of the manuscript; I.H. performed experiments; M.L. supervised research and contributed to writing of the manuscript; N.P. collected clinical data and contributed to writing of the manuscript; M.S. collected clinical data; J.T. supervised research and contributed to writing of the manuscript; I.V. supervised research and contributed to writing of the manuscript.
References
- Law RHP, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, D'Angelo ME, Orlova EV, Coulibaly F, Verschoor S et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 2010; 468:447-U277; PMID:21037563; http://dx.doi.org/10.1038/nature09518
- de Saint-Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol 2010; 10:568-79; PMID:20634814; http://dx.doi.org/10.1038/nri2803
- Lopez JA, Susanto O, Jenkins MR, Lukoyanova N, Sutton VR, Law RHP, Johnston A, Bird CH, Bird PI, Whisstock JC et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013; 121:2659-68; PMID:23377437; http://dx.doi.org/10.1182/blood-2012-07-446146
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999; 286:1957-9; PMID:10583959; http://dx.doi.org/10.1126/science.286.5446.1957
- Molleran Lee S, Villanueva J, Sumegi J, Zhang K, Kogawa K, Davis J, Filipovich AH. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet 2004; 41:137-44; PMID:14757862; http://dx.doi.org/10.1136/jmg.2003.013003
- Voskoboinik I, Trapani JA. Perforinopathy: a spectrum of human immune disease caused by defective perforin delivery or function. Front Immunol 2013; 4:441-1; PMID:24376445; http://dx.doi.org/10.3389/fimmu.2013.00441
- Nagafuji K, Nonami A, Kumano T, Kikushige Y, Yoshimoto G, Takenaka K, Shimoda K, Ohga S, Yasukawa M, Horiuchi H et al. Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica 2007; 92:978-81; PMID:17606450; http://dx.doi.org/10.3324/haematol.11233
- Clementi R, Locatelli F, Dupré L, Garaventa A, Emmi L, Bregni M, Cefalo G, Moretta A, Danesino C, Comis M et al. A proportion of patients with lymphoma may harbor mutations of the perforin gene. Blood 2005; 105:4424-8; PMID:15728124; http://dx.doi.org/10.1182/blood-2004-04-1477
- Muralitharan S, Wali Y, Pathare AV. Perforin A91V polymorphism and putative susceptibility to hematological malignancies. Leukemia 2006; 20:2178; PMID:17039227; http://dx.doi.org/10.1038/sj.leu.2404433
- Cannella S, Santoro A, Bruno G, Pillon M, Mussolin L, Mangili G, Rosolen A, Aric∫ M. Germline mutations of the perforin gene are a frequent occurrence in childhood anaplastic large cell lymphoma. Cancer 2007; 109:2566-71; PMID:17477373; http://dx.doi.org/10.1002/cncr.22718
- Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci USA 2009; 106:9809-14; PMID:19487666; http://dx.doi.org/10.1073/pnas.0903815106
- Trapani JA, Thia KYT, Andrews M, Davis ID, Gedye C, Parente P, Svobodova S, Chia J, Browne K, Campbell IG et al. Human perforin mutations and susceptibility to multiple primary cancers. Oncoimmunology 2013; 2:e24185; PMID:23734337; http://dx.doi.org/10.4161/onci.24185
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. J Immunol 2012; 188:2046-56; PMID:22345701; http://dx.oi.org/10.1016/0092-8674(94)90169-4
- Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med 2005; 353:2477-90; PMID:16339096; http://dx.doi.org/10.1056/NEJMra043172
- Vauléon E, Tony A, Hamlat A, Etcheverry A, Chiforeanu DC, Menei P, Mosser J, Quillien V, Aubry M. Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med Genomics 2012; 5:41; PMID:22980038; http://dx.doi.org/10.1186/1755-8794-5-41
- Trambas C, Gallo F, Pende D, Marcenaro S, Moretta L, De Fusco C, Santoro A, Notarangelo L, Aric∫ M, Griffiths GM. A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin. Blood 2005; 106:932-7; PMID:15741215; http://dx.doi.org/10.1182/blood-2004-09-3713
- House IG, Thia K, Brennan AJ, Tothill R, Dobrovic A, Yeh WZ, Saffery R, Chatterton Z, Trapani JA, Voskoboinik I. Heterozygosity for the common perforin mutation, p.A91V, impairs the cytotoxicity of primary natural killer cells from healthy individuals. Immunol Cell Biol 2015; 93:575-80; PMID:25776844; http://dx.doi.org/10.1038/icb.2015.1
- Mancebo E, Allende LM, Guzmán M, Paz-Artal E, Gil J, Urrea-Moreno R, Fernández-Cruz E, Gayà A, Calvo J, Arbós A et al. Familial hemophagocytic lymphohistiocytosis in an adult patient homozygous for A91V in the perforin gene, with tuberculosis infection. Haematologica 2006; 91:1257-60; PMID:16956828
- Burnet FM. Immunological Surveillance in Neoplasia. Immunol Rev 1971; 7:3-25; PMID:5146537; http://dx.doi.org/10.1111/j.1600-065X.1971.tb00461.x
- Smyth MJ, Thia KY, Cretney E, Kelly JM, Snook MB, Forbes CA, Scalzo AA. Perforin is a major contributor to NK cell control of tumor metastasis. J Immunol 1999; 162:6658-62; PMID:10352283
- Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 2001; 97:192-7; PMID:11133760; http://dx.doi.org/10.1182/blood.V97.1.192
- Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol 2005; 23:276-92; PMID:15637391; http://dx.doi.org/10.1200/JCO.2005.10.042
- Löfstedt A, Chiang SCC, Onelöv E, Bryceson YT, Meeths M, Henter J-I. Cancer risk in relatives of patients with a primary disorder of lymphocyte cytotoxicity: a retrospective cohort study. Lancet Haematol 2015; 2:e536-42; PMID:26686408; http://dx.doi.org/10.1016/S2352-3026(15)00223-9
- Voskoboinik I, Sutton VR, Ciccone A, House CM, Chia J, Darcy PK, Yagita H, Trapani JA. Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood 2007; 110:1184-90; PMID:17475905; http://dx.doi.org/10.1182/blood-2007-02-072850
- Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, Johnson JA, Filipovich AH. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood 2014; 124:1331-4; PMID:24916509; http://dx.doi.org/10.1182/blood-2014-05-573105