
ABSTRACT
Natural killer (NK)-cell count is predictive of chronic lymphoid leukemia (CLL) disease progression and their dysfunction is well documented, but the etiology of this is currently lacking. CLL cells have been shown to over-express HLA-E, the natural ligand for NKG2A expressed on NK-cells that generates a distinct negative signal relative to direct NK-cell cytotoxicity in other disease models. Utilizing a novel anti-NKG2A monoclonal blocking antibody (mAb), monalizumab, we explored the in vitro preclinical activity of targeting the NKG2A receptor, and the NKG2A/HLA-E interaction as a mechanism of tumor evasion in CLL patients. Our work confirmed overexpression of HLA-E on CLL B-cells and demonstrated NKG2A expression on CD56+/16+ NK-cells from CLL patients. We also demonstrate that blocking NKG2A on CLL NK-cells was sufficient to restore direct cytotoxicity ability of NK-cells against HLA-E-expressing targets without impacting NK-cell mediated antibody-dependent cellular cytotoxicity. Additionally, we proved the specificity of monalizumab in blocking NKG2A through Fc-blocking mechanisms. This paper provides justification for the potential clinical utility of monalizumab in the treatment of patients with CLL.
Introduction
Chronic lymphoid leukemia (CLL) is a malignant proliferation of morphologically mature CD5+, CD19+, CD20+, CD23+ B lymphocytes and is the most frequent leukemia diagnosed in adults, with 18,960 new cases estimated in the US in 2016.Citation1 Despite significant advances in therapy, CLL remains incurable with current standard therapies. The B cell receptor signaling is essential for proliferation and survival of malignant CLL B-cells.Citation2 Conversely, cytotoxic T-cells (CTLs) as well as NK-cells may play an important role in the control of the disease. NK-cell count is predictive of disease progression in newly diagnosed CLL.Citation3 NK-cells, although functionally competent, appear to exert weak cytotoxicity against CLL cells, potentially resulting from both upregulation of HLA-E and low expression of NK-cell activating ligands.Citation4
Monalizumab (IPH2201) is a humanized monoclonal antibody (mAb) of the immunoglobulin-4 (IgG4) subtype produced by recombinant technology in Chinese Hamster Ovary cells. It has a non-depleting and purely blocking activity directed with high affinity and specificity against the NKG2A subunit of the inhibitory, heterodimeric CD94/NKG2A receptor with its ligand HLA-E. CD94/NKG2A is expressed by large subsets of NK-cells as well as CTLs-like activated αβ CD8+ T-cells, γδ T-cells, and NKT-cells.Citation5 The role of NKG2A on these cells appears to be a negative feedback loop to limit T-cell receptor (TCR) activation. TCR engagement leads to increased expression of NKG2A on CTLs, subsequently, ligand engagement with NKG2A limits TCR activation, and this regulatory loop is specific to the NKG2 family of inhibitory receptors.Citation6 Likewise, NKG2A is found highly expressed on in the intestinal micro-environment indicating that the NKG2A/HLA-E negative feedback loop is utilized to regulate T-cells exposed to high antigenic load.Citation7
The natural ligand of CD94/NKG2A is HLA-E, a non-classical major histocompatibility complex (MHC) class I molecule, which is over-expressed by malignant cells in a variety of cancers, including in CLL.Citation4 Higher levels of HLA-E on primary B-CLL cells, as compared to normal B-cells, were also suggested by earlier studies.Citation8 Binding of HLA-E to CD94/NKG2A induces inhibitory signals that suppress the cytokine secretion and direct cytotoxicity of effector cells against malignant cells and this mechanism plays a significant role in the immune escape of certain tumor cells.Citation9-11 Conversely, by suppressing the inhibitory signal transduced by NKG2A, monalizumab enhances the antitumor functions, including lytic activity of these immune effector cells, as shown ex vivo and in vivo in several experimental models.Citation9-11 Herein, we report the increase of HLA-E on CLL tumor cells and demonstrate promising pre-clinical activity of monalizumab to enhance NK-cell activity by specifically blocking the NKG2A/HLA-E interaction in CLL patients.
Materials and methods
Cells and culture
Blood samples were obtained from normal donors (NDs) or CLL patients in accordance with the Declaration of Helsinki. All subjects provided written, informed consent under an Ohio State University Institutional Review Board—approved protocol. All patients had immunophenotypically defined CLLCitation12 and had been without prior therapy for a minimum of 30 d. Peripheral blood mononuclear cells were isolated by the Ficoll density gradient centrifugation (Ficoll-Paque Plus, GE Healthcare, Uppsala, Sweden). Enriched CLL and ND fractions were prepared via negative selection for B cells or NK cells with RosetteSep (Stem Cell Technologies, Vancouver, BC, Canada) according to the manufacturer's protocol. NK-cell purity was >80% and utilizing this negative selection methods ensured <5% contamination with CD3+ NK-T cells. This procedure also allows isolation of B-cells with >95% purity. Purity was assessed by immunophenotyping prior to the specific experiments. Cells were cultured in RPMI 1640 (Life Technologies, Grand Island, NY, USA) media supplemented with 10% heat-inactivated fetal bovine serum (Sigma, St. Louis, MO, USA), 2 mM L-glutamine (Invitrogen, Carlsbad, CA, USA), and 56 U/mL penicillin with 56 μg/mL streptomycin (Invitrogen), and cells were maintained at 37° Celsius with 5% CO2 atmospheric conditions.
Flow Cytometry, HLA-E & NKG2a surface expression
1×106 cells of the following types of cells were used per reaction tube: K562 cell line (CLL244, ATCC, Manassas, VA, USA), K562-E6 clone cell line (provided by Innate Pharma, S.A.), tumor cells from CLL patients treated at The Ohio State University Medical Center James Cancer Hospital, or whole blood from leukopacks (American Red Cross, SER-BC, Zen-Bio, Research Triangle Park, NC, USA). CLL samples were collected after obtaining written informed consent as part of an institutional review board (IRB) approved clinical trial, whereas normal leukopaks were obtained as part of an exempt IRB approved protocol. Patient cells were enriched from whole blood using Rosette Sep (Stem Cell technologies, Inc.) and Ficoll separation method, where whole blood is diluted with PBS, layered over Ficoll, and centrifuged for 30 min at 1500 rpm. The leukocyte layer is then pulled, washed with RPMI media, and re-pelleted and re-suspended in media for counting. Cells are pelleted at 1800 rpm for 10 min and washed with PBS. Cells for HLA-E staining are stained for 30 min at 4°C with the following: Live Dead Near IR (L010119, Life Technologies), CD45 Pacific Blue (A74765, Beckman Coulter, Brea, CA), CD3 PC7 (6607100, Beckman Coulter), CD19 FITC (555412, BD Bioscience, San Jose, CA, USA), and HLA-E PE (12-9953-42, eBiosciences, San Diego, CA, USA). Cells for NKG2A staining were stained for 30 min at 4°C with the following: Live Dead Near IR (L010119, Life Technologies), CD45 Pacific Blue (A74765, Beckman Coulter), CD3 PC7 (6607100, Beckman Coulter), CD16 FITC (IM0814U, Beckman Coulter), CD56 APC (555518, BD Bioscience), and CD159a PE (IM3291U, Beckman Coulter). Cells were pelleted again, washed with PBS and fixed with 2% paraformaldehyde. Fixed cells were run on Gallios flow cytometer (Beckman Coulter) and Kaluza software (Beckman Coulter) was used for analysis.
ELISA assay
A 96-well plate was pre-coated with PBS, isotype control, or monalizumab (each provided by Innate Pharma, S.A.) overnight at 4°C. NK-cells (1×105 cells/well) from CLL patients cultured in RPMI media with 20% FBS and 200,000 IU recombinant human IL-2 (200–02, PeproTech, Rocky Hill, NJ, USA) per mL of media were calculated for the total number of cells needed. The NK-cells were then plated into the corresponding wells and incubated for 24 h at 37°C in a 5% CO2 environment. Cells were collected at the reported time point and pelleted at 1800 rpm for 10 min. Supernatants were transferred to labeled tubes and frozen at −80°C and ran in triplicate by ELISA for human IFNγ Immunoassay following manufacturer's instructions (DIF50, R&D Systems).
Real-time PCR
RNA was isolated from the respective selected NK or B cells using a RNA Easy mini kit (74106, Qiagen, Valencia, CA, USA) and following the manufacturer's instructions a total of 2 µg of RNA per reaction was used to make cDNA by adding random hexamer to the RNA, incubating for 2 min at 70°C and then adding the cDNA master mix containing 6 µL of 5X buffer, 3 µL of 0.1M DTT, 1.5 µL of 10 mM dNTP, 1.5 µL of M-MLV, and 0.9 µL of RNAse-Out per reaction from the TaqMan Reverse Transcription Reagent kit (N8080234, Life Technologies). Tubes were placed on the thermocycler for 42°C for 1 h and 5 min at 95°C before going to 4°C. The cDNA (1 µL) was added to a qPCR master mix of 5 µL 2X TaqMan Fast Advanced universal PCR Master Mix (4444557, Life Technologies) with 3.5 µL of nuclease-free water and 0.5 µL of TaqMan Gene Expression Primers HLA-E (Hs03045171_m1, Invitrogen), NKG2A (Hs00970273_g1, Invitrogen), Beta-Actin or GAPDH (4331182, Life Technologies) per reaction. The plate is then run for 40 cycles of 95°C for 15 sec and 60°C for 1 min after a 10-min 95°C pre-amplification step.
NK-cell killing assays (Direct Cytotoxicity and ADCC)
Assessment of NK-cell killing activity was performed using standard 4 h 51Chromium release (CR) assay. Briefly, target K562 parental or K562-E6 cloned cells were labeled with radioactive 51Cr for 1 h at 37°C, washed, and plated on 96 well flat bottom plates. Antibodies monalizumab, isotype, or PBS were added to NKs and then co-cultured with 51Cr-labeled target cells at a 12:1 and/or 6:1 effector to target (E: T) ratio as indicated in each experiment. For blocking experiments, the target cells were treated with CD32 block (AF1330-SP, R&D Systems, Minneapolis, MN, USA) and CD64 block (MAB1257-SP, R&D Systems) at 10 μg/mL each for 30 min at 4°C. Supernatants were collected after 4 h of co-culture and counted on a Perkin Elmer (Waltham, MA) Wizard γ-counter. Specific lysis was determined by % lysis = 100 × (ER – SR)/(MR –SR) where ER, SR, and MR are experimental, spontaneous, and maximum release, respectively.
Statistical considerations
The difference in the percent of positive HLAE and NKG2A between groups was compared by using Mann–Whitney tests.Citation13 The difference in mRNA expression of HLA-E and NKG2A was compared by t-tests. The associations of the percent of HLAE or NKG2A and its ΔMFI and the percentage of HLAE and NKG2A were evaluated by using Spearman's rank correlation coefficient (ρ). Additionally, for the experiments that treated sample from the same subject with various conditions, mixed-effect models were used for analysis to take into consideration the dependency of these observations.Citation14 Holm's procedureCitation15 was used to control the family-wise error rate at 0.05.
Results
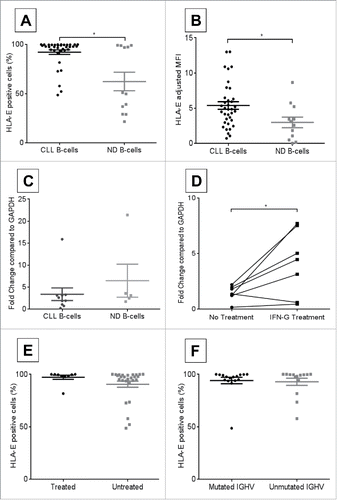
Previous studies have demonstrated ubiquitous HLA-E expression across a number of primary cells and cell lines and upregulation in various cancers (reviewed inCitation16). First, we compared surface HLA-E expression on freshly isolated CLL patient to ND B-cells. CLL B-cells appeared to have variable expression of HLA-E, possibly as a reflection of their immune deregulation, as compared to ND B-cells as shown by the percentage of cells positive for HLA-E (, CLL B-cells median = 97.34%, n = 36; ND B-cells median = 50.45%, n = 12; p = 0.005), or intensity of HLA-E as determined by adjusted mean fluorescence intensity (MFI) (, CLL B-cells median = 4.87, n = 36; ND B-cells median = 2.86 n = 12; p = 0.017). Similarly, CLL B-cells had similar levels of HLA-E mRNA compared to ND B-cells (, CLL HLA-E median = 2.29, n = 10; ND HLA-E median = 3.15, n = 5; p = 0.24) suggesting that the regulation of this gene expression between normal and CLL patients was predominately with post-transcriptional mechanism. The increased surface expression hints at a putative role of HLA-E in allowing CLL B-cells to evade NK-cell mediated lysis. Induction of HLA-E mRNA was also confirmed by interferon (IFN)γ treatment of CLL B-cells (, no treatment adjusted mean fold change = 1.44, treated mean fold change = 4.13; n = 6, p = 0.032), which has been shown to occur through distinct STAT1α and GATA3 response elements.Citation17,18 HLA-E expression was also found to be unrelated to patient treatment status (, untreated median = 99.5%, n = 9; treated median = 97.4%, n = 27; p = 0.06) or IGHV mutation status (, unmutated median = 99.3%, n = 14; mutated median = 97.5%, n = 16; p = 0.4).
Figure 1. HLA-E and NKG2A landscape in CLL.
Continued
Continued
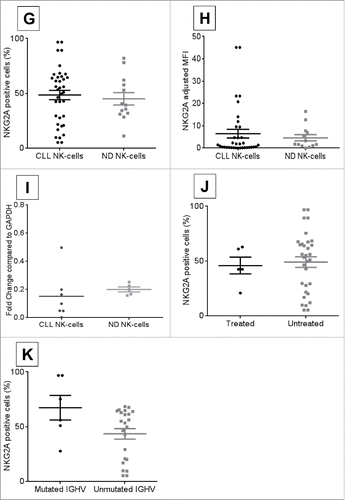
Surface NKG2A expression was confirmed on CLL patient NK-cells and compared to normal donors. No significant differences were observed between CLL and ND NK-cells with wide variability in percent positive cells (, CLL NK-cell median = 53.4%, n = 37; ND NK-cells median = 39.3%, n = 13; p = 0.59) and MFI (, CLL NK-cells median = 1.12, n = 36; ND NK-cells median = 1.75, n = 13; p = 0.33). CLL patient NK-cells tended to have a similar expression of NKG2A mRNA compared to normal donor NK-cells (, ND NKG2A median = 0.2, n = 5; CLL NKG2A median = 0.1, n = 7; p = 0.19). NKG2A expression was not related to patient treatment status (, untreated median = 54.3%, n = 32; mutated median = 42.5%, n = 5; p = 0.60) or IGHV mutation status (, unmutated median = 53.4%, n = 23; mutated median = 65.7%, n = 6; p = 0.10).
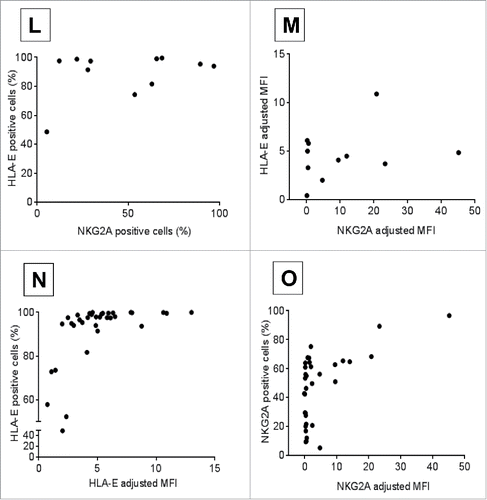
Moreover, no significant correlation was observed between HLA-E and NKG2A expression within matched CLL patient samples by percent cells positive for HLA-E vs. NKG2A (, n = 11, rho-ρ = 0.23, p = 0.48) or MFI (, n = 11, rho-ρ = 0.15, p = 0.65). As anticipated, patients who had higher percentage of positive cells had higher MFI for HLA-E (, n = 36, ρ = 0.74, p < 0.0001) on CLL B-cells and NKG2A (, n = 37, ρ = 0.5, p = 0.002) on CLL NK-cells. Collectively, these data suggest that CLL B-cells express more HLA-E, which could potentially contribute to and enhance immune evasion by their NK-cells.
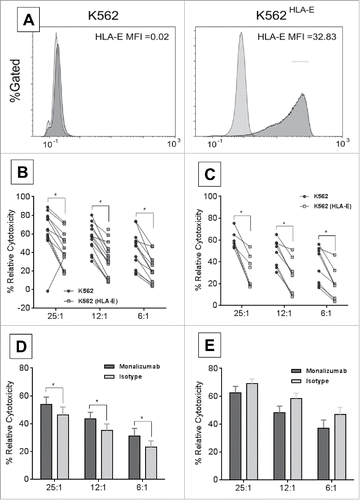
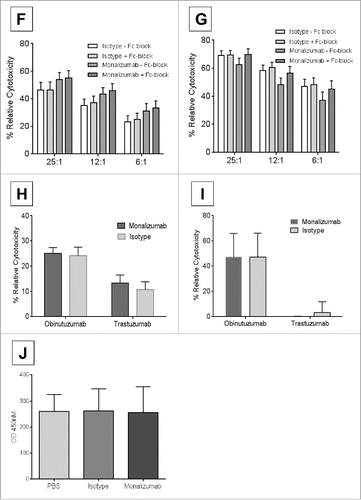
Monalizumab is a first-in-class humanized IgG4 antibody targeting NKG2A and blocking its interaction with HLA-E thus preventing the propagation of inhibitory signals that is utilized by certain cancer cells as immune escape mechanism. In order to determine the role of tumor cell's expressing HLA-E on NK-cell function, a HLA-E overexpressing K562 cell line (K562-E6) was utilized (, Adjusted MFI K562-E6 vs. K562 = 32.81) to allow serial comparison of CLL patient NK-cell function with blockade of NKG2A. Additionally, CLL tumor cells have variable numerous other immune checkpoint inhibitors in addition to HLA-E expression in varied amounts that would prevent dissecting out the influence of this ligand receptor interaction. CLL patient NK-cells showed diminished direct cytotoxicity against K562-E6 cell line as compared to the parental K562 cell line that lacks HLA-E expression (, mean % relative cytotoxicity: K562 vs. K562-E6 at 25:1 = 63% vs. 43.3%, 12:1 = 54.9% vs. 32.9%; 6:1 = 43.5% vs. 21.7%; n = 15, p = 0.03, <0.01, <0.01 respectively). Similar effect was also observed when utilizing ND NK-cells confirming the impact of overexpression of HLA-E on diminishing NK-cell mediated direct cytotoxicity (, mean % relative cytotoxicity: K562 vs. K562-E6 25:1 = 60.6% vs. 32.5%, 12:1 = 48.1% vs. 26.6%; 6:1 = 36.7% vs. 18.8%. n = 8, p = <0.01, <0.01, 0.03 respectively. The addition of monalizumab significantly increased CLL NK-cell mediated direct cytotoxicity against HLA-E-expressing targets (K562-E6) (, monalizumab vs. isotype at 25:1 = 54.0% vs. 46.3%, n = 12, p = 0.04; at 12:1 = 43.3% vs. 35.1%, n = 14, p = 0.02; at 6:1 = 31.2% vs. 23.2%. n = 12, p = 0.05). This effect was specific to the blockade of NKG2A/HLA-E interaction since monalizumab was unable to enhance direct cytotoxicity in non-HLA-E expressing target cells (, mean % relative cytotoxicity of monalizumab vs. isotype at 25:1 = 62.5% vs. 69.0%, n = 12, p = 0.26; 12:1 = 48.1% vs. 58.4%, n = 13, p = 0.1; at 6:1 = 37.2% vs. 46.8%. n = 12, p = 0.2). To further establish the specificity of monalizumab, Fc-gamma receptor blocking antibodies were utilized in direct cytotoxicity experiments with Fc-blocking-treated K562-E6 targets cultured with CLL NK-cells (, CLL NK-cells, mean % relative cytotoxicity of monalizumab–Fc-block vs. + Fc-block at E:T 25:1 = 54% vs. 55.1%, n = 12, p = 0.6, at 12:1 = 43.3% vs. 46.1%, n = 14, p = 0.8, at 6:1 = 31.2% vs. 33.2%, n = 12, p = 0.87). Similarly, mixed effect interaction tests proved no interaction of adding Fc-blocker on monalizumab or isotype on CLL NK-cells relative cytotoxicity when cultured with K562 targets (, CLL NK-cells, mean % relative cytotoxicity of monalizumab–Fc-block vs. + Fc-block at 25:1 = 62.5% vs. 69.8%, n = 12, p = 0.09, at 12:1 = 48.1% vs. 56.3%, n = 13, p = 0.19, at 6:1 = 37.2% vs. 45.2%, n = 12, p = 0.17). We further demonstrate that monalizumab does not impair NK-cell mediated antibody-dependent cellular cytotoxicity (ADCC). CLL B-cells treated with obinutuzumab or trastuzumab control and ND NK-cells treated with monalizumab or isotype at E:T ratio of 25:1 showed similar mean % relative cytotoxicity as compared to isotype (25% vs. 24.1%, respectively, n = 15, p = 0.83, ). Similar effect was also observed when autologous conditions were utilized where B-cells and NK-cells were derived from the same CLL patient (mean % relative cytotoxicity of monalizumab vs. isotype with obinutuzumab = 47.2% vs. 47%, n = 3, p = 0.99, ). Specificity of monalizumab to enhance DC without increasing cytokine production of NK-cells was determined by utilizing plate bound antibodies and measuring interferon-γ concentration in supernatant after 24-h of incubation (optical density (OD) by ELISA of PBS = 260.6 vs. isotype = 261 vs. monalizumab = 255.4, n = 5, p = 0.9). These experiments confirm our hypothesis that the impact of monalizumab in enhancing CLL NK-cell mediated direct cytotoxicity is not mediated through engagement with Fc-gamma receptors on B-cells. Monalizumab is shown here to be specific in blocking NKG2A and enhancing CLL NK-cell lytic activity against HLA-E expressing targets without impacting ADCC.
Figure 2. Monalizumab blocks NKG2A and enhances CLL NK-cell mediated cytotoxicity.
Continued
Discussion
CLL NK-cells exhibit profound immune deregulation and mediate weak killing of tumor target.Citation19-21 In an effort to elucidate the role of NKG2A/HLA-E in CLL immune evasion from NK-cell-mediated killing, we performed surface expression and transcript analysis on CLL patient samples. Various in vitro functional assays using CLL patient NK-cells were carried out to demonstrate the deregulated immune effector activity mediated by NKG2A/HLA-E.
Within this report, we describe the variable expression of HLA-E and NKG2A on CLL B- and NK-cells, respectively, and demonstrate that this variability is not correlated with either treatment status or IGHV mutational status and that no correlation was found between HLA-E and NKG2A surface expression. In agreement with previous reports by Veiullen et al.Citation22 HLA-E was be higher on CLL compared to normal B-cells, but again CLL patients showed variable expression. Additionally, no correlation was found between both HLA-E and NKG2A expression with treatment or IGHV mutational status implying that immune evasion via NKG2A/HLA-E deactivating NK-cells does not discriminate subsets of CLL patients. Furthermore, we show for the first time that blocking NKG2A/HLA-E interaction was sufficient to restore CLL NK-cells' ability to lyse HLA-E-expressing target cells. The primary role of HLA-E, a non-classical HLA-class I molecule, is to identify “self” cells and tissue to surveying NK-cells, protecting the cell-expressing HLA-E from NK-cell killing.Citation23 As compared to classical HLA molecules like HLA-A, -B, and -C that have restricted expression to specific cells and tissues, HLA-E is expressed on all human cells and tissues with significant expression on endothelial and immune cells and the highest mRNA expression levels reported in resting T-cells.Citation24 HLA-E is overexpressed in various cancers and overexpressed in CLL as others have demonstrated and we have shown within this report.Citation22 We found no correlation between prior treatment status or IGHV mutation change versus germ-line with expression of HLA-E or NKG2A. This implies that regardless of a patient's treatment history or disease stage, NKG2A is actively involved in interacting with HLA-E and deactivating NK-cell lytic activity. Additionally, we showed that IFNγ induced expression of HLA-E transcript in six of seven patients tested. IFNγ is reported to be abundant in CLL patient serumCitation25 and may potentially limit CLL B-cell apoptosis in vitro through both autocrine and paracrine pathways. Here, we add that IFNγ can up regulate HLA-E transcript in CLL and demonstrates that IFNγ is more tumorigenic than previously reported and acts through multiple mechanisms to protect CLL B-cells.
Human NKG2A expresses two ITIMs, which become phosphorylated upon NKG2A binding with HLA-E. The phosphorylated ITIMs recruit and activate phosphatase SHP-1, which suppresses the signals generated from ITAM containing NK-cell activating receptors.Citation26 Functional studies have showed that HLA-E binding NKG2A interferes with CD16 association with spleen tyrosine kinase (SYK) and inhibits activation of SYK as well as extracellular regulated kinases therefore decreasing NK-cell activation, direct killing, and ADCC.Citation27 Maintenance of inhibitory receptor expression is critical to prevent self-destruction and by overexpressing NKG2A, the cancer cell can protect itself from NK-cell lytic activity. Using NKL and NK92 cells lines, it has been shown that IFN-α can decrease NKG2A surface expression leading to increased lysis of MICA+ targets and by this same measure, IFNγ increased surface expression of NKG2A and thus reduced MICA+ target killing.Citation28 Here, we demonstrate that this mechanism of evading NK-cell detection and killing is active in CLL patients and that the NKG2A/HLA-E interaction can be blocked leading to increased lytic activity of CLL NK-cells without compromising NK-cell mediated ADCC. Furthermore, high IFNγ production by CLL cells may explain our observed increased surface expression though transcripts between normal and CLL are comparable.
In acute myeloid leukemia (AML) where HLA-E is expressed by all blasts, blocking NKG2A restores lysis against AML blasts.Citation29 In contrast to this study, which utilized an IgG2b anti-NKG2A mAb, monalizumab is of IgG4 subtype and thus would not activate complement or innate immune effector functions; it would act purely as a blocking antibodyCitation30 and we postulate would mediate the same effects on restoring NK-cell-mediated lysis against AML. CLL NK-cells, which exert weak lytic activity against HLA-E targets, have improved function by blocking NKG2A with monalizumab. The studies described above have specifically addressed the efficacy of blocking NKG2A on CLL NK-cells, however, activated αβ CD8+ T cells, γδ-T cells, and NKT cells also express NKG2ACitation5,31 and the efficacy of blocking NKG2A on these cells remains to be studied. Monalizumab has previously been evaluated in patients with rheumatoid arthritis based on the proposed ability of eliminating auto-reactive CD4+ T-cells. Preliminary results from early phase clinical trials show excellent tolerability without increase in autoimmune complications secondary to uncontrolled immune activation.Citation32 Based upon the data with monalizumab restoration of NK-cell direct cytotoxic function, clinical trials with this agent as monotherapy or combination with other immune agents seems warranted.
Disclosure of potential conflicts of interest
FTA received research funding from Innate Pharma, Inc.
Funding
This work was supported by a Career Development Award to FTA from the Lymphoma Research Foundation, P01-CA095426 from the National Cancer Institute, and The D. Warren Brown Foundation.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA 2016; 66:7-30; PMID:26742998; http://dx.doi.org/10.3322/caac.21332
- Awan FT, Byrd JC. New strategies in chronic lymphocytic leukemia: shifting treatment paradigms. Clin Cancer Res 2014; 20:5869-74; PMID:25294898; http://dx.doi.org/10.1158/1078-0432.CCR-14-1889
- Palmer S, Hanson CA, Zent CS, Porrata LF, Laplant B, Geyer SM, Markovic SN, Call TG, Bowen DA, Jelinek DF et al. Prognostic importance of T and NK-cells in a consecutive series of newly diagnosed patients with chronic lymphocytic leukaemia. Br J Haematol 2008; 141:607-14; PMID:18384436; http://dx.doi.org/10.1111/j.1365-2141.2008.07070.x
- Veuillen C, Aurran-Schleinitz T, Castellano R, Rey J, Mallet F, Orlanducci F, Pouyet L, Just-Landi S, Coso D, Ivanov V et al. Primary B-CLL resistance to NK cell cytotoxicity can be overcome in vitro and in vivo by priming NK cells and monoclonal antibody therapy. J Clin Immunol 2012; 32:632-46; PMID:22318393; http://dx.doi.org/10.1007/s10875-011-9624-5
- Arlettaz L, Villard J, de Rham C, Degermann S, Chapuis B, Huard B, Roosnek E. Activating CD94:NKG2C and inhibitory CD94:NKG2A receptors are expressed by distinct subsets of committed CD8+ TCR αβ lymphocytes. Eur J Immunol 2004; 34:3456-64; PMID:15517612; http://dx.doi.org/10.1002/eji.200425210
- Jabri B, Selby JM, Negulescu H, Lee L, Roberts AI, Beavis A, Lopez-Botet M, Ebert EC, Winchester RJ. TCR specificity dictates CD94/NKG2A expression by human CTL. Immunity 2002; 17:487-99; PMID:12387742; http://dx.doi.org/10.1016/S1074-7613(02)00427-2
- Jabri B, De Serre NPM, Cellier C, Evans K, Gache C, Carvalho C, Mougenot JF, Allez M et al. Selective expansion of intraepithelial lymphocytes expressing the HLA-E-specific natural killer receptor CD94 in celiac disease. Gastroenterology 2000; 118:867-79; PMID:10784586; http://dx.doi.org/10.1016/S0016-5085(00)70173-9
- Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu TM, Chang BY, Larkin KM et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013; 122:2539-49; PMID:23886836; http://dx.doi.org/10.1182/blood-2013-06-507947
- Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI, Phillips JH et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998; 391:795-9; PMID:9486650; http://dx.doi.org/10.1038/35869
- Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med 1998; 187:813-8; PMID:9480992; http://dx.doi.org/10.1084/jem.187.5.813
- Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M, Geraghty DE. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci USA 1998; 95:5199-204; PMID:9560253; http://dx.doi.org/10.1073/pnas.95.9.5199
- Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood 2008; 111:5446-56; PMID:18216293; http://dx.doi.org/10.1182/blood-2007-06-093906
- Mann HB, Whitney DR. On a test of whether one of two random variables is stochastically larger than the other. Ann Math Statist; 18(1):50-60; http://dx.doi.org/10.1214/aoms/1177730491
- Verbeke G, Molenberghs G. Linear Mixed Models for Longitudinal Data. Springer-Verlag New York, 2000; 570:1; http://dx.doi.org/10.1007/978-1-4419-0300-6
- Hsu J. Multiple comparisons: theory and methods. CRC Press Boca Raton, FL, 1996; http://dx.doi.org/10.1007/978-1-4899-7180-7
- Wieten L, Mahaweni NM, Voorter CEM, Bos GMJ, Tilanus MGJ. Clinical and immunological significance of HLA-E in stem cell transplantation and cancer. Tissue Antigens 2014; 84:523-35; PMID:25413103; http://dx.doi.org/10.1111/tan.12478
- Gustafson KS, Ginder GD. Interferon-γ induction of the human leukocyte antigen-E gene is mediated through binding of a complex containing STAT1α to a distinct interferon-γ-responsive element. J Biol Chem 1996; 271:20035-46; PMID:8702722; http://dx.doi.org/10.1074/jbc.271.33.20035
- Marusina AI, Kim D-K, Lieto LD, Borrego F, Coligan JE. GATA-3 is an important transcription factor for regulating human NKG2A gene expression. J Immunol 2005; 174:2152-9; PMID:15699146; http://dx.doi.org/10.4049/jimmunol.174.4.2152
- Ziegler H-W, Kay NE, Zarling JM. Deficiency of natural killer cell activity in patients with chronic lymphocytic leukemia. Int J Cancer 1981; 27:321-7; PMID:6169660; http://dx.doi.org/10.1002/ijc.2910270310
- Kay NE, Zarling JM. Impaired natural killer activity in patients with chronic lymphocytic leukemia is associated with a deficiency of azurophilic cytoplasmic granules in putative NK cells. Blood 1984; 63:305-9; PMID:6607080
- Jaglowski SM, Alinari L, Lapalombella R, Muthusamy N, Byrd JC. The clinical application of monoclonal antibodies in chronic lymphocytic leukemia. Blood 2010; 116:3705-14; PMID:20610811; http://dx.doi.org/10.1182/blood-2010-04-001230
- Veuillen C, Aurran-Schleinitz T, Castellano R, Rey J, Mallet F, Orlanducci F, Pouyet L, Just-Landi S, Coso D, Ivanov V et al. Primary B-CLL resistance to NK Cell cytotoxicity can be overcome in vitro and in vivo by priming NK cells and monoclonal antibody therapy. J Clin Immunol 2012; 32:632-46; PMID:22318393; http://dx.doi.org/10.1007/s10875-011-9624-5
- Rodgers JR, Cook RG. MHC class Ib molecules bridge innate and acquired immunity. Nat Rev Immunol 2005; 5:459-71; PMID:15928678; http://dx.doi.org/10.1038/nri1635
- Koller BH, Geraghty DE, Shimizu Y, DeMars R, Orr HT. HLA-E. A novel HLA class I gene expressed in resting T lymphocytes. J Immunol 1988; 141:897-904; PMID:3260916
- Buschle M, Campana D, Carding SR, Richard C, Hoffbrand AV, Brenner MK. Interferon gamma inhibits apoptotic cell death in B cell chronic lymphocytic leukemia. J Exp Med 1993; 177:213-8; PMID:7678114; http://dx.doi.org/10.1084/jem.177.1.213
- Carretero M, Palmieri G, Llano M, Tullio V, Santoni A, Geraghty DE, López-Botet M. Specific engagement of the CD94/NKG2-A killer inhibitory receptor by the HLA-E class Ib molecule induces SHP-1 phosphatase recruitment to tyrosine-phosphorylated NKG2-A: evidence for receptor function in heterologous transfectants. Eur J Immunol 1998; 28:1280-91; PMID:9565368; http://dx.doi.org/10.1002/(SICI)1521-4141(199804)28:04%3c1280::AID-IMMU1280%3e3.0.CO;2-O
- Palmieri G, Tullio V, Zingoni A, Piccoli M, Frati L, Lopez-Botet M, Santoni A. CD94/NKG2-A inhibitory complex blocks CD16-triggered Syk and extracellular regulated kinase activation, leading to cytotoxic function of human NK cells. J Immunol 1999; 162:7181-8; PMID:10358164
- Zhang C, Zhang J, Sun R, Feng J, Wei H, Tian Z. Opposing effect of IFNγ and IFNα on expression of NKG2 receptors: Negative regulation of IFNγ on NK cells. Int Immuno Pharmacol 2005; 5:1057-67; PMID:15829421; http://dx.doi.org/10.1016/j.intimp.2005.02.003
- Nguyen S, Dhedin N, Vernant J-P, Kuentz M, Jijakli AA, Rouas-Freiss N, Carosella ED, Boudifa A, Debré P, Vieillard V. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 2005; 105:4135-42; PMID:15687235; http://dx.doi.org/10.1182/blood-2004-10-4113
- Irani V, Guy AJ, Andrew D, Beeson JG, Ramsland PA, Richards JS. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol Immunol 2015; 67:171-82; PMID:25900877; http://dx.doi.org/10.1016/j.molimm.2015.03.255
- Gunturi A, Berg RE, Crossley E, Murray S, Forman J. The role of TCR stimulation and TGF-β in controlling the expression of CD94/NKG2A receptors on CD8 T cells. Eur J Immunol 2005; 35:766-75; PMID:15714583; http://dx.doi.org/10.1002/eji.200425735
- Seymour L, Tinker A, Hirte H, Wagtmann N, Dodion P. O3.2Phase I and dose ranging, phase II studies with IPH2201, a humanized monoclonal antibody targeting HLA-E receptor CD94/NKG2A. Annals Oncol 2015; 26:ii3; http://dx.doi.org/10.1093/annonc/mdv081.2