
ABSTRACT
IL-10 has been classically defined as a broad-spectrum immunosuppressant and is thought to facilitate the development of regulatory CD4+ T cells. IL-10 is believed to represent one of the major suppressive factors secreted by IDO+FoxP3+CD4+ Tregs. Contrary to this view, we have previously reported that PEGylated recombinant IL-10 (PEG-rIL-10) treatment of mice induces potent IFNγ and CD8+ T-cell-dependent antitumor immunity. This hypothesis is currently being tested in clinical trials and we have reported that treatment of cancer patients with PEG-rHuIL-10 results in inhibition and regression of tumor growth as well as increased serum IFNγ. We have continued to assess PEG-rIL-10's pleiotropic effects and report that treatment of tumor-bearing mice and humans with PEG-rIL-10 increases intratumoral indoleamine 2, 3-dioxygenase (IDO) in an IFNγ-dependent manner. This should result in an increase in Tregs, but paradoxically our data illustrate that PEG-rIL-10 treatment of mice reduces intratumoral FoxP3+CD4+ T cells in an IDO-independent manner. Additional investigation indicates that PEG-rIL-10 inhibits TGFβ/IL-2-dependent in vitro polarization of FoxP3+CD4+ Tregs and potentiates IFNγ+T-bet+CD4+ T cells. These data suggest that rather than acting as an immunosuppressant, PEG-rIL-10 may counteract the FoxP3+CD4+ Treg suppressive milieu in tumor-bearing mice and humans, thereby further facilitating PEG-rIL-10's potent antitumor immunity.
Introduction
A conundrum exists concerning Interleukin-10's (IL-10) pleiotropic regulation of the immune system. Discovered in 1991, it was initially reported to suppress cytokine secretion, antigen presentation and CD4+ T cell activation.Citation1-4 Further investigation showed that IL-10 predominantly inhibits lipopolysaccharide (LPS) and bacterial product-mediated induction of the pro-inflammatory cytokines TNFα, IL-1β,Citation5 IL-12Citation6 and IFNγ.Citation7 Conversely, IL-10 was also shown to be immunostimulatory in the context of tumor immunotherapy and to exert immune-mediated inhibition of tumor metastasis.Citation8 In these reports, expression of IL-10 from transfected tumor cells,Citation9,10 expression of IL-10 in transgenic mice,Citation11 or of mice dosed with IL-10,Citation12,13 the exposure of mice to IL-10 lead to the control of primary tumors and decreased metastatic burden, whereas expression of viral IL-10 from immunogenic tumors reduced their immunogenicity.Citation14 More recently, PEGylated recombinant mouse IL-10 (PEG-rMuIL-10) has been shown to induce IFNγ and CD8+ T-cell-dependent antitumor immunity.Citation15,16 These data collectively demonstrate both IL-10s immunoinhibitory and immunostimulatory effects. These seemingly opposite effects were further shown in (Chan et al.) where the authors report that exposure of peripheral blood mononuclear cells (PBMCs) to PEG-rHuIL-10 results in the concomitant inhibition of IFNγ expression in monocyte/macrophages, but the priming of CD8+ T cells, (to secrete IFNγ upon T cell receptor (TCR) ligation) within the same culture.
Based on these data, we are currently assessing the therapeutic potential of PEGylated recombinant human IL-10 (AM0010) in a Phase I immune oncology trial.Citation17 In this setting of late stage, multi-indication, heavily pretreated patients, we have thus far observed 45% stable disease for at least 2 mo and 18% objective response in both renal cell carcinoma and melanoma. One colorectal patient and one melanoma patient have continued treatment for 18+ and 8+ weeks, respectively. We have observed dose-titratable increase of immunostimulatory serum cytokines, (IL-4, IL-7, IL-18, GM-CSF and IFNγ), as well as fold increases of PD1+ and LAG3+ peripheral CD8+ T cells and a decrease of serum TGFβ17. Consistent with previously published preclinical reports, therapeutic doses of AM0010 are potently immunostimulatory. Taken together, however, these data suggest AM0010 regulates the peripheral immune system more broadly than previously considered.
Recently, significant interest has been generated by inhibitors to indoleamine 2, 3-dioxygenase (IDO). IDO is a heme-containing, monomeric oxidoreductase that is ubiquitously distributed in the cytosol of mammalian tissues and cells. This enzyme converts tryptophan to N-formylkynurenine that is further catabolized to kynurenine and its terminal metabolites picolinic or quinolinic acid.Citation18,19 IDO expression is induced by influenza infection, LPS and cytokines.Citation20-22 IDO-derived tryptophan catabolites have been shown to directly inhibit T and natural killer cell proliferation in vitro.Citation23 Due to IDO's broadly immunosuppressive function, its role in tumor immunity has been investigated and IDO inhibitors have shown therapeutic antitumor function, predominantly through reducing FoxP3+ CD4+ T regulatory cells.Citation24-26
We have previously reportedCitation16 that PEG-rMuIL-10's antitumor function is CD8+ T cell and IFNγ dependent. Furthermore, treatment of CD8+ T cells with PEG-rIL-10 significantly increases TCR ligation-mediated IFNγ secretion.Citation27 Since IFNγ directly induces IDO expression,Citation28-30 we have investigated PEG-rIL-10's potential IFNγ-dependent regulation of IDO.
Results
PEG-rMuIL-10 treatment induces intratumoral IDO expression
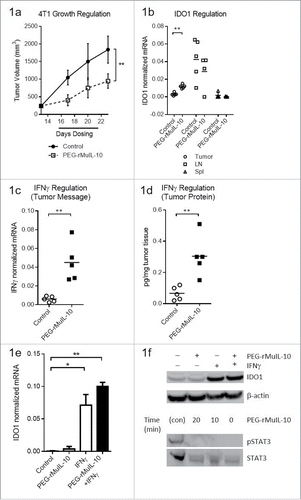
To determine whether PEG-IL-10 regulates the expression of intratumoral IDO expression and function in vivo, we dosed 4T1 tumor-bearing mice daily with the non-optimal 1 mg/kg dose of PEG-rMuIL-10, the murine surrogate for AM0010 () in order to subsequently combine PEG-rMuIL-10 with an IDO inhibitor and potentially observe combinatorial or synergistic antitumor effects. We began dosing after 14 days of tumor growth in order to have sufficient tissue for TIL isolation. We assessed IDO expression levels in the tumor, spleen and tumor-draining lymph nodes (). Consistent with the intratumoral induction of IFNγ (), therapeutic levels of PEG-rMuIL-10 increased IDO1 expression within the tumor, but not in the spleen or lymph nodes (). We determined that IFNγ and not PEG-rMuIL-10 induces IDO1 expression in 4T1 tumor cells () and that PEG-rMuIL-10 does not induce STAT3 phosphorylation in tumor cells (). These data suggest PEG-rMuIL-10 regulation of IDO1 is IFNγ dependent.
Figure 1. Treatment with PEG-rMuIL-10-induced IFNγ-dependent intratumoral IDO expression. For 1(A)–1(D), the tissue was harvested at day 24, 24 h after the last dose of PEG-rMuIL-10. (A) 4T1 tumor-bearing mice were treated for 9 d with 1 mg/kg PEG-rMuIL-10 s.c. daily and tumor growth inhibition is shown vs. control-treated mice. Closed circles represent control-treated mice, open squares represent PEG-rMuIL-10-treated mice. (B) IDO mRNA expression analysis from tumor, spleen and lymph node of mice described in (A). Open circles represent tumor tissue, open squares represent lymph node (LN), open triangles represent spleen tissue (Spl). (C) Intratumoral mRNA levels of IFNγ of mice in (A). Open circles represent control-treated mice, closed squares represent PEG-rMuIL-10-treated mice. (D) Intratumoral IFNγ protein levels of mice in (A). Open circles represent control-treated mice, closed squares represent PEG-rMuIL-10-treated mice. (E) IDO mRNA levels of in vitro cultured 4T1 tumor cells treated with PEG-rMuIL-10 alone or in combination with IFNγ. (F) Intracellular IDO1 and STAT3/pSTAT3 protein levels from AM0010 and IFNγ-treated 4T1 cells as detected by protein gel blot. IDO1 was detected after 48 h incubation with 50 ng/mL IFNγ and/or 100 ng/mL PEG-rMuIL-10. pSTAT3 and STAT3 were assessed in a time course from 0 to 20 min incubation with 100 ng/mL PEG-rMuIL-10. The protein control for pSTAT3 and STAT3 (con) was a STAT3 control cell extract purchased from Cell Signaling Technology. Statistics were determined by Students t-test where p < 0.05 is denoted by * and p < 0.01 is denoted by **.
PEG-rIL-10 treatment changes the serum/plasma kynurenine: tryptophan ratio but does not exhibit combinatorial antitumor effects with an IDO inhibitor
To determine if the observed intratumoral changes to IDO expression could be monitored in the plasma, we assessed the kynurenine:tryptophan (kyn:trp) plasma ratio in PEG-rMuIL-10-treated mice (). These data suggest that the induction of intratumoral IFNγ correlates with changes in the plasma kyn:trp ratio. In 42 of our AM0010-treated Phase I patients, we observed similar kyn:trp ratio changes (). The most significant difference occurred at the therapeutic target dose of 20 μg/kg/d (). Kynurenine is toxic to CD8+ T cellsCitation23 and we determined that it inhibited AM0010 activation of CD8+ T cells in vitro at concentrations between 250 μM and 1000 µM (), with the later resulting in greater than 95% cell death (data not shown).
Figure 2. Treatment with an IDO inhibitor and PEG-rMuIL-10 did not exhibit combined antitumor efficacy. (A) Treatment of 4T1 tumor-bearing mice with 1mg/kg PEG-rMuIL-10 s.c. daily alters the plasma kynurenine to tryptophan ratio. Open circles represent control-treated mice, open squares represent PEG-rMuIL-10-treated mice. (B) Treatment of 42 cancer patients with AM0010 increases the serum kynurenine to tryptophan ratio. Open circles represent patients prior to treatment (Pre). Open squares represent patients after treatment (Post). (C) The cancer patient 20 μg/kg dose group serum kynurenine to tryptophan ratio. Open circles represent patients prior to treatment (Pre). Open squares represent patients after treatment (Post). (D) In vitro cell culture media IFNγ levels of activated CD8+ T cells exposed to 100 ng/mL AM0010 and 62.5, 250 or 1000 μM kynurenine for 3 d and triggered for 4 h with anti-CD3. (E) In vitro cell culture media Granzyme B levels of activated CD8+ T cells exposed to 100 ng/mL AM0010 and 62.5, 250 or 1000 μM kynurenine for 3 d and triggered for 4 h with anti-CD3. Data in (D) and (E) are representative of three donor responses. (F) Antitumor efficacy of PEG-rMuIL-10 with or without IDO inhibitor (n = 5 mice/cohort). Closed circles represent control-treated mice, open squares represent PEG-rMuIL-10-treated mice, open triangles represent IDO-inhibitor-treated mice and upside down open triangles represent PEG-rMuIL-10 and IDO inhibitor combined treated mice. Mice were treated with 1 mg/kg PEG-rMuIL-10 s.c. daily, control, IDO inhibitor (1-methyl-D-tryptophan) or the combination of the same dose of PEG-rMuIL-10 with the IDO inhibitor for 28 d. Data are representative of four total in vivo experiments. (G) Intratumoral IFNγ protein levels from two–four 4T1 tumor-bearing mice treated in (2F). (H) Intratumoral Granzyme B of the mice described in (2F). Statistics for were determined by Student's t-test where p < 0.05, p < 0.01 and p < 0.001 is denoted by *, **, ***, respectively. Statistics for was determine by use of ANOVA multiple comparisons where p < 0.05 is denoted by *.
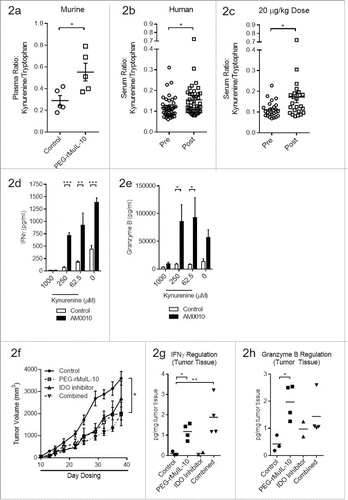
The serum concentration of kynurenine in our patients was between 0.86 μM and 5.1 μM with a pre-dose average concentration of 1.85 μM and post dose average of 2.03 μM. While these concentrations are well below the 250 μM concentration toxic to CD8+ T cells in vitro, it is possible that the intratumoral kynurenine concentration is increased with PEG-rIL-10 treatment and that IDO inhibition concurrent with PEG-rIL-10 treatment may exhibit additive or synergistic antitumor function. To investigate this, we treated 4T1 tumor-bearing mice with PEG-rMuIL-10 and the IDO inhibitor, 1-methyl-D-tryptophan, formulated as a time release lipid encapsulated pellet. Unexpectedly, this combination did not exhibit enhanced antitumor function (). Consistent with PEG-IL-10's antitumor function, the combination also did not result in an increased induction of intratumoral IFNγ () or Granzyme B () vs. PEG-rMuIL-10 alone.
PEG-rIL-10 treatment reduces FoxP3+ CD4+ T cells
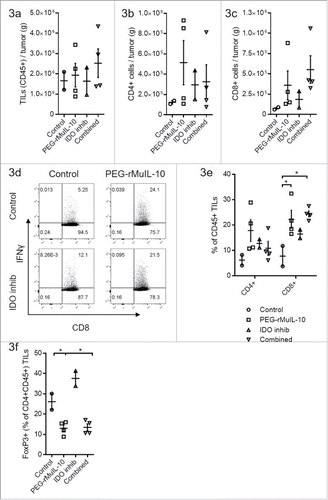
We have previously reported that treatment with PEG-rMuIL-10 increases the number of CD8+ T cells within the tumor as well as their cytotoxic function.Citation16 Given this T cell biology, and IDO's inhibitory effects on CD8+ T cells, we fully expected the two compounds to exert at least additive antitumor function. Therefore, to further explore the lack of combinatorial antitumor effects with PEG-rMuIL-10 and IDO inhibition, we investigated the tumor-infiltrating lymphocytes (TILs) phenotype. Neither treatment alone or in combination significantly altered the number of lymphocytes infiltrating the tumor (), CD4+ T cells infiltrating the tumor () or CD8+ T cells infiltrating the tumor (). There is a trend toward more CD8+ T cells in that is consistent with both the non-optimal dosing strategy and previous reports.Citation16 When administered individually or in combination, both treatments increased the percentage of IFNγ+ CD8+ TILs (). The percentage of intratumoral CD45+ CD8+ T cells was increased with PEG-rMuIL-10 treatment alone or in combination (). Unexpectedly, PEG-rMuIL-10 treatment decreased the percentage of intratumoral FoxP3+ CD4+ T cells alone or in combination with the IDO inhibitor ().
Figure 3. PEG-rMuIL-10 and AM0010 treatment decrease FoxP3+ CD4+ T cells. (A) Flow cytometric analysis of CD45+ tumor-infiltrating lymphocytes (TILs) from 4T1 tumors treated for 28 d with control, 1 mg/kg PEG-rMuIL-10 s.c. daily, IDO inhibitor (1-methyl-D-tryptophan) or the combination of the same dose of PEG-rMuIL-10 with the IDO inhibitor (n = 5 mice/cohort). Total TILs (CD45+) per gram tumor were isolated from two–four tumors per group. (B) Total CD4+ TILs from (A) per gram tumor. (C) Total CD8+ TILs from (A) per gram tumor. (D) Flow cytometric analysis of intracellular IFNγ in CD8+ TILs from (A). (E) Percentage of CD4+ or CD8+ cells within the CD45+ TIL population from (A). (F) Percentage of FoxP3+ cells within the CD4+ CD45+ TIL population from (A). Statistics for Fig. 3(E)–(F) was determine by use of ANOVA multiple comparisons, where p < 0.05 is denoted by *. Data is representative of three in vivo studies.
IDO inhibition regulates FoxP3+ CD4+ T cells independently of AM0010
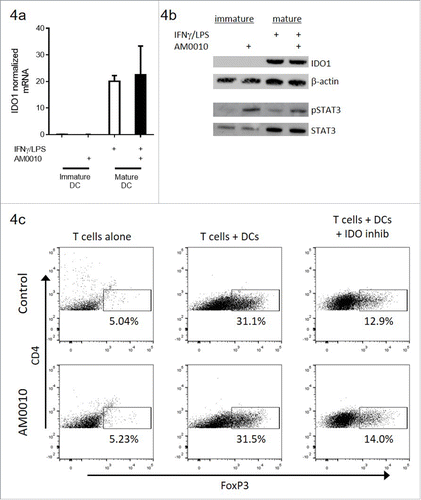
Given the in vivo results, we assessed whether IDO regulation of FoxP3+ CD4+ T cells was augmented or inhibited by AM0010. AM0010 did not directly alter IDO mRNA () or protein () levels in immature or mature dendritic cells. We then used mature IDO-expressing dendritic cells to induce TregsCitation31 () and consistent with previous reports, the addition of an IDO inhibitor to this system reduced FoxP3+CD4+ T cells (compare middle upper panel with right upper panel). AM0010 did not reduce FoxP3+ CD4+ Tregs in this system (middle lower panel). Similarly, IDO inhibition of FoxP3+ CD4+ Tregs was not altered by the presence of AM0010 (right panels). AM0010's lack of effect in IDO-mediated polarization of Tregs was consistent across five out of five donors and IDO-mediated induction of Tregs was consistent in four out of five donors.
Figure 4. AM0010 exerts no effect on IDO-mediated induction of FoxP3+ CD4+ T cells in vitro. (A) IDO1 mRNA message regulation in immature or IFNγ/LPS-matured dendritic cells treated with or without 100 ng/mL AM0010 for 2 d. (B) Protein gel blot for IDO1 and STAT3/phospho-STAT3 in cells from (A). (C) Induction of FoxP3+ CD4+ T cells as per referenceCitation29 with mature dendritic cells. Human CD4+ CD25− T cells were cocultured with mature DCs for 6 d with 10 ng/mL IL-2, 100 ng/mL IFNγ and 5 µg/mL LPS with or without 100 ng/mL AM0010 and with or without 2 µM IDO inhibitor, INCB024360. T cells were then collected and analyzed by flow cytometry. Percentage indicates FoxP3+ T cells. Data is representative of four out of five donor responses.
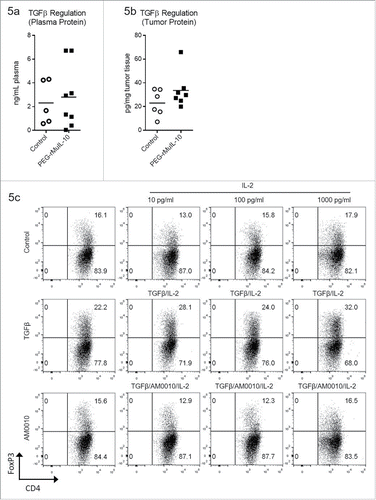
AM0010 inhibits TGFβ/IL-2 induced Treg polarization
Since AM0010 did not exert any effect in the IDO-dependent induction of FoxP3+ CD4+ Tregs, we investigated alternative means of inducing Tregs. TGFβ is an immunoregulatory cytokine with pleiotropic biology. In context of T helper CD4+ cells, TGFβ induces FoxP3+ CD4+ TregsCitation32-34 and IL-2 stabilizes FoxP3+ expression.Citation35 IL-10 has been show to regulate some TGFβ−mediated effects,Citation36,37 and we have also reported that cancer patients treated with AM0010 exhibit decreased serum TGFβ levels.Citation17 We therefore investigated PEG-rMuIL-10's regulation of TGFβ in vivo. Unlike treatment of cancer patients with AM0010,Citation17 PEG-rMuIL-10 treatment did not decrease plasma TGFβ levels in mice (). Similarly, PEG-rMuIL-10 treatment did not alter intratumoral TGFβ expression (not shown) or protein levels (). We therefore investigated AM0010's effect on TGFβ/IL-2 polarization of Tregs in vitro. Concomitant exposure of CD4+ T cells to AM0010, TGFβ and IL-2 inhibits TGFβ/IL-2-mediated induction of CD4+ Tregs (). AM0010's inhibition of TGFβ/IL-2-mediated polarization of Tregs was consistent across six out of nine donors.
Figure 5. AM0010 treatment inhibits TGFβ/IL-2 induction of FoxP3+ CD4+ Tregs. (A) Quantitation of TGFβ plasma protein from tumor-bearing mice treated s.c. daily with 1 mg/kg PEG-rMuIL-10 or control for 10–28 d. Open circles represent control-treated mice, filled squares represent PEG-rMuIL-10-treated mice. (n = 5–8 mice/cohort) (B) Quantitation of intratumoral TGFβ protein from 4T1 tumor-bearing mice treated as in (A). Open circles represent control-treated mice, filled squares represent PEG-rMuIL-10-treated mice. (C) Assessment of human CD4+ T cells treated with 5 ng/mL TGFβ and 1000, 100 and 10 ng/mL IL-2 with or without AM0010 in vitro for 5 d. Data is representative of six of nine donor responses.
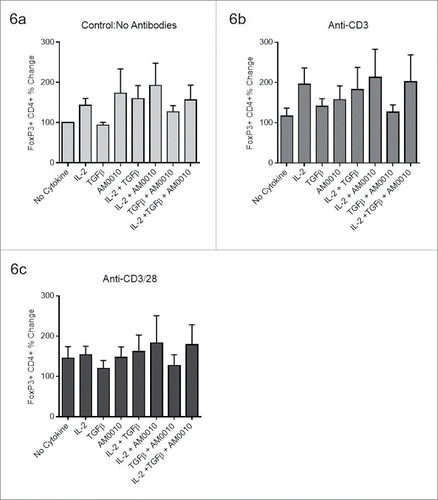
AM0010 does not suppress pre-polarized Tregs
Using the same system to induce FoxP3 CD4+ Tregs, we investigated AM0010's effects on TGFβ/IL-2 pre-polarized Tregs. Tregs were first induced with TGFβ/IL-2, and then treated as indicated. Data in are normalized to each donor's no cytokine/no antibody control presented in (No Cytokine). The data for each plot represents the median and variation in response for each donor under the same stimulatory conditions. It is important to note that none of the changes reflected in are statistically significant, suggesting these changes are within normal variance of donor response and the assay. However, contrary to AM0010's inhibition of Treg polarization, AM0010 exposure in the absence of TCR engagement, slightly increased Treg numbers of pre-polarized FoxP3+ CD4+ Tregs as did IL-2 (). Interestingly, the stimulatory effects of both AM0010 and IL-2 appear to be contextual. Providing the same cytokine stimuli in context of anti-CD3, to mimic what the T cell might encounter within the tumor microenvironment, revealed that IL-2 maintains its stimulatory effect, but AM0010 did not (). Exposure of the same pre-polarized population to anti-CD3/anti-CD28, to mimic engagement with antigen-presenting cells, and AM0010 results in no alteration to Treg numbers (). These variable results were reproducible across six donors.
Figure 6. AM0010 does not suppress pre-polarized FoxP3+ CD4+ Tregs. The data for each plot represents the median and variation in response for each donor under the same stimulatory conditions. (A) Human CD4+ peripheral T cells were polarized for 5–7 d (with TGFβ, IL-2, anti-CD3, anti-CD28), then exposed to the stated combination of cytokines for 2–3 d and then analyzed by flow cytometry for CD4+ and FoxP3+ expression. (B) Pre-polarized cells from (A) were exposed to the identical conditions except with the addition of 2 µg/mL immobilized anti-CD3 for 2–3 d. Cells were analyzed as in (A). (C) Pre-polarized cells from (A) were exposed to the identical conditions except with the addition of both 2 µg/mL immobilized anti-CD3 and 1 µg/mL soluble anti-CD28 for 2–3 d. Cells were analyzed as in (A). Data is representative of six out of 6 donor responses. One-way ANOVA statistical analysis was performed where each treatment was compared to the No Cytokine control values for each plot. None of the differences were determined to be statistically different. In addition, each treatment group was analyzed as an independent Students t-test where the treatment group was compared to the control. In this analysis, no treatments were determined to be statistically significant.
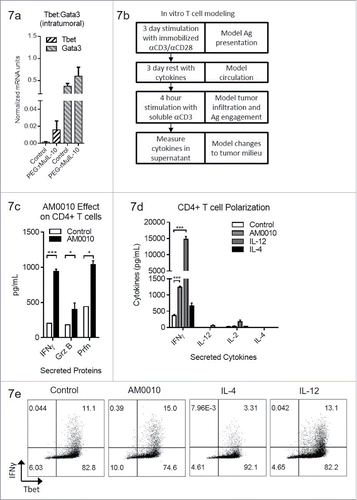
AM0010 potentiates IFNγ+, Th1-like CD4+ T cells
We have previously shown that exposure of activated CD8+ T cells to AM0010 potentiates the expression of IFNγ, Granzyme B and Perforin upon subsequent TCR engagement.Citation27 These data explain the underlying IFNγ and CD8+ T-cell-dependent antitumor mechanism of action of PEG-rMuIL-10/AM0010.Citation15,16 In our previous investigation, we discovered that intratumoral MHC I upregulation precedes intratumoral MHC II upregulation. Consistent with these data, the PEG-rMuIL-10 antitumor response requires CD8+ but not CD4+ T cells. However, given the observation that treatment of tumor-bearing mice and human CD4+ T cells with PEG-rMuIL-10/AM0010 decreases FoxP3+CD4+ T cells, we further investigated AM0010's effect on CD4+ T cell polarization in vivo. Expression analysis of tumors from mice treated with PEG-rMuIL-10 reveal that Tbet is moderately increased (). We previously developed an in vitro model to recapitulate AM0010's in vivo biological effect on CD8+ T cells () and now subjected CD4+ T cells to this same model. The cells are first activated by immobilized anti-CD3/anti-CD28 to mimic antigen presentation. The cells are then removed from stimulus and exposed to cytokines for 3 days to model exposure to AM0010 or other cytokines during systemic circulation. After this incubation period, the cells are re-stimulated with low concentration, soluble anti-CD3 for 4 hours to model a T cells re-entry into the tumor microenvironment and engagement of MHC I+ antigen. Surprisingly, in vitro modeling of CD4+ T cell activation and stimulation with AM0010 potentiates IFNγ, Granzyme B and Perforin secretion (). The potentiation of IFNγ, Granzyme B and Perforin are very similar to AM0010's effect on CD8+ T cells when treated in vitro in a similar manner.Citation27 However, AM0010 treatment of CD4+ T cells does not elicit classically defined Th1 or Th2 cells. While AM0010 treatment induces IFNγ, the prototypic Th1 cytokine, the magnitude is substantially less than the levels induced by IL-12, the classic mediator of Th1 development (). In addition, AM0010 does not induce the secretion of IL-12, IL-2 or IL-4, the prototypic Th2 cytokine (). AM0010 treatment of CD4+ T cells also promotes the generation of Tbet+ IFNγ–expressing CD4+ T cells (). These data suggest AM0010 promotes Th1-like CD4+ T cell polarization, but as expected, not as efficiently as IL-12. Therefore, we conclude that activated CD4+ T cells when rested in the presence of AM0010, maintain a heightened expression level of IFNγ, Perforin and Granzyme B.
Figure 7. AM0010 treatment potentiates IFNγ+ Th1 like CD4+ T cells. (A) Quantitation of Tbet and Gata3 mRNA from tumors of mice treated s.c. daily with 1 mg/kg PEG-rMuIL-10 or control for 10–28 d. (n = 5 mice/cohort). (B) Schematic depicting in vitro experimental setup for data shown in (C–E). (C) Quantitation of secreted IFNγ, Granzyme B and Perforin from human CD4+ T cells activated for 3 d with immobilized anti-CD3/anti-CD28, then exposed for 3 d to control or 100 ng/mL AM0010. Cells were washed and then stimulated with 1 μg/mL soluble anti-CD3 for 4 h at 37°C and the proteins in the supernatant were quantified by ELISA. Data is representative of three out of three donor responses. (D) Quantitation of secreted cytokines from activated CD4+ T cells treated as in (C) with 20 ng/mL IL-12, 10 ng/mL IL-4 or 100 ng/mL AM0010. (E) Intracellular flow cytometric analysis from cells as in (D) treated with 20 ng/mL IL-12, 10 ng/mL IL-4 or 100 ng/mL AM0010. Data is representative of three out of three donor responses. Statistics in (C) were determined by Student's t-test where p < 0.05, p < 0.01 and p < 0.001 is denoted by *, **, ***, respectively. Statistics in (D) were determined by one-way ANOVA where p < 0.05, p < 0.01 and p < 0.001 is denoted by *, **, ***, respectively.
Discussion
We have previously observed that treatment of tumor-bearing mice with PEG-rMuIL-10 leads to inhibition and even cure of tumors in an IFNγ and CD8+ T-cell-dependent manner. We have also observed the induction of IFNγ in the serum of AM0010-treated cancer patients.Citation17 Since IFNγ is known to upregulate IDO expression, we investigated and found that treatment of tumor-bearing mice with PEG-rMuIL-10 induced IFNγ-dependent intratumoral IDO expression, leading to changes in the plasma kynurenine/tryptophan ratio. Similar changes to the kynurenine/tryptophan ratio were also detected in the serum of AM0010-treated cancer patients. These data suggest that treatment of cancer patients with AM0010 induces intratumoral IFNγ leading to increased intratumoral IDO expression. Surprisingly, however, the combination of PEG-rMuIL-10 and the IDO inhibitor, 1-methyl-D-tryptophan, did not exert combinatorial antitumor function in mice. Furthermore, in contrast to expectations, treatment with PEG-rMuIL-10 leads to a decrease in intratumoral FoxP3 Tregs. In vitro analysis of these phenomena revealed that AM0010 inhibits the TGFβ/IL-2 driven polarization of FoxP3+ CD4+ Tregs, but had no effect on IDO-mediated induction of Tregs. However, treatment of TGFβ/IL-2 pre-polarized Tregs with AM0010 did not dramatically alter the percentage of FoxP3+ Tregs. In fact, AM0010 moderately, but not significantly, increased Treg numbers under some conditions. Our interpretation of these data is that AM0010 does not directly impact TGFβ production by the tumor. Rather, the chronic presence of AM0010 at therapeutic levels will limit the generation of new Tregs elicited by TGFβ in the tumor microenvironment.
We investigated AM0010's further regulation of CD4+ T cell polarization biology. Again, contrary to expectations, AM0010 treatment of activated CD4+ T cells lead to a potentiation of IFNγ as well as Granzyme B and Perforin. Continued investigation revealed that treatment of activated CD4+ T cells with AM0010 does not elicit canonical Th1 polarized cells, but rather AM0010 treatment potentiated the number of Tbet+ IFNγ+ CD4+ cells.
We have previously investigated the serum trough concentration necessary to elicit antitumor effects in mice and discovered that a chronic trough concentration of approximately 1–2 ng/mL is required to elicit PEG-rMuIL-10s antitumor effect (data not shown). We have also provided data in previous publications that suggests the activation of CD8+ T cells with AM0010 requires continual IL-10 receptor engagement.Citation27 We have interpreted these data to suggest that PEG-rMuIL-10/AM0010 must be provided at a therapeutic trough concentration to exert its most significant antitumor effects. Analysis of serum trough levels from cancer patients dosed with AM0010 also supports these conclusions (data not shown).
Therefore, chronic treatment with AM0010 should lead to two primary effects on CD4+ T cells. First, the generation of new Tregs will be suppressed. Second, the chronic presence of AM0010 within the tumor microenvironment will also serve to elicit new Tbet+ IFNγ+ CD4+ T cells. Therefore over time, the relative percentage of FoxP3+ CD4+ T cells should decrease due to the inhibition of new Treg polarized (FoxP3+) cells and the increase of IFNγ potentiated CD4+ T cells.
While IL-10 has been previously defined as a broadly immunosuppressive cytokine, its regulatory function is context specific. IL-10 inhibits the secretion of LPS induced pro-inflammatory cytokines from myeloid cells suggesting its use in treating human autoimmune diseases that exhibit a significant bacteria/bacterial product induced inflammatory pathology. To determine whether this in vitro and in vivo biology translated to humans, Schering-Plough investigated the use of IL-10 in multiple Phase I–III studies treating autoimmune patients. In contrast to expectations, IL-10 did not exert dose titratable anti-inflammatory effects in the treatment of Crohn's diseaseCitation38,39 or psoriasis.Citation40 In fact, Crohn's patients treated with IL-10 exhibited an induction of serum IFNγ and neopterin.Citation41 Consistent with these results, healthy patients treated with LPS first, followed after an hour with IL-10 treatment, exhibited increased serum IFNγ and granzymes relative to patients treated with LPS alone.Citation42 Patients subjected to IL-10 treatment prior to LPS treatment exhibited a muted response to LPS. These data collectively suggest that IL-10 is in fact not a pan-immunosuppressive cytokine. Rather, consistent with our previous publications and the data presented herein, IL-10s immunoregulatory biology is context dependent. If IL-10 is present at high levels prior to an LPS-mediated inflammatory insult, the resulting pro-inflammatory cytokine burst is moderated. However, under conditions lacking LPS or the bacterial infection it mimics, such as those found in cancer patients, IL-10 treatment activates CD8+ T cells while concomitantly inhibiting the generation of FoxP3+ CD4+ Tregs. These data are consistent with the systemic phenomena previously reported for IL-10. However, as we treat more patients with AM0010, we are uncovering more immunoncology potentiating aspects of this intriguing cytokine.
Our current model of AM0010's effects are presented in . Our functional hypothesis is that treatment of the lymphoid compartment potentiates the cytotoxic- and cytokine-secreting capacity of CD8+ T cells that is quiescent until the CD8+ T cell TCR engages MHC I+ antigen. Upon engagement of MHC I+ antigen, the AM0010-primed CD8+ T cell response is significantly greater than in the absence of AM0010 stimulation, leading to the effective delivery of IFNγ to the tumor microenvironment. The increase in IFNγ leads to enhanced MHC I and eventually MHC II expression. The AM0010-primed CD8+ T cells also release increased amounts of both granzyme and perforin, leading to increased tumor cell cytolysis. This, in turn, provides more antigens for tumor-infiltrating antigen-presenting cells, leading to increased tumor antigen presentation, and enhanced immune surveillance.
Figure 8. The pleiotropic effects of AM0010. This is a schematic cartoon of AM0010's pleiotropic effects. AM0010 treatment concomitantly and directly affects both CD4+ and CD8+ T cells. AM0010's effect on CD4+ T cells is to block IL-2/TGFβ-mediated polarization of CD4+ T cells into new FoxP3+ Tregs. This leads to a decrease in Tregs over time and decrease in TGFβ mediate immune suppression. AM0010 also facilitates the development of a Th1 IFNγ+-like CD4+ T cell. AM0010 potentiates IFNγ, granzyme and perforin production in CD8+ T cells upon TCR engagement. The potentiation of these factors then leads to a cascade of effects, not directly induced by AM0010 treatment. The IFNγ drives MHCI/II presentation on all cells in the tumor microenvironment. Therefore, both tumor cells and antigen-presenting cells express more MHCI/II+ antigen. The increased secretion of granzyme and perforin leads to more tumor cell death, increasing the antigen pool for intratumoral antigen-presenting cells to take up and present. This becomes a reiterative cycle where more tumor cell killing leads to more antigen presentation, where both immune-dominant and, we hypothesize, cryptic tumor antigens are presented. This cascade of events permits enhanced tumor immune surveillance.
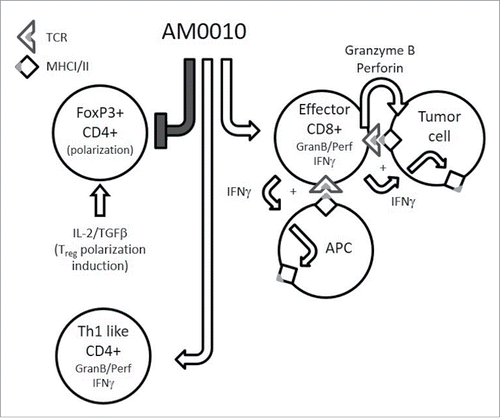
The data presented in this manuscript detail the other half of AM0010's function (), specifically that it inhibits the generation of new FoxP3+ CD4+ Treg suppressor cells by virtue of blocking their polarization. We had previously underappreciated the role of AM0010 on CD4+ T cells due to their comparatively small role in PEG-rMuIL-10s antitumor function in mice.Citation16 However, due to our expanding clinical understanding of AM0010's effect on the peripheral immune systemCitation17 (and data not shown) it is becoming clearer that AM0010 controls many aspects of the systemic human immune response.
While PEG-rMuIL-10 treatment did not exhibit combined antitumor function when dosed with the IDO inhibitor, 1-methyl-D-tryptophan, these data show that treatment with PEG-rMuIL-10 unexpectedly reduces FoxP3+ CD4+ Tregs in tumor-bearing mice. These data suggest a possible combination with immunoncology therapeutics, such as Ipilimumab, which exhibit enhanced function in combination with FoxP3+ CD4+ T cell limiting therapeutics.Citation43 These results warrant further investigation to determine the extent to which AM0010 can control and even dramatically reduce tumor burdens in cancer patients both as a monotherapy and in combination with standard of care chemotherapy as well as the new wave of exciting immunoncology compounds.
Materials and methods
Human subjects and study approval
Open label, dose-escalation study evaluating the safety and tolerability of AM0010 in heavily pretreated patients with advanced solid tumors, (limited to melanoma, prostate, and ovarian, renal, colorectal, pancreatic, or non-small cell lung cancer) with a life expectance of > 16 weeks and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Fully consented male and female patients, 18–80 y old suffering from terminal, surgically non-operable, late-stage neoplastic disease of non-lymphatic origin, were subjected to weekly peripheral blood collection by trained phlebotomists under ARMO Biosciences' Institutional Review Board (IRB) approved protocol; FDA study ID# NCT02009449. Patients in this study were enrolled under the IRB approved protocol at the following five institutes: (i.) Sarah Cannon Research Institute (SCRI), (ii.) Dan Farber Cancer Institute (DFCI), (iii.) MD Anderson Cancer Center (MDACC), (iv.) Memorial Sloan Kettering Cancer Center (MSKCC) and (v.) South Texas Accelerated Research Therapeutics (START). More than 50% of patients were non-immune sensitive tumors such as colorectal and pancreatic. Most patients received four or more prior therapies. Three–six patients were enrolled per cohort using a standard 3+3 dose-escalation format, where patients dose themselves subcutaneously, daily with 1, 2.5, 5, 10, 20 or 40 μg/kgCitation17.
Tumor challenge and treatment experiments
Female BALB/c mice, at 5 to 7 weeks of age, were implanted with 1 × 104 4T1 cells (ATCC, Manassas, VA) in RPMI (Life Technologies; Carlsbad, CA) subcutaneously on day 0. Tumors were measured by digital caliper and volumes were calculated with the following formula: π/6 × L × W × H. Once tumor volumes reached approximately 100 mm3, mice were stratified and dosed for the duration indicated in respective mg/kg PEG-rMuIL-10Citation16 (ARMO BioSciences, Redwood City, CA) or vehicle control (10 mM HEPES pH 6.5, 100 mM NaCl and 0.05% mouse serum albumin) was dosed daily, subcutaneously. 1-methyl-D-tryptophan administration was performed by implanting 28-d release pellets (140 mg/pellet or control pellets without the active product; Innovative Research of America, Sarasota, FL) under the dorsal skin when PEG-rMuIL-10 dosing was initiated according to manufacturer's instructions. These studies were performed at Aragen Biosciences (Gilroy, CA) in accordance with standard operating procedures approved by their Institutional Animal Care and Use Committee (IACUC).
Tumor-infiltrating lymphocyte isolation
To isolate TILs, tumors were minced with 5 mL of digest buffer (RPMI (Life Technologies), 10% Fetal Bovine Serum (Hyclone Thermo Fisher Scientific, Waltham, MA), 10 mM HEPES (Life Technologies), 2 mg/mL Collagenase Type I (Worthington Biochemical Corporation, Lakewood, NJ), 30 U/mL DNaseI (Worthington Biochemical Corporation)) and brought to a final volume of 35 mL with digest buffer. The tumor slurry was rotated at 37°C for 45 min. The tumor slurry was then mechanically disrupted by forcing the material through a 70-micron cell strainer. Cells were washed with RPMI twice and then resuspended with 25 mL of HBSS (Life Technologies). Cell suspensions were underlayed with 15 mL Histopaque (Sigma-Aldrich, St. Louis, MO) and centrifuged at 1000 rpm for 30 min at room temperature with the brakes turned off. After centrifugation, the cell interface, containing TILs, was collected and washed twice with complete RPMI. An aliquot of TILs was stained for flow cytometry. CD8+ T cells were then isolated from the remaining TILs using MACS cell separation technology from Miltenyi, following the manufacturer's protocol, and then stained for flow cytometry on a LSR II (BD Biosciences) and analyzed with FlowJo software v10 (Ashland, OR). Gating strategy: A lymphocyte gate was first created based on forward vs. side scatter. Next, a single cell gate was created based on forward scatter area vs. forward scatter height. This was followed by a live/dead cell gate. Live cells were then gated on CD45+ cells and subsequently gated for either CD4+ or CD8+ cells. CD4+ cells were graphed vs. FoxP3. CD8+ cells were graphed vs IFNγ.
Gene-expression analysis
RNA extraction, first strand synthesis, and quantitative PCR was performed using the RNeasy kit, RTCitation2 First Strand Kit and RT2 SYBR Green qPCR Mastermix from Qiagen (Germantown, MD, USA), respectively. All primers were obtained from Qiagen; mouse IFNγ: ifnγ, mouse Indoleamine 2,3-dioxygenase 1: ido1, mouse Tbet: tbx21, mouse Gata3: gata3, human Indoleamine 2,3-dioxygenase 1: IDO1. CT values were normalized to the average CT value of at least two housekeeping genes (GUSB and GAPDH).
Kynurenine and tryptophan quantitation
Analysis of kynurenine and tryptophan was performed by LakePharma (Belmont, CA). Mouse or human plasma samples were diluted and analyzed using LC-MS/MS (Shimadzu 20LC and ABscix API4000). The standard curves of tryptophan and kynurenine were generated by serial dilution followed by LC-MS/MS analysis. The final tryptophan and kynurenine concentrations of the mouse plasma samples were calculated based on the mass spectrum and standard curve.
Human T cell sourcing
Human PBMC's were obtained by Ficoll density gradient centrifugation of buffy coats (negligible amount of anticoagulant (CPD or CPDA-1)) acquired from anonymous donors who provided their tissue to the Stanford Blood Bank, (Palo Alto, CA). Buffy coats were stored at 2–8°C for up to 24 h prior to PBMC isolation. Cells were counted by Trypan blue exclusion for live vs. dead cells. All experiments were conducted in an exploratory research laboratory setting.
In vitro cell assays
CD8+ T cell IFNγ experiments: CD8+ T cells were isolated from PBMCs using CD8+ microbeads (Miltenyi Biotec, Auburn CA). Isolated CD8+ T cells (3 × 106 cells/mL, 3 × 106 cells per well in a 24-well plate) were activated with plate-bound anti-CD3 and anti-CD28 (pre-coated with 10 µg/mL anti-CD3 and 2 µg/mL anti-CD28; Affymetrix eBioscience) for 3 d. Following activation, cells were collected, replated (2 × 106 cells/mL, 5 × 105 cells per well in a 96-well plate) and treated with AM0010Citation16 for 3 d; in indicated experiments, cells were also concurrently treated with L-Kynurenine (Sigma Aldrich, St. Louis, MO). Following treatment, cells were collected, replated (2 × 106 cells/mL, 5 × 105 cells per well in a 96-well plate) and treated with 1 µg/mL soluble anti-CD3 for 4 h. After treatment, cell culture media was collected and assayed. Cell viability was determined using CellTiter-Glo (Promega, Madison, WI) according to the manufacturer's instructions. CD4+ T cell IFNγ experiments: CD4+ T cells were isolated with CD4+ microbeads (Miltenyi Biotec) and experiments were performed as described above. CD8+ and CD4+ T cell experiments were cultured in AIM V media. Induction of IDO1 in 4T1 cells: 1 × 105 4T1 cells/mL (2.5 × 105 cells per well in a 6-well plate) were cultured in complete RPMI (cRPMI: RPMI, 10 mM HEPES (Life Technologies), 10% Fetal Calf Serum (Hyclone Thermo Fisher Scientific, Waltham, MA) and Penicillin/Streptomycin cocktail (Life Technologies)) with 100 ng/mL PEG-rMuIL-10 and/or 50 ng/mL IFNγ for 2 d. After treatment, cells were collected for IDO1 gene expression. Induction of IDO1 in human dendritic cells: Human monocytes were isolated from PBMCs using CD14 microbeads (Miltenyi Biotec). Monocytes were differentiated to immature DCs with 10 ng/mL IL-4 and 40 ng/mL GM-CSF for 6 d in cRPMI. Immature DCs were treated with 100 ng/mL AM0010 for 2 d and cells were collected for IDO1 gene expression. To mature the DCs, cells were cultured with 5 µg/mL LPS and 100 ng/mL IFNγ with or without 100 ng/mL AM0010 for 2 d. After treatment, cells were collected for IDO1 gene expression. Treg cell induction with DCs: Mature DCs were isolated and induced as above. CD4+CD25− T cells were isolated through consecutive rounds of CD4+ selection and CD25 depletion using CD4+ and CD25 microbeads, respectively, according to manufacturer's instructions (Miltenyi Biotec). CD4+CD25− T cells (2 × 106 cells per well in a 6-well plate) were cocultured with mature DCs (4 × 105 cells per well in a 6-well plate) for 6 d with 10 ng/mL IL-2, 100 ng/mL IFNγ and 5 µg/mL LPS (Sigma-Aldrich) with or without 100 ng/mL AM0010 and with or without 2 µM IDO inhibitor, INCB024360 (Selleck Chemicals, Houston, TX) in cRPMI. T cells were then collected by flow cytometry on a FACScan or LSR II (BD Biosciences) and analyzed with FlowJo software v10 (Ashland, OR). Gating strategy: A single cell gate was first created based on forward scatter area vs. forward scatter height. Next, a lymphocyte gate was created based on forward vs. side scatter. CD4+ cells were gated via histogram plot and then graphed vs. FoxP3 or graphed as Tbet vs. IFNγ.
TGFβ/IL-2 Treg induction
CD4+ T cells were isolated with CD4+ microbeads (Miltenyi Biotec) and cultured for 5–6 d in AIMV media containing 10, 100, 1000 or 10,000 pg/mL IL-2 (Miltenyi Biotec), 10 ng/mL TGFβ ( Cell Signaling Technology) or 100 ng/mL AM0010 and 2 μg/mL immobilized anti-CD3 (Affymetrix eBioscience) and 1 μg/mL anti-CD28 (Affymetrix eBioscience).Citation33-35 Cells were then analyzed for FoxP3 expression by flow cytometric analysis.
Th1-like polarization
CD4+ T cells were isolated with CD4+ microbeads (Miltenyi Biotec) and activated for 3 d with 10 μg/mL anti-CD3 (Affymetrix eBioscience), 2 μg/mL anti-CD28 (Affymetrix eBioscience) in AIMV media and then rested in AIMV media containing 10 ng/mL IL-4 (BioLegend), 20 ng/mL IL-12 (BioLegend) or 100 ng/mL AM0010. Cells were then triggered with anti-CD3 at 1 μg/mL for 4 h in the presence of 1 µl/mL BD GolgiPlug (Becton Dickinson). Cells were then analyzed for Tbet and IFNγ expression by flow cytometric analysis.
Cytokine quantitation and protein gel blot analysis
To determine protein levels in murine tumor samples, tumors were weighed and homogenized in a fixed volume of water containing complete protease inhibitor cocktail (Sigma Aldrich). Samples were centrifuge to collect the supernatant. Supernatants were assayed by ELISA and concentrations were normalized to milligrams of tumor sample. Commercial ELISAs were used to detect mouse IFNγ (Meso Scale Discovery, Rockville, MD), mouse Granzyme B (Affymetrix eBiosciene), mouse TGFβ (Affymetrix eBioscience), human IFNγ (Affymetrix eBioscience), human Granzyme B (Mabtech, Cincinnati, OH) from mouse tumor supernatant, mouse plasma, and human cell culture media. For protein gel blots, cells were collected and lysed with RIPA buffer (Thermo Fisher Scientific). Protein concentrations were determined by BCA assay (Thermo Fisher Scientific) and normalized prior to gel loading. pSTAT3/STAT3 analysis: STAT3 Control Cell Extracts (Cell Signaling Technology) were used to verify protein gel blot reagents.
Antibodies
Protein gel blot antibodies were used according to manufacturer's instructions and obtained from Cell Signaling Technology (Danvers, MA): human IDO1, human/mouse STAT3 and human/mouse phospho-STAT3 antibodies; from Abcam (Cambridge, MA): Actin antibodies; and from BioLegend (San Diego, CA): mouse IDO1 antibody. Flow cytometry antibodies were used according to manufacturer's instructions and obtained from eBioscience Affymetrix: human Tbet or BioLegend: mouse CD45, CD8a, CD4+, FoxP3 and IFNγ; human CD4+ and FoxP3.
Statistical analysis
Data was analyzed for statistical significance by Student's t-test or ANOVA using GraphPad Prism software (La Jolla, CA) as indicated.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1197458_s02.pdf
Download PDF (779.7 KB)Acknowledgements
All authors are current employees of ARMO BioSciences and have no other financials interests.
References
- de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 1991; 174:1209-20; PMID:1940799; http://dx.doi.org/10.1084/jem.174.5.1209
- de Waal Malefyt R, Haanen J, Spits H, Roncarolo MG, te Velde A, Figdor C, Johnson K, Kastelein R, Yssel H, de Vries JE. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med 1991; 174:915-24; PMID:1655948; http://dx.doi.org/10.1084/jem.174.4.915
- Akdis CA, Joss A, Akdis M, Faith A, Blaser K. A molecular basis for T cell suppression by IL-10: CD28-associated IL-10 receptor inhibits CD28 tyrosine phosphorylation and phosphatidylinositol 3-kinase binding. FASEB J: Off Pub Federat Am Soc Exp Biol 2000; 14:1666-8; PMID:10973911; http://dx.doi.org/10.1096/fj.99-0874fje
- Joss A, Akdis M, Faith A, Blaser K, Akdis CA. IL-10 directly acts on T cells by specifically altering the CD28 co-stimulation pathway. Euro J Immunol 2000; 30:1683-90; PMID:10898505; http://dx.doi.org/10.1002/1521-4141(200006)30:6%3c1683::AID-IMMU1683%3e3.0.CO;2-A
- Opp MR, Smith EM, Hughes TK, Jr. Interleukin-10 (cytokine synthesis inhibitory factor) acts in the central nervous system of rats to reduce sleep. J Neuroimmunol 1995; 60:165-8; PMID:7642744; http://dx.doi.org/10.1016/0165-5728(95)00066-B
- Aste-Amezaga M, Ma X, Sartori A, Trinchieri G. Molecular mechanisms of the induction of IL-12 and its inhibition by IL-10. J Immunol 1998; 160:5936-44; PMID:9637507
- Varma TK, Toliver-Kinsky TE, Lin CY, Koutrouvelis AP, Nichols JE, Sherwood ER. Cellular mechanisms that cause suppressed gamma interferon secretion in endotoxin-tolerant mice. Infect Immun 2001; 69:5249-63; PMID:11500393; http://dx.doi.org/10.1128/IAI.69.9.5249-5263.2001
- Zheng LM, Ojcius DM, Garaud F, Roth C, Maxwell E, Li Z, Rong H, Chen J, Wang XY, Catino JJ et al. Interleukin-10 inhibits tumor metastasis through an NK cell-dependent mechanism. J Exp Med 1996; 184:579-84; PMID:8760811; http://dx.doi.org/10.1084/jem.184.2.579
- Sun H, Gutierrez P, Jackson MJ, Kundu N, Fulton AM. Essential role of nitric oxide and interferon-gamma for tumor immunotherapy with interleukin-10. J Immunother 2000; 23:208-14; PMID:10746547; http://dx.doi.org/10.1097/00002371-200003000-00005
- Sun H, Jackson MJ, Kundu N, Fulton AM. Interleukin-10 gene transfer activates interferon-gamma and the interferon-gamma-inducible genes Gbp-1/Mag-1 and Mig-1 in mammary tumors. Int J Cancer J Int du Cancer 1999; 80:624-9; PMID:9935167; http://dx.doi.org/10.1002/(SICI)1097-0215(19990209)80:4%3c624::AID-IJC23%3e3.0.CO;2-9
- Groux H, Cottrez F, Rouleau M, Mauze S, Antonenko S, Hurst S, McNeil T, Bigler M, Roncarolo MG, Coffman RL. A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J Immunol 1999; 162:1723-9; PMID:9973435
- Fujii S, Shimizu K, Shimizu T, Lotze MT. Interleukin-10 promotes the maintenance of antitumor CD8(+) T-cell effector function in situ. Blood 2001; 98:2143-51; PMID:11568001; http://dx.doi.org/10.1182/blood.V98.7.2143
- Berman RM, Suzuki T, Tahara H, Robbins PD, Narula SK, Lotze MT. Systemic administration of cellular IL-10 induces an effective, specific, and long-lived immune response against established tumors in mice. J Immunol 1996; 157:231-8; PMID:8683120
- Suzuki T, Tahara H, Narula S, Moore KW, Robbins PD, Lotze MT. Viral interleukin 10 (IL-10), the human herpes virus 4 cellular IL-10 homologue, induces local anergy to allogeneic and syngeneic tumors. J Exp Med 1995; 182:477-86; PMID:7629507; http://dx.doi.org/10.1084/jem.182.2.477
- Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, Gorman DM, Oft M. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res 2012; 72:3570-81; PMID:22581824; http://dx.doi.org/10.1158/0008-5472.CAN-12-0721
- Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 2011; 20:781-96; PMID:22172723; http://dx.doi.org/10.1016/j.ccr.2011.11.003
- Infante JR, Naing A, Papadopoulos KP, Autio KA, Ott PA, Wong DJL, Falchook GS, Patel MR, Pant S, Whiteside M et al. A first-in-human dose escalation study of PEGylated recombinant human IL-10 (AM0010) in advanced solid tumors. ASCO Meeting Abstracts 2015; 33:3017
- Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 2004; 4:762-74; PMID:15459668; http://dx.doi.org/10.1038/nri1457
- Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med 2004; 10:15-8; PMID:14720581; http://dx.doi.org/10.1016/j.molmed.2003.11.003
- Huang L, Li L, Klonowski KD, Tompkins SM, Tripp RA, Mellor AL. Induction and role of indoleamine 2,3 dioxygenase in mouse models of influenza a virus infection. PloS One 2013; 8:e66546; PMID:23785507; http://dx.doi.org/10.1371/journal.pone.0066546
- Sakash JB, Byrne GI, Lichtman A, Libby P. Cytokines induce indoleamine 2,3-dioxygenase expression in human atheroma-associated cells: implications for persistent Chlamydophila pneumoniae infection. Infect Immun 2002; 70:3959-61; PMID:12065543; http://dx.doi.org/10.1128/IAI.70.7.3959-3961.2002
- Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, Wada H, Noma A, Seishima M. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Euro J Immunol 2001; 31:2313-8; PMID:11477543; http://dx.doi.org/10.1002/1521-4141(200108)31:8%3c2313::AID-IMMU2313%3e3.0.CO;2-S
- Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med 2002; 196:459-68; PMID:12186838; http://dx.doi.org/10.1084/jem.20020121
- Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, Salvestrini V, Bonanno G, Rutella S, Durelli I et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007; 109:2871-7; PMID:17164341; http://dx.doi.org/10.1182/blood-2006-07-036863
- Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting tregs in malignant brain cancer: overcoming IDO. Front Immunol 2013; 4:116; PMID:23720663; http://dx.doi.org/10.3389/fimmu.2013.00116
- Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol 2009; 183:2475-83; PMID:19635913; http://dx.doi.org/10.4049/jimmunol.0900986
- Chan IH, Wu V, Bilardello M, Mar E, Oft M, Van Vlasselaer P, Mumm JB. The potentiation of IFN-gamma and induction of cytotoxic proteins by pegylated IL-10 in human CD8 T cells. J Interferon Cytokine Res 2015; PMID:26309093; http://dx.doi.org/10.1089/jir.2014.0221
- Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J: Off Pub Federat Am Soc Exp Biol 1991; 5:2516-22; PMID:1907934
- Jurgens B, Hainz U, Fuchs D, Felzmann T, Heitger A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood 2009; 114:3235-43; PMID:19625705; http://dx.doi.org/10.1182/blood-2008-12-195073
- Sarkar SA, Wong R, Hackl SI, Moua O, Gill RG, Wiseman A, Davidson HW, Hutton JC. Induction of indoleamine 2,3-dioxygenase by interferon-gamma in human islets. Diabetes 2007; 56:72-9; PMID:17192467; http://dx.doi.org/10.2337/db06-0617
- Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010; 115:3520-30; PMID:20197554; http://dx.doi.org/10.1182/blood-2009-09-246124
- Jana S, Jailwala P, Haribhai D, Waukau J, Glisic S, Grossman W, Mishra M, Wen R, Wang D, Williams CB et al. The role of NF-kappaB and Smad3 in TGF-beta-mediated Foxp3 expression. Euro J Immunol 2009; 39:2571-83; PMID:19701891; http://dx.doi.org/10.1002/eji.200939201
- Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D, Zhang H, Ding Y, Bromberg JS. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am J Transplant 2004; 4:1614-27; PMID:15367216; http://dx.doi.org/10.1111/j.1600-6143.2004.00566.x
- Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood 2007; 110:2983-90; PMID:17644734; http://dx.doi.org/10.1182/blood-2007-06-094656
- Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol 2011; 186:6329-37; PMID:21525380; http://dx.doi.org/10.4049/jimmunol.1100061
- Van Vlasselaer P, Borremans B, van Gorp U, Dasch JR, De Waal-Malefyt R. Interleukin 10 inhibits transforming growth factor-beta (TGF-beta) synthesis required for osteogenic commitment of mouse bone marrow cells. J Cell Biol 1994; 124:569-77; PMID:8106554; http://dx.doi.org/10.1083/jcb.124.4.569
- Nakagome K, Dohi M, Okunishi K, Tanaka R, Miyazaki J, Yamamoto K. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax 2006; 61:886-94; PMID:16809410; http://dx.doi.org/10.1136/thx.2005.056317
- Fedorak RN, Gangl A, Elson CO, Rutgeerts P, Schreiber S, Wild G, Hanauer SB, Kilian A, Cohard M, LeBeaut A et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn's disease. The interleukin 10 inflammatory bowel disease cooperative study group. Gastroenterology 2000; 119:1473-82; PMID:11113068; http://dx.doi.org/10.1053/gast.2000.20229
- Schreiber S, Fedorak RN, Nielsen OH, Wild G, Williams CN, Nikolaus S, Jacyna M, Lashner BA, Gangl A, Rutgeerts P et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn's disease. Crohn's disease IL-10 cooperative study group. Gastroenterology 2000; 119:1461-72; PMID:11113067; http://dx.doi.org/10.1053/gast.2000.20196
- Asadullah K, Docke WD, Ebeling M, Friedrich M, Belbe G, Audring H, Volk HD, Sterry W. Interleukin 10 treatment of psoriasis: clinical results of a phase 2 trial. Arch Dermatol 1999; 135:187-92; PMID:10052405; http://dx.doi.org/10.1001/archderm.135.2.187
- Tilg H, van Montfrans C, van den Ende A, Kaser A, van Deventer SJ, Schreiber S, Gregor M, Ludwiczek O, Rutgeerts P, Gasche C et al. Treatment of Crohn's disease with recombinant human interleukin 10 induces the proinflammatory cytokine interferon gamma. Gut 2002; 50:191-5; PMID:11788558; http://dx.doi.org/10.1136/gut.50.2.191
- Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJ, van der Poll T. Proinflammatory effects of IL-10 during human endotoxemia. J Immunol 2000; 165:2783-9; PMID:10946310; http://dx.doi.org/10.4049/jimmunol.165.5.2783
- Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med 2013; 210:1389-402; PMID:23752227; http://dx.doi.org/10.1084/jem.20130066