
ABSTRACT
Activated and in vitro expanded natural killer (NK) cells have substantial cytotoxicity against many tumor cells, but their in vivo efficacy to eliminate solid cancers is limited. Here, we used chimeric antigen receptors (CARs) to enhance the activity of NK cells against Ewing sarcomas (EwS) in a tumor antigen-specific manner. Expression of CARs directed against the ganglioside antigen GD2 in activated NK cells increased their responses to GD2+ allogeneic EwS cells in vitro and overcame resistance of individual cell lines to NK cell lysis. Second-generation CARs with 4-1BB and 2B4 co-stimulatory signaling and third-generation CARs combining both co-stimulatory domains were all equally effective. By contrast, adoptive transfer of GD2-specific CAR gene-modified NK cells both by intratumoral and intraperitoneal delivery failed to eliminate GD2-expressing EwS xenografts. Histopathology review revealed upregulation of the immunosuppressive ligand HLA-G in tumor autopsies from mice treated with NK cells compared to untreated control mice. Supporting the relevance of this finding, in vitro co-incubation of NK cells with allogeneic EwS cells induced upregulation of the HLA-G receptor CD85j, and HLA-G1 expressed by EwS cells suppressed the activity of NK cells from three of five allogeneic donors against the tumor cells in vitro.
We conclude that HLA-G is a candidate immune checkpoint in EwS where it can contribute to resistance to NK cell therapy. HLA-G deserves evaluation as a potential target for more effective immunotherapeutic combination regimens in this and other cancers.
Abbreviations
| BLI | = | bioluminescence imaging |
| CAR | = | chimeric antigen receptor |
| CD | = | cluster of differentiation |
| DNAM-1 | = | DNAX accessory molecule-1 |
| eGFP | = | enhanced green fluorescence protein |
| EwS | = | Ewing sarcoma |
| FFPE | = | formalin-fixed paraffin-embedded |
| HLA | = | human leukocyte antigen |
| IFNγ | = | interferon gamma |
| IL-2 | = | -12, -15, -18, interleukin-2, -12, -15, -18 |
| ILT2 | = | immunoglobulin-like transcript 2 |
| i.p. | = | intraperitoneal |
| i.t. | = | intratumoral |
| KIR | = | killer-cell immunoglobulin-like receptors |
| MHC | = | major histocompatibility complex |
| MICA | = | MHC class I polypeptide-related sequence A |
| MICB | = | MHC class I polypeptide-related sequence B |
| NK cell | = | natural killer cell |
| NSG mice | = | NOD scid gamma mice |
| PBMC | = | peripheral blood mononuclear cells |
| PD-L1 | = | programmed death ligand 1 |
| PD-1 | = | programmed cell death protein 1 |
| rhIL-2 | = | recombinant human IL-2 |
| s.c. | = | subcutaneous |
| TNF-α | = | tumor necrosis factor-α |
| ULBP1 | = | 2, 3, UL16 binding protein 1, 2, 3 |
Introduction
We aim to develop effective cellular immunotherapies for Ewing sarcoma (EwS), an aggressive and often incurable solid cancer of bone and soft tissues.Citation1 EwS cells have been found to be exceptionally susceptible to lysis by in vitro activated and expanded natural killer (NK) cells.Citation2,3 EwS cells interact with NK cells by engaging activating NK cell receptors, most notably NKG2D and DNAX accessory molecule-1 (DNAM-1).Citation2,4 Moreover, EwS cells often downregulate surface MHC class I and thereby fail to bind to inhibitory NK cell receptors, further shifting the balance toward NK cell activation.Citation5,6 But within a multicellular tumor architecture, EwS cells can develop NK cell resistance,Citation6 and reports of patients with relapsed or refractory EwS receiving haploidentical stem cell grafts with high numbers of NK cells do not support substantial therapeutic activity of this cell population.Citation7,8 Thus, additional strategies must be incorporated to enhance the antitumor activity of NK cells and allow effective targeting of this and further solid cancers.
We and others have previously shown that engineering of activated human NK cells with recombinant chimeric antigen receptors (CARs) can enhance their function against various cancer targets in an antigen-dependent manner.Citation9-14 CARs are single molecules that link antibody-derived antigen recognition to activating intracellular signals. Their use has been most widely explored in T cells. Adoptive therapy with T cells gene-modified to express CARs against the B cell antigen CD19 was highly active to induce remissions in patients with B cell malignancies.Citation15,16 EwS is a candidate tumor for CAR targeting by surface expression of the ganglioside antigen GD2.Citation17,18 But whereas B cell antigens are reliable surface markers of B-lymphoid cancers, GD2 expression in EwS is heterogeneous and varies not only among individual tumors but also within single biopsies.Citation17 Since effective CAR-mediated cytolysis depends on at least moderate expression of the target antigen,Citation19 tumor cells with low GD2 antigen density are likely to escape. Adjusting the affinity of CARs to detect lower levels of antigen is potentially hazardous by toxic on-target/off-tumor interactions with normal tissues expressing GD2, especially in the central nervous system that CAR T cells can effectively access.Citation15 Here, we proposed to exploit the native activity of NK cells against EwS cells to extend the CAR-mediated effector response toward GD2 antigen-negative/low tumor cells while sparing normal cells. We hypothesized that expression of GD2-specific CARs in in vitro activated NK cells would overcome limitations of both strategies and translate into potent anti-EwS activity in vivo.
Results
Optimal culture conditions for in vitro activation and expansion of human NK cells
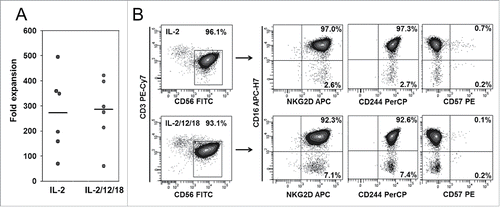
To optimize in vitro activation and expansion of human NK cells, we compared co-cultures of peripheral blood mononuclear cells (PBMCs) with irradiated K562-mb15-41BBL cells in the presence of low-dose interleukin-2 (IL-2) alone, as established by Imai et al.Citation12 and with additional IL-12 and IL-18. These cytokines were reported to promote high proliferation rates and an increased proportion of NK cells with enhanced functional capacity.Citation20 IL-15 signals are provided to both types of cultures by membrane-bound IL-15 on the stimulator cells. NK cell expansion was highly efficient and was comparable in culture medium containing either IL-2 alone or the IL-2/IL-12/IL-18 cytokine cocktail (). The activated and expanded NK cells, regardless of the type of cytokine stimulation, had a homogenous phenotype co-expressing CD56high and CD16high along with the activating NK cell receptor NKG2D and the (co)stimulatory receptor 2B4 (CD244) while lacking expression of the terminal differentiation marker CD57 (). Thus, stimulation of PBMC with K562-mb15-41BBL and low-dose IL-2 induces efficient in vitro expansion of NK cells with a highly activated phenotype. Additional cytokine support with IL-12 and IL-18 does not affect NK cell expansion or phenotype under these conditions.
Figure 1. In vitro expansion and phenotype of NK cells activated with K562-mb15-41BBL stimulator cells and either IL-2 alone or a cytokine cocktail of IL-2, IL-12, and IL-18. (A) Absolute NK cell numbers of six individual NK cell lines were determined on day 14 of in vitro expansion by staining viable cells with CD3- and CD56-specific antibodies and calculating %CD3−/CD56+ NK cells × viable cell number. (B) Phenotypes were analyzed by six color-flow cytometry on day 14, determining expression of NKG2D, CD16, CD244, and CD57 within a gate on CD3−/CD56+ NK cells. Shown is one representative example of six individual donors.
In vitro antitumor responses of activated human NK cells against EwS cells
To assess the capacity of EwS cells to interact with NK cells via engagement of the NK cell activation receptors NKG2D and/or DNAM-1, we analyzed the expression of respective ligands in three EwS cell lines, VH-64, DC-ES-6, and MS-PES-4. Although EwS cells express only low levels of the NKG2D ligands MICA, MICB, ULBP1, ULBP2, or ULBP3, all cell lines had intermediate-to-high level expression of the DNAM-1 ligands, CD112 and CD155 (). Thus, EwS cells have the potential to induce activating signals in NK cells via the DNAM-1 pathway. Since NK cell activation can be outbalanced by inhibitory signals via KIRs (killer cell immunoglobulin-like receptors) and further (co)inhibitory NK cell receptors, including CD85j (ILT2, immunoglobulin-like transcript) and PD (programmed cell death protein)-1, we also analyzed the expression of respective ligands in EwS cells. Cells from the three lines expressed surface MHC class I (HLA-ABC), which can engage inhibitory KIRs, at variable densities. Neither the CD85j ligand HLA-G1 nor PD-L1 was detected on the surface of EwS cells.
Figure 2. In vitro activated NK cells can functionally interact with EwS cells. (A) Flow cytometry analysis of EwS tumor cell lines for expression of DNAM-1 (CD112, CD155) and NKG2D (MICA, MICB, ULBP-1, -2, -3) ligands, and HLA-ABC, HLA-G, and PD-L1. Shown is one representative experiment of two. (B) Intracellular IFNγ and TNF-α expression of CD3−/CD56+ NK cells by flow cytometry following 6 h of co-incubation with EwS cell lines, medium alone, or K562 cells (n = 4). (C) Degranulation responses by CD107a staining of CD3−/CD56+ NK cells after 3 h coincubation with EwS cell lines, medium alone, or K562 cells (n = 4). (D) CD25 upregulation by NK cells after 24 h of co-incubation with tumor cells. Shown are triplicates of the two donors. (E) Viability of EwS cell lines within 24 h of co-culture with NK cells. (F) NK cell lysis of VH-64 spheres. Intact VH-64 spheres on days 8 and 9 were transferred into individual wells of 96-well round bottom plates in sphere culture medium, and maximum perpendicular sphere diameters were quantified. Spheres were co-incubated with NK cells at the indicated effector-to-target (E:T) ratios for 24 h, followed by photodocumentation (left panel). After manual dissociation of spheres, the percentages of viable tumor cells were determined by co-staining of cells with fluorescence-labeled CD56, CD99, and CD45 specific mAbs and subsequent flow cytometry quantification of viable cells in the CD99+CD56−CD45− tumor cell gate. Shown are triplicates of one representative experiment of three.
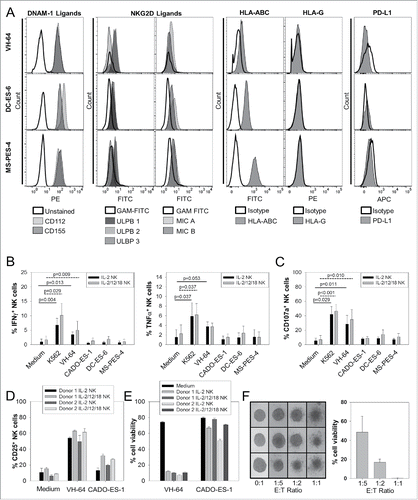
Next, we compared NK cell responses to individual EwS cell lines in vitro by quantifying the intracellular expression of the cytokines IFNγ and TNF-α, and upregulation of CD107a, a marker of degranulation and prerequisite for perforin-mediated toxicity. The NK-cell sensitive leukemia cell line K562 was used as a positive control. The EwS cell line VH-64 induced significant cytokine secretion (), degranulation (), and CD25 upregulation () by in vitro activated allogeneic NK cells, whereas responses to other EwS targets did not exceed the background. NK cells effectively reduced the viability of VH-64, and, less effectively, CADO-ES-1 EwS cells (). Additional cytokine pre-activation with IL-12 and IL-18 did not noticeably affect NK cell responses against tumor targets (). Consequently, we decided to proceed with NK cells expanded in the presence of IL-2 and membrane-bound IL-15 alone.
Since EwS cells within a multicellular tumor structure in vitro were shown to develop NK cell resistance,Citation6 we further addressed the sensitivity of EwS spheres to lysis by activated NK cells. Intact VH-64 spheres were co-cultured with NK cells from an allogeneic donor for 20–24 h in sphere medium. Cell numbers were derived from sphere diameters by correlation with the corresponding cell numbers.Citation21 At effector-to-target ratios of 1:5–1:1, co-incubation with NK cells resulted in disintegration of VH-64 spheres (). Tumor cytolysis was further reflected by elimination of viable CD99+CD56−CD45− tumor cells from the sphere co-cultures with NK cells (). Thus, activated allogeneic NK cells can effectively lyse both single tumor cells and intact EwS spheres from an individual cell line, but their in vitro activity against several other EwS targets is limited despite tumor surface expression of DNAM-1 receptor ligands.
Expression of second- and third-generation CARs in activated human NK cells can enhance their in vitro antitumor activity in an antigen-dependent manner
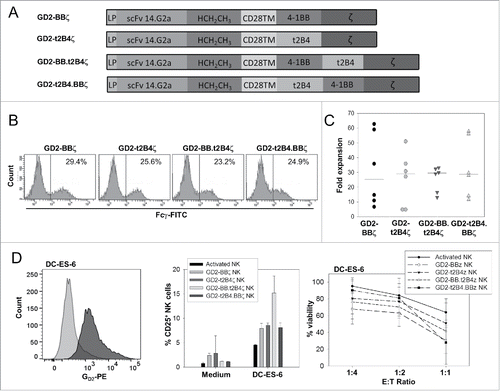
To overcome resistance of EwS cells to targeting by activated NK cells, we engineered allogeneic NK cells to express CARs directed against the surface ganglioside GD2 expressed in neuroblastomas and many EwS.Citation17 We have previously shown that NK cell lysis of neuroblastoma cells can be enhanced by genetic modification with a GD2-specific CAR containing a co-stimulatory domain derived from truncated 2B4 (t2B4).Citation9 To determine whether alternative or additional co-stimulatory signals further enhance the antigen-specific functionality of CAR-engineered NK cells, we generated a second-generation CAR with integrated 4-1BB co-stimulation, as described by Imai et al.,Citation12 and two third-generation CARs containing both co-stimulatory domains, with either 4-1BB or t2B4 upstream of the other (). Retroviral transfer of the individual CAR genes into K562-mb15-41BBL-activated human NK cells resulted in comparable CAR gene expression in >20% of NK cells (), and expansion was not different among NK cells transduced with the individual CARs (). Compared to unmodified activated NK cells, expression of both second- and third-generation CARs noticeably increased the specific activation response to tumor cells from the GD2+ EwS cell line DC-ES-6 as well as the capacity to reduce the viability of DC-ES-6 cells (). CARs with 4-1BB and 2B4 co-stimulatory signaling were equally effective, and third-generation CARs were not functionally superior to CARs with single co-stimulatory domains. We therefore precede with GD2-BBζ CAR-engineered NK cells in the subsequent in vivo experiments.
Figure 3. In vitro activated human NK cells transduced with second- and third-generation CAR genes effectively respond to and lyse target-expressing tumor cells. (A) Schematic of second- and third-generation CAR constructs. (B) CAR surface expression, quantified by staining CAR gene-transduced NK cells with FITC-labeled anti-Fcγ antibody. (C) Absolute NK cell numbers of six individual NK cell lines were determined on day 14 of in vitro expansion by staining viable cells with CD3- and CD56-specific antibodies and calculating %CD3−/CD56+ NK cells × viable cell number. (D) GD2 surface expression of DC-ES-6 cells in comparison to isotype control (left panel, representative experiment of two), CD25 upregulation by NK cells (center panel, means of two donors and triplicate values for each donor) and tumor cell viability (right panel, means of three donors and duplicate values for each donor) after 20–24 h of co-incubation of unmodified activated NK cells and NK cells transduced to express the various GD2-specific CARs with the GD2+ EwS cell line DC-ES-6 or incubation of NK cells with medium alone, as indicated. Significant differences were found between activated NK cells and NK cells transduced with GD2-BBζ at 1:2 (p = 0.021) and 1:4 (p = 0.044) E:T ratios, but not between activated NK cells and NK cells expressing alternative CARs (paired t-test). A comparison between NK cells expressing the various CARs by one-way analysis of variance (ANOVA) did not show any significant differences.
CAR-engineered human NK cells fail to eliminate EwS xenografts in vivo
To investigate the potential of CAR-modified NK cells to prevent growth of EwS xenografts in vivo, we injected 5 × 106 DC-ES-6 cells/mouse subcutaneously (s.c.) into NSG mice. Of 14 xenografted mice, only 7 developed localized tumors, limiting the size of experimental cohorts to 3 mice per cohort. One mouse was left untreated since it had to be sacrificed prior to NK cell transfer due to rapid tumor growth. Mice with palpable tumors received two intratumoral doses of 5 × 106 GD2-BBζ gene-modified human NK cells or unmodified activated NK cells. In contrast to the in vitro findings and against our hypothesis, CAR gene-modified, GD2-specific NK cells failed to control tumor growth in this model (Fig. S1). All mice had to be sacrificed due to uncontrolled tumor growth until day 16. Lack of activity was not explained by immediate clearance of NK cells from the tumor microenvironment, as NK cells remained detectable by flow cytometry for at least 6 d post-NK cell transfer in post-therapeutic tumor autopsies (Fig. S1). Moreover, DC-ES-6 xenografts growing despite the presence of either unmodified activated or GD2-BBζ gene-modified NK cells continued to express the target antigen (Fig. S1); thus, the lack of activity of GD2-redirected NK cells cannot be attributed to the in vivo downregulation of the target antigen.
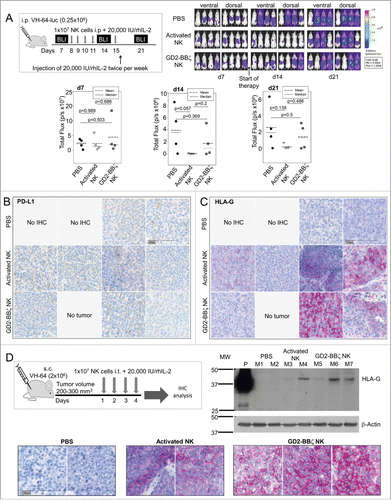
Although the low number of mice per cohort was inadequate to ultimately confirm lack of efficacy of NK cells in vivo, the lack of antitumor activity was so clear that we decided not to reproduce the same experiment with higher numbers of mice. Instead, we further explored NK cell therapy in an alternative EwS model based on intraperitoneal (i.p.) xenografts of the cell line VH-64 that more reliably engrafts in immunodeficient mice. Of 15 NSG mice receiving i.p. injections of 2.5 × 105 VH-64-luc cells/mouse, 12 engrafted (80%). Five i.p. doses of 1 × 107 human GD2-BBζ-engineered NK cells or unmodified activated NK cells or phosphate-buffered saline (PBS) were administered to cohorts of four mice each (). IL-2 was given i.p. twice weekly. Tumor growth was monitored by weekly bioluminescence imaging (BLI). VH-64-luc cells maintained expression of the CAR target antigen GD2 (Fig. S2A), and CAR NK cells used in this experiment were transduced at 43% efficiency (Fig. S2B). BLI on day 14 after tumor cell injection showed marginal tumor responses to both unmodified activated and GD2-BBζ transduced NK cells, but NK cell therapy failed to ultimately control VH-64 tumor growth in vivo, and CAR expression did not increase the activity of activated NK cells against VH-64 xenografts (). We conclude that the in vitro and in vivo activities of CAR-gene-modified NK cells against EwS targets are highly discrepant. GD2-redirected NK cells are unable to effectively eliminate EwS in their microenvironment despite high GD2 target antigen expression.
Figure 4. VH-64 EwS xenografts are resistant to NK cell therapy and respond by upregulation of HLA-G. (A) Experimental design (upper left panel) and bioluminescence imaging of tumors in three groups of four mice each, receiving either GD2-BBζ gene-modified NK cells, unmodified activated NK cells, or PBS alone (upper right panel). The lower panel shows a summary of the bioluminescence intensities in each cohort (dorsal and ventral intensities were combined) on day 7 (prior to therapy) and days 14 and 21 post-therapy. (B, C) Expression of PD-L1 (B) and HLA-G (C) in FFPE tumor tissue sections obtained on day 29 post-therapy from mice receiving either PBS alone, or either unmodified activated or GD2-BBζ-transduced NK cells, as indicated. Shown is one tumor section of each mouse arranged in the same order as the individual mice are in (A). (D) Experimental design (upper left panel) and HLA-G expression by Western Blot analysis (upper right panel) and by immunohistochemistry analysis (lower panel) of VH-64 tumor xenografts from mice receiving PBS or either unmodified activated or GD2-BBζ CAR NK cells, as indicated. P = Placenta, positive control for HLA-G expression. The lower panel shows one tumor section per mouse.
NK cell therapy of EwS xenografts is associated with upregulation of immune-inhibitory HLA-G
We hypothesized that immune-inhibitory receptor/ligand interactions in the tumor microenvironment may have prevented functional NK cell responses to tumor cells. Therefore, we analyzed the expression of PD-L1 and HLA-G, which can engage inhibitory NK cell receptors, in post-therapeutic tumors. Consistent with the lack of PD-L1 in single cell suspensions of VH-64 cells in vitro (), none of the treated tumor xenografts expressed PD-L1 (). By contrast, we found moderate to high expression of HLA-G in 5 of 7 tumor xenografts growing in mice treated with either unmodified activated or CAR-transduced NK cells, but not in the tumors from control mice (). Thus, VH-64 EwS cells can upregulate HLA-G in response to immune targeting under in vivo growth conditions. Retrospective analysis of autopsies from DC-ES-6 xenografts from the experiment shown in Fig. S1 also demonstrated both lack of PD-L1 and moderate HLA-G expression in all six post-therapeutic tumors but not in the untreated tumor (Fig. S3).
Finally, we analyzed if HLA-G upregulation is inducible by repeated injection of NK cells directly into localized tumors. Xenografts growing after s.c. injection of VH-64 cells were treated with four intratumoral doses of GD2-BBζ gene-modified human NK cells transduced at 71% efficiency (Fig. S4) (n = 3) or unmodified activated NK cells (n = 2), or with PBS alone (n = 2). IL-2 was given intratumorally along with NK cells and also to controls. Immunohistochemical analysis of HLA-G in this model confirmed moderate to strong expression in all VH-64 xenografts treated with NK cells, whereas tumors from control mice remained HLA-G negative (). The exclusive presence of HLA-G protein in xenografts injected with NK cells, and not in control tumors receiving IL-2 alone, was confirmed by Western Blot analysis (, upper right panel).
Together we have shown that both i.p. and localized tumor xenografts from two independent EwS cell lines respond to NK cell therapy by upregulation of HLA-G.
EwS cells can induce CD85j receptor upregulation in NK cells and functionally suppress NK cell responses via HLA-G
We hypothesized that HLA-G upregulation in response to infiltration by activated NK cells is a consequence of resulting inflammatory immune response in the tumor microenvironment. To mimic inflammatory conditions in vitro, we stimulated both VH-64 and DC-ES-6 cells with IFNγ and studied upregulation of HLA. As expected, both cell lines upregulated classical HLA molecules (MHC class I and HLA-ABC) in response to incubation with IFNγ (). HLA-G1 expression remained negative by flow cytometry (), whereas the choriocarcinoma cell line JEG-3 clearly expressed HLA-G (Fig. S5). By contrast, Western Blot analysis of protein lysates from VH-64 and DC-ES-6 cells, which allows analysis of all HLA-G isoforms beyond HLA-G1, showed induction of HLA-G in IFNγ stimulated cells (). Since the only HLA-G antibody validated for flow cytometry is restricted to the detection of the HLA-G1 isoform bound to β2-microglobulin, flow cytometry staining could fail to detect surface HLA-G expression. Moreover, HLA-G expression in EwS could be limited to secreted isoforms detectable by Western Blot analysis but not by flow cytometry. As a first step to understanding whether activated NK cells can functionally respond to HLA-G, we analyzed the expression of the NK-cell inhibitory HLA-G receptor CD85j (ILT2) in both activated unmodified and GD2.BBζ transduced NK cells. CD85j was indeed expressed on a proportion of NK cells, with noticeably higher expression on GD2-BBζ gene-transduced than non-transduced activated NK cells directly compared in four donors without additional IL-2 (median 25.1%, range 16.7–33.8% versus median 17.6%, range 11.7–24.7%; p = 0.025). In a preincubation assay containing 500 IU IL-2/mL, EwS cells led to further upregulation of CD85j in both unmodified and GD2-BBζ transduced NK cells (). These in vitro experiments support our hypothesis that adoptively transferred NK cells can interact with EwS xenografts via HLA-G.
Figure 5. NK cells and EwS cells can interact via HLA-G. (A, B) Flow cytometric analysis of HLA-ABC (A) and HLA-G1 (B) surface expression by untreated and IFNγ treated VH-64 and DC-ES-6 EwS cells. Shown is one representative experiment of two. (C) Total HLA-G protein expression by Western Blot analysis of untreated and IFNγ treated VH-64 and DC-ES-6 EwS cells. Shown is one representative experiment of two. JEG-3 cells served as a HLA-G positive control. (D) Flow cytometric analysis of HLA-G receptor CD85j (ILT2) expression on unmodified activated and GD2-BBζ transduced NK cells after 5 d of co-incubation with VH-64 or DC-ES-6 EwS cells in medium containing 500 IU IL-2/mL, or in medium containing 500 IU IL-2/mL alone. Analyzed were four NK cell lines from four healthy donors (circle = donor 1, triangle = donor 2, diamond = donor 3, square = donor 4). Gates were set on CD3−/CD56+/living (7AAD−) cells, with a subsequent gate on Fcγ− (non-transduced) or Fcγ+ (GD2-BBζ) cells, as indicated, and p-values were derived with a paired t-test (n.t., not tested). (E) Apoptotic cell death of HLA-G1 and mock-transduced tumor cells co-incubated with allogeneic VPD450-labeled NK cells derived from five healthy donors by 7AAD staining (left panel, VH-64; right panel WE-68). The gate was set on VPD450− cells, and p-values were derived with a Mann–Whitney U test where test of normality has failed (VH-64 donor 5), or in cases of normal distribution with the t-test (all other datasets).
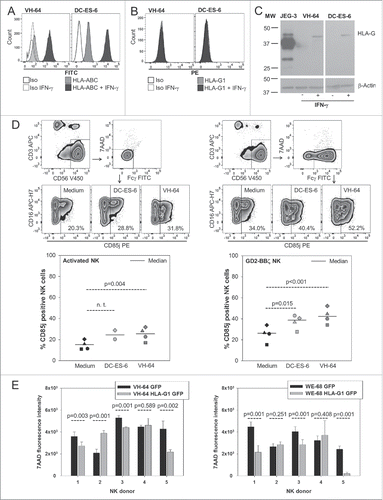
To study the general relevance of HLA-G interactions with NK cells, HLA-G isoform G1 was stably introduced into the EwS cell lines VH-64 and WE-68, another NK-cell susceptible EwS cell line,Citation22 by retroviral gene transfer. HLA-G1 was detected at high densities on the tumor cell surface of transduced EwS cells (Fig. S6). When HLA-G1-negative mock-transduced control EwS cells were co-cultured with freshly isolated allogeneic NK cells, tumor cell apoptosis was observed in all EwS cell lines. HLA-G1 surface expression reduced EwS cell apoptosis by the same NK cells in three of five NK cell donors (), with consistent results across the two cell lines. Non-responders in this assay are most likely explained by variable activating and inhibitory NK cell receptor interactions contributing to individual alloreactivity. Thus, HLA-G upregulation by EwS cells, as found in response to NK cell therapy in our in vivo models, can contribute to NK cell resistance of EwS.
Discussion
Our rationale for using activated NK cells in combination with CARs was to simultaneously overcome the variable NK cell susceptibility of EwS cells and the limited and often heterogenous expression of the CAR target GD2 in this cancer.
We have incorporated several levels of optimization, all aiming to enhance the potency of therapeutic NK cells. First, we have extended the cytokine support during in vitro expansion of NK cells. Romee et al. found that culture of human NK cells with a cytokine cocktail including IL-12 and IL-18 or IL-15 in addition to IL-2 supports expansion of an NK cell population with enhanced functionality.Citation20 The Campana method of NK cell expansion that we used relies on a strong cell-based stimulus that includes IL-15 stimulation along with IL-2.Citation12 Adding IL-18 and IL-12 did not affect NK cell phenotype and in vitro function. Thus, these cytokines are unlikely to improve the functional properties of NK cells generated with this method after adoptive transfer. Second, to enhance the quality and strength of the activation signal induced by engagement of CARs, we have modified their intracellular signaling domains. The T cell receptor (TCR) CD3ζ chain provides a strong activation signal to NK cells,Citation9,12, Citation13 but additional co-stimulatory signals, which are vital to optimal CAR-induced activation of T cells, can improve CAR function also in NK cells. Imai et al. found that in vitro NK cell lysis of leukemia cells was substantially improved by adding the 4-1BB co-stimulatory signaling domain to an anti-CD19-CD3ζ CAR.Citation12 Our own group reported that signaling via lymphocyte activation molecule-related receptor 2B4 (CD244) along with a CAR-induced CD3ζ signal enhances NK cell activation responses to leukemia and neuroblastoma cells.Citation9 In T cells, combined signals from two co-stimulatory molecules have further augmented CAR-mediated T cell activation in some models.Citation23,24 Such third-generation CARs have not yet been evaluated in NK cells. Here, two second-generation CARs as well as two third-generation CAR designs integrating both types of co-stimulation were all equally effective to overcome resistance of GD2+ tumor cell targets against lysis by NK cells and to induce robust NK cell activation responses to EwS cells in vitro. Since dual co-stimulation bears a risk of overstimulation with accelerated terminal maturation and activation-induced cell death,Citation25 we chose the second-generation CAR design for translational development.
We report highly discrepant antitumor activities of CAR-engineered NK cells against EwS in both in vitro and in vivo tumor models. Although expression of GD2-specific CARs was effective to enhance NK cell responses to EwS cells in vitro in a tumor-antigen specific manner, CAR-redirected NK cells were unable to prevent or reduce tumor growth in two xenograft models. Similar failures to translate in vitro tumor lysis by NK cells into effective disease control in animal models or in first clinical studies have been previously described.Citation26,27 Clearly, EwS and other solid tumors can evade immune attack by NK cells in vivo.
A potential explanation for the lack of in vivo activity of NK cells is their functional inhibition by immunosuppressive ligands in the tumor microenvironment. PD-1 pathway inhibition was a key obstacle to CAR T cell therapy of solid tumors in preclinical models.Citation26,27 NK cells can also induce PD-L1 upregulation on tumor cells, and this results in a substantial reduction of their capacity to lyse tumor cells.Citation28 In our study, none of the NK-cell treated xenograft tumors expressed PD-L1, arguing against the hypothesis that the PD-L1/PD-1 immune checkpoint is involved in EwS immune escape from NK cell recognition in our model. Instead, we suggest that EwS evade NK cell targeting in vivo by an increased expression of immunosuppressive HLA molecules. Tumor immune editing by NK cells via HLA molecules was previously described by Balsamo et al.Citation29. In an in vitro co-culture model, melanoma cells were found to acquire resistance to NK-cell mediated lysis by upregulating both classical and non-classical HLA class I molecules by an IFNγ-mediated mechanism. Since IFNγ increases HLA class I expression also in EwS, interactions with inhibitory KIRs very likely have contributed to the observed immune resistance to NK cells in vivo. Here, we focus on the non-classical MHC class I molecule HLA-G and provide evidence that this molecule also has a relevant role in NK cell immune escape of EwS cells.
We detected HLA-G in EwS xenografts from two individual cell lines that had failed to respond to the treatment with activated NK cells, in both a subcutaneous and an intraperitoneal model, but not in untreated tumors. The observation that CAR expression by NK cells was not an essential prerequisite for induction of HLA-G is explained by the activity of NK cells against EwS cells even without genetic modification. HLA-G expression in our models is not caused by exogenous IL-2 stimulation since control mice in the s.c. model received intratumoral IL-2 analogous to the treatment cohort and still failed to upregulate HLA-G. Instead, we hypothesize that HLA-G upregulation is a consequence of the activation response of adoptively transferred NK cells to EwS cells in their microenvironment. This is supported by our findings that (i) NK cells can respond to interaction with EwS cells with IFNγ cytokine release, (ii) prestimulation with IFNγ upregulates HLA-G, (iii) substantial proportions of GD2-BBζ gene-modified NK cells express the HLA-G receptor CD85j (ILT2), (iv) CD85j expression in NK cells is further upregulated upon co-incubation with EwS cells, resulting in up to >50% of NK cells expressing CD85j, and (v) EwS gene-modified to express HLA-G1 can have reduced apoptotic responses to NK cells.
HLA-G is a strong negative regulator of both allogeneic and autologous immune responses.Citation30-34 In various cancers, HLA-G expression was associated with high risk parameters and even predictive of outcome,Citation35-39 indicating a role of this molecule in tumor immune escape. Expression of HLA-G in EwS biopsies from patients with individual disease manifestations and outcomes has not yet been studied. Indirect support for a potential relevance of HLA-G in EwS is its physiological role in immune tolerance of mesenchymal stem/stroma cellsCitation40,41 from which EwS is thought to originate.Citation42 Our findings add evidence that upregulation of HLA-G may be a potent mechanism by which EwS cells escape from the patient's immune system and resist therapeutic immune targeting.
The question remains how this finding could translate into more effective CAR NK cell therapy of EwS. Blockade of HLA-G or its receptor may not completely restore NK cell sensitivity to CAR targets since classical HLA antigens and further negative regulators of NK cell function contribute to NK cell resistance and may keep the balance toward functional inhibition. Understanding the detailed mechanism by which NK cell interaction with tumor cells leads to HLA-G expression may provide clues to effective intervention. Although we show that IFNγ cytokine stimulation can induce production of HLA-G in EwS cells, we do not yet know which cytokines mediate HLA-G upregulation in the tumor microenvironment under in vivo conditions. Although the pattern of staining observed in the tissue sections argues against intracellular retention of HLA-G, future experiments will have to exclude this possibility that would be incompatible with our hypothesis that HLA-G in our model interacts with immune effector cells. We further need to understand in detail which HLA-G isoforms are upregulated in EwS. Besides membrane-bound HLA-G, soluble HLA-G, which is secreted or shedded by either tumor cells or myeloid bystander cells, may contribute to HLA-G mediated immune escape, as shown in neuroblastoma.Citation43
Importantly, beyond the scope of NK cell targeting, HLA-G upregulation in EwS and other solid cancers could be an important barrier also to alternative cellular immunotherapies. We suggest to include non-classical HLA molecules into analyses of inhibitory immune checkpoints in scenarios where antitumor immune therapies fail. An example could be CAR T cell targeting of solid tumors that so far has been less successful than of lymphoid malignancies.Citation44,45 To ultimately clarify the relevance of HLA-G in adoptive cell therapy of EwS, experiments with downregulated HLA-G by antibody blockade or gene editing are needed.
In conclusion, effective NK cell therapy of EwS remains a challenge. Even antigen-specific NK cell activation by highly potent second- and third-generation CARs can fail to overcome resistance of tumor cells within their in vivo microenvironment. Strategies that modulate the tumor niche by overcoming the relevant immune-inhibitory pathways will be needed to prevent functional disability of adoptively transferred NK cells.
Materials and methods
Cell lines
K562 (ATCC) is a human erythroleukemia line that is sensitive to lysis by NK cells. JEG-3 is a choriocarcinoma cell line. The EwS cell lines VH-64, CADO-ES-1, and WE-68Citation46 were gifts from the Frans van Valen Laboratory, Institute of Experimental Orthopedics, University of Muenster, Germany. MS-PES-4 and DC-ES-6 were generated in our own institution.Citation21 The identity of the cell lines was confirmed by short tandem repeat (STR) profiling (Table S1). For standard adherent growth, tumor cells were cultured in collagen-coated 25 or 75 cm2 tissue culture flasks (VH-64, DC-ES-6, MS-PES-4, CADO-ES-1, and WE-68) or in uncoated flasks (K562) in RPMI 1640 medium (Invitrogen, 21875-034), supplemented with 10% heat-inactivated fetal calf serum (FCS; Thermo Fisher, 10270-106) and 2 mM L-glutamine, and maintained at 37 °C and 5% CO2. For some experiments, VH-64 and WE-68 cells were transduced with the HLA-G1 gene (NCBI NM_002127.5) upstream of an IRES site and the eGFP gene using the retroviral vector SFG. HEK293T cells were co-transfected with SFG/HLA-G1.eGFP and helper plasmids containing gagpol and RD114 sequences. Retroviral supernatant harvested after 48 h and enriched by centrifugation was used to transduce the tumor cells in 24-well plates coated with recombinant fibronectin fragment FN CH-296Citation47 (Retronectin, Takara Shuzo, T100B-TK) by 48 h co-incubation. For in vivo experiments, VH-64 cells were lentivirally transduced to express an eGFP-luciferase fusion protein as described elsewhere.Citation48 VH-64 sphere cultures were obtained by seeding cells from monolayer cultures at 2,000 cells/well (1 cell/μL, cell lines) in Ultra-low attachment six-well plates (Costar Corning, 3741) in a total volume of 2 mL. The serum-free sphere culture medium consisted of DMEM/F12 (1:1) (Gibco, 11320033) supplemented with 4% B27 (Invitrogen, 17504-044), 20 ng/mL recombinant human epidermal growth factor (rhEGF) (Gibco, PH60311), 20 ng/mL leukemia inhibitory factor (LIF; 20 ng/mL) (Sigma-Aldrich, L5283), and 10 IE/mL (5 μg/mL) heparin (Roche). rhEGF was added once on day 4.
Expansion of human NK cells
Peripheral blood samples were obtained from 17 buffy coats (gender and age not provided) obtained from the local blood bank and 5 healthy donors (3 females, 2 males, mean age 36 y) upon consent. Heparinized or EDTA whole blood was collected from buffy coat or healthy donors by venipuncture under a procedure approved by our Institutional Review Board. It was stored at room temperature (RT) prior to isolation of PBMC via Ficoll density gradient separation (Ficoll-Plaque). Processing was completed within 4 h for all sample specimens. Isolated PBMCs were washed once with RPMI medium and counted using trypan blue staining and a microscope. The median PBMC number obtained from buffy coats was 8.1 Mio cells/mL blood, and from peripheral blood of healthy donors was 2.6 Mio cells/mL blood. The median cell viability was not determined. For the expansion of human NK cells, freshly isolated PBMC were seeded at 1.5 × 106/well in 24-well tissue-culture plates and stimulated once with irradiated (120 Gy) K562-mb15-41BBL stimulator cells (0.5 × 106/well) in a total volume of 2 mL. Citation12. Culture medium during stimulation consisted of RPMI 1640 or equal portions of RPMI 1640 and AIM V® (Invitrogen, 12055-083) with 10% FCS, supplemented with 40 IU/mL rhIL-2 (Proleukin, Novartis Pharma, 730523), or with a three-cytokine cocktail of rhIL-2 (40 IU/mL), rhIL-12 (10 ng/mL; Immunotools, 11349123), and rhIL-18 (50 ng/mL; MBL, B001-5). After 48 h, 1 mL of the medium was exchanged with 1 mL medium containing rhIL-2 (40 IU/mL). For in vivo experiments, 10 × 106 activated NK cells were super-expanded in gas-permeable culture devices with 500 mL capacity (Wilson Wolf Manufacturing, 80500S-3) in 400 mL of medium and restimulated with 20 × 106 K562-mb15-41BBL cells and rhIL-2 at final concentration of 40 IU/mL for another 6 d. For in vitro experiments, 5 × 106 activated NK cells were transferred to gas-permeable culture devices with 50 mL capacity (Wilson Wolf Manufacturing, 80040S) in 40 mL of medium and restimulated with 10 × 106 K562-mb15-41BBL cells and rhIL-2 at a final concentration of 40 IU/mL. For retroviral transductions, NK cells on day 5 were transferred to 24-well non-tissue culture-treated plates coated with Retronectin and co-incubated with viral supernatant for 48 h, as described.Citation9 All functional assays were done in RPMI medium supplemented with 10% heat-inactivated FCS, 2 mM L-glutamine and 40 IU IL-2/mL if not indicated otherwise, and maintained at 37 °C and 5% CO2. FCS was from a single lot selected for optimal NK cell expansion and functionality.
General lab operation
All work was performed using general research investigative, but pretested assays. The assays were performed by experienced individuals throughout the course of the study. This study was performed using established laboratory protocols covering the processing, freezing, storage, and thawing of cells as well as the staining procedure, data acquisition, and gating strategy. Response was determined after data analysis with reference to the applied controls. Raw data can be provided per request.
Constructs
All GD2-specific CARs contain the single-chain antibody domain (scFv) of the monoclonal antibody (mAb) 14.G2a.Citation49 14.G2a-t2B4ζ contains a transmembrane and a truncated cytoplasmic domain of the co-stimulatory receptor 2B4 upstream of the cytoplasmic domain of the TCR ζ gene and was previously described.Citation9 To generate second- and third-generation CAR gene constructs, we recloned the CAR by inserting restriction sites that allow to exchange transmembrane and (co)stimulatory signaling domains. First, we replaced the transmembrane domain with the one derived from CD28 resulting in a new construct named GD2-t2B4ζ. Next, we generated an alternative CAR, GD2-BBζ, by exchanging the gene fragment encoding for the cytoplasmic t2B4 domain by the respective signaling domain derived from the co-stimulatory receptor 4-1BB. To generate GD2-BB.t2B4ζ, the cytoplasmic signaling domain of t2B4 was cloned upstream of the ζ chain gene into GD2-BBζ, and to obtain GD2-t2B4.BBζ, the cytoplasmic domain of 4-1BB was cloned upstream of TCRζ into GD2-t2B4ζ. All CAR genes were codon-optimized, and then subcloned into the AgeI and XhoI sites of the retroviral vector SFG.Citation9 Generation of stable retroviral producer cell lines and production of recombinant retrovirus for transduction of NK cells was performed as described.Citation9
Flow cytometry
Tumor cells were stained with fluorescein-conjugated mAbs (BioLegend) against CD112 (clone TX31, 337410), CD155 (clone SKII-4, 337610), and GD2 (clone 14.G2a, 357304). NKG2D ligand expression was analyzed using unconjugated antibodies (all from BAMOMAB) against MICA (clone AMO1, AMO1-100), MICB (clone BMO1, BMO1-100), ULBP1 (clone AUMO2, AUMO2-100), ULBP2 (clone BUMO1, BUMO1-100), and ULBP3 (clone CUMO3, CUMO3-100) and APC-labeled goat-anti-mouse-IgG (405308, BioLegend). HLA-G1 expression was detected with MEM-G/9 mAb (Exbio, 11-292-C100), HLA-ABC with clone W6/32 (MCA81F, Serotec), and PD-L1 expression was detected with clone 5H1 (kindly provided by Lieping Chen, Yale School of Medicine, New Haven, USA). NK cells were characterized using mAbs from BD Pharmingen against CD3 (clone SK-7, 557851), CD45 (clone HI30, 555483), CD56 (clone HCD-56, 345811), CD314 (NKG2D, clone 1D11, 558071), CD57 (clone NK-1, 560844) CD226 (DNAM-1, clone DX-11, 559789), from eBioscience against CD244 (clone eBioC1-7, 13-5838-82) and CD85j (ILT2, clone HP-F1, 12-5129-42), and from BioLegend against CD279 (PD-1, clone EH12.2H7, 329905). Surface expression of CARs was determined with a biotinylated goat anti-mouse mAb specific for IgG F(ab’)2 fragment (Dianova, 115-065-072) and secondary phycoerythrin (PE)-labeled streptavidin antibody (BD Pharmingen, 349023), or alternatively with a fluorescein isothiocyanate (FITC)-labeled goat-anti-human Fcγ Ab recognizing the hinge domain (Jackson ImmunoResearch, 109-095-098). All antibodies were titrated and tested on known positive and negative cells prior to use, and all samples were either directly acquired or for fixed samples not later than 24 h after staining. For each sample, at least 10,000 cells were analyzed within the respective gates with FACS Calibur and BD Cell Quest Software or with FACS Canto and FACS Diva Software (BD Biosciences), subsequent analysis and figure creation was done with FlowJo. For each experiment, PMT voltages and compensation were adjusted using unstained cells and stained BD CompBeads (552843). Analysis was done on single cells and gates were set as mentioned in the respective figure legends.
Intracellular cytokine assay
3 × 105 NK cells (day 14 after activation) and 3 × 105 target cells were seeded in tubes in a total volume of 300 μL medium and co-incubated for 2 h at 37 °C and 5% CO2. To block cytokine secretion, 10 μg Brefeldin A in 40 μL PBS (Sigma-Aldrich, B7651-5MG) was added after 2 h. After washing with PBS+0.5% BSA, CD3- and CD56-specific antibodies were added in 100 μL of PBS+0.5% BSA and incubated for 15 min at RT. Following another washing step, cells were incubated for 10 min at RT with 1 mL FACS Lysing Solution (BD Biosciences, 349202), diluted 1:10 in dH2O, then washed and treated for another 10 min at RT with 500 μL of FACS Permeabilizing Solution (BD, 347692) diluted 1:10 in dH2O. Cells were washed again, resuspended in 100 μL of PBS+0.5% BSA containing the antibodies specific for intracellular IFNγ and TNF-α, and incubated for another 30 min at RT. After a final washing step, cells were fixed in 250 μL of 1% paraformaldehyde (PFA) prior to FACS analysis. Co-incubation of NK cells in medium served as a background control, whereas co-incubation with K562 cells served as a positive control.
CD25 activation assay
NK cells (1 × 105/well) were incubated with an equal number of target cells in a total volume of 200 μL in a 96-well round bottom plate at 37 °C and 5% CO2. After 24 h, the cells were stained with CD3-, CD56-, and CD25-specific antibodies (clone M-A251, 555432 BD Pharmingen) in the experimental wells for 15 min, then transferred into FACS tubes, fixed in 1% PFA, and analyzed by flow cytometry.
CD107a degranulation assay
NK cells (1 × 105/well) were incubated with an equal number of target cells in a total volume of 200 μL in eppendorf tubes in the presence of Monensin (eBioscience, 00-4505-51) (1 μL/mL) and CD107a-PE antibody (BioLegend, 328608) (100 ng/mL) for 3 h at 37 °C and 5% CO2. The cells were washed, then incubated with CD3- and CD56-specific antibodies for 15 min, fixed in 1% PFA, and analyzed by flow cytometry. Co-incubation of NK cells in medium served as a background control, whereas co-incubation with K562 cells served as a positive control.
CD85j (ILT2) upregulation
1 × 106 VH-64 and DC-ES-6 tumor cells, respectively, were seeded in 2 mL medium into one well per cell line of a collagen-coated tissue culture six-well plate and grown to 100% confluence for 48 h at 37 °C, 5% CO2. On day 3, the medium was removed and 4.5 × 105 NK cells (on day 14 after activation) in 1.5 mL medium with 500 IU rhIL-2/mL were added to each well. After co-incubation for 48 h, the NK cells were harvested, washed by centrifugation, resuspended in 1.5 mL fresh medium with 500 IU rhIL-2/mL, and added to newly grown EwS monolayers as described above. After co-incubation for additional 72 h, NK cells were harvested and stained with monoclonal antibodies against CD3, CD56, CD16, CD85j, and goat-anti-human Fcγ (to allow gating on CAR-expressing NK cells). Viability was determined by exclusion of 7AAD+ cells after incubation with 150 ng 7AAD for 10 min.
IFNγ stimulation assay
VH-64 and DC-ES-6 cells were grown to 30% confluence in 25 cm2 tissue culture flasks. The cell culture medium was changed with 5 mL RPMI 10% FCS and 2 mM L-Glutamine without phenol red containing 500 IU rhIFNγ/mL. After 48 h (VH-64) to 72 h (DC-ES-6), cells were either collected directly in 100 μL RIPA buffer for Western Blot analysis, or collected and washed once in PBS 0.5% BSA followed by staining with antibodies for HLA-G1 (clone MEM-G/9), HLA-ABC (clone W6/32) and the respective isotypes (clone X40, 345816, BD Pharmingen and clone PPV-04, 21275523, Immunotools).
Viability assays
NK cells were labeled with Violet Proliferation Dye 450 according to the manufacturer's recommendations (BD, 562158) and then co-cultured with target cells for 20 h in ultra-low attachment plates. After staining with 150 ng 7AAD (BD, 559925) for 10 min, viable tumor cells were quantified by flow cytometry by gating on VPD450- and 7AAD-negative cells. For analysis of HLA-G suppression, freshly isolated NK cells from five healthy donors (two females, three males, mean age 31 y) were obtained from heparinized or EDTA whole blood by Ficoll density gradient centrifugation. NK cells were isolated by magnetic untouched separation according to the manufacturer's recommendation (Miltenyi Biotec, 130-092-657). NK cells were VPD450 labeled and co-incubated with EwS cells for 20–24 h in ultra-low attachment 96-well plates with an effector to target ratio of 1:1 (1 × 105 NK cells per 1 × 105 EwS cells in a total volume of 200 μL medium). 7AAD intensity was measured by gating on VPD450 negative and 7AAD positive cells. To assess the sensitivity of intact spheres to NK cell lysis, spheres on day 8 or 9 were transferred into individual wells of 96-well ultra-low attachment plates (Costar Corning, CLS7007) in sphere culture medium. For each experiment, nine similarly sized spheres were selected and co-incubated with NK cells at various effector-to-target ratios for 20–24 h. Triplicates of three pooled spheres each were used for analysis. Spheres were manually dissociated in an enzyme-free solution containing 1 mM EDTA, 40 mM Tris-HCl, and 150 mM NaCl, and viable CD99+ EwS cells were quantified by co-staining with fluorescence-labeled mAbs against CD56, CD99 (clone HCD99 318008, BioLegend), and CD45 and subsequently analyzed by flow cytometry.
Mouse model
Mouse experiments were approved by the animal care committee of the local government (Bezirksregierung Muenster, Az. 87-51.04.2010.A117). Eight- to twelve-week-old NSG mice (The Jackson Laboratory, 005557) were i.p. injected with 0.25 × 106 luciferase-expressing VH-64 (VH-64-luc) cells per mouse. Five consecutive doses of 1 × 107 GD2-BBζ, CAR transduced or non-transduced NK cells were administered i.p. on days 8, 9, 10, 11, and 15, along with 20,000 IU rhIL-2. The NK cell dose was chosen on the basis of a previously reported model for the use of activated NK cells to treat i.p. EwS xenografts.Citation2 Additional 20,000 IU rhIL-2 were given i.p. twice weekly starting on day 15 after tumor inoculation. EwS engraftment and growth were monitored weekly prior to NK cell administration and on days 7, 14, and 21 by BLI using an IVIS Spectrum Imaging System (PerkinElmer). Mice were injected i.p. with D-luciferin (150 mg/kg; Synchem OHG, s039). About 3 min after injection, mice were anesthetized with isoflurane (2% isoflurane, 0.5 L/min oxygen), and images were acquired after 6 min in a dorsal and a ventral position (f/stop: 1, binning: 4, exposure time: automatic). Photon emission from luciferase-expressing cells was quantified using the Living Image 4.2 software (Caliper Life Sciences). Grayscale photographic images and bioluminescence color images were superimposed. Regions of interest (ROI) were drawn over the torso of the mouse to determine the signal intensity emitted by the tumors (total flux, p/s). 24–29 d post-therapy, mice were sacrificed and the tumors used for histopathological analysis.
For s.c. tumor growth, 2 × 106 VH-64 cells or 5 × 106 DC-ES-6 cells, respectively, were injected s.c. into the right flanks of NSG mice. Upon a tumor volume of 200–300 mm3, 5 × 106 (DC-ES-6) or 1 × 107 (VH-64) GD2-BBζ CAR transduced or non-transduced activated NK cells were injected intratumorally along with 20,000 IU rhIL-2 on days 3 and 7 (DC-ES-6) or on four consecutive days (VH-64). A lower dose of NK cells than for i.p. treatment was used on the basis of the rationale that direct intratumoral application bypasses the need for tumor homing. NK cells from single cultures and individual donors per experiment were used to ensure intra-experimental comparability, and were administered freshly to avoid any impairment of viability or cell loss due to freezing. Tumor growth was monitored by caliper quantification of diameters. Mice were sacrificed and the tumors used for histopathological analysis as indicated in the respective figure legends.
Isolation of NK cells from xenografts
Tumor sections from xenografts were placed in a 100 μm nylon cell strainer (BD Falcon, 352360) and mashed with the end of the plunger from a 5 mL syringe (BD, 309050). After adding 1 mL PBS, the homogenized cell suspension was transferred into a universal tube (BD Falcon, 352070) and centrifuged for 5 min. The cell pellet was resuspended in 1–2 mL of Erythrocyte Lysis Buffer (Qiagen, 79217) and incubated for 5–10 min at RT. After another centrifugation, the pellet was resuspended in 200 μL PBS+0.5% BSA followed by staining with anti-CD45 antibody for 10 min at RT. The cells were washed, resuspended in 300 μL 1% PFA, and analyzed by flow cytometry.
Immunohistochemistry and immunofluorescence analysis
Formalin-fixed paraffin-embedded (FFPE) sections were stained with the HLA-G specific mAb clone 4H84 (Exbio, 11-499-C100), which detects all HLA-G isoforms, or the PD-L1-specific mAb clone E1L3N (NEB, 13684) using either Autostainer (Dako, 3400-9580-03) or Bench Mark Ultrastainer (Ventana, N750-BMKU-FS). Immunoreactions were visualized with a biotinylated secondary antibody (Dako, E0432, E0433) including the Red chromogen for HLA-G or Peroxidase/DAB for PD-L1 detection, according to the manufacturer. Slides were counterstained with hematoxylin and covered with Cytoseal (Thermo Scientific). GD2 expression was analyzed on 4–5 μm cryostat-frozen tumor sections from xenografts fixed in 2% paraformaldehyde (Sigma-Aldrich, P6148-500G) for 10 min, then permeabilized for 3 h in PBS (Sigma-Aldrich, D8537-500ML) containing 0.075% Tween (Sigma-Aldrich, P9416-100ML) and 1% bovine serum albumin (Roth, 8076.2) at RT. After washing with PBS (3 × 5 min) sections were incubated overnight at 4 °C with unconjugated 14.G2a mAb (BD Pharmingen, 554272), diluted 1:50 in PBS supplemented with 0.01% Tween. After washing with PBS sections were incubated with FITC-conjugated goat-anti-mouse antibody (BD Pharmingen, 554001) diluted 1:500 for 1 h at RT. After another washing step, sections were covered with Vectashield mounting medium containing DAPI (Vector, H-1200) and kept at 4 °C until analyzed.
Western Blot analysis
Tumor xenograft tissues or cultured cells were homogenized in 100 μL ice-cold RIPA-buffer (0.1% DTT (Sigma-Aldrich, 43815-1G) with fresh protease inhibitor cocktail (Roche, 11873580001), shortly fractured in liquid nitrogen, thawed on ice, and then clarified by spinning for 15 min at 4 °C and 20,000g. The protein containing supernatant was separated by electrophoresis in a SDS/10% polyacrylamide gel and then electroblotted onto a nitrocellulose membrane (Bio-Rad). Blocking was done in TBST-buffer (5% nonfat dry milk) for 1 h, followed by incubation with mAb 4H84 diluted 1:1,000 in TBST (5% BSA) for 12 h at 4 °C. After washing, the membrane was incubated with HRP-linked anti-mouse IgG whole Ab (GE Healthcare, NA931) 1:2,000 in TBST 5% milk for 1 h at RT, followed by treatment with enhanced chemiluminescence reagent (ECL Plus Western Blotting Detection Systems, GE Healthcare) and exposed to Hyperfilm ECL film (GE Healthcare) for 1 min. Equal protein loading was determined by Ponceau staining. Equal protein loading was determined by washing the membrane with 20 mL TBST buffer for 10 min at RT, followed by incubation with 7 mL RestoreTM Plus WB stripping buffer (46430, Thermo Scientific) for 15 min at RT. The membrane was then washed twice in 20 mL TBST buffer for 10 min at RT, followed by blocking with TBST (5% nonfat dry milk) for 1 h. Detection of β-actin antibody (4967, Cell Signaling) diluted 1:5,000 in TBST (5% BSA) for 12 h at 4 °C. After washing, the membrane was incubated with HRP-linked anti-rabbit antibody (donkey, NA934V, GE Healthcare) 1:2,000 in TBST 5% milk for 1 h at RT, followed by detection as described above.
Statistics
The paired student's t-test was used with paired samples and the unpaired student's t-test with independent samples to test whether the means in each set of parametric distributed values differ significantly, and the Mann–Whitney U test was used to compare nonparametric mean values. One-way analysis of variance (ANOVA) was performed to compare the means of three or more samples. Test used are indicated in the figure legends and p values of less than 0.05 were considered to indicate statistically significant differences. Statistical analyses were performed using Sigmaplot 11 (Systat Software, San Jose, CA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KONI_A_1250050_supplementarymaterials.zip
Download Zip (6.6 MB)Funding
This work was funded by grant DKH 109566 from Deutsche Krebshilfe (to CR), by faculty funding (IMF, AL1 2 10 05, to BA and RÖ 2 2 1109 to CR and SK), and by funding from “Löwenkinder® Verein zur Unterstützung krebskranker Kinder e.V”.
References
- Ladenstein R, Potschger U, Le Deley MC, Whelan J, Paulussen M, Oberlin O, van den Berg H, Dirksen U, Hjorth L, Michon J et al. Primary disseminated multifocal ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 2010; 28:3284–91; PMID:20547982; http://dx.doi.org/10.1200/JCO.2009.22.9864
- Cho D, Shook DR, Shimasaki N, Chang YH, Fujisaki H, Campana D. Cytotoxicity of activated natural killer cells against pediatric solid tumors. Clin Cancer Res 2010; 16:3901–9; PMID:20542985; http://dx.doi.org/10.1158/1078-0432.CCR-10-0735
- Schlegel P, Feuchtinger T, Nitschke-Gerard C, Seidel UJ, Lang AM, Kyzirakos C, Teltschik HM, Ebinger M, Schumm M, Koscielniak E et al. Favorable NK cell activity after haploidentical hematopoietic stem cell transplantation in stage IV relapsed Ewing's sarcoma patients. Bone Marrow Transpl 2015; 50 Suppl 2:S72–6; PMID:26039213; http://dx.doi.org/10.1038/bmt.2015.100
- Verhoeven DH, de Hooge AS, Mooiman EC, Santos SJ, ten Dam MM, Gelderblom H, Melief CJ, Hogendoorn PC, Egeler RM, van Tol MJ et al. NK cells recognize and lyse Ewing sarcoma cells through NKG2D and DNAM-1 receptor dependent pathways. Mol Immunol 2008; 45:3917–25; PMID:18657862; http://dx.doi.org/10.1016/j.molimm.2008.06.016
- Berghuis D, de Hooge AS, Santos SJ, Horst D, Wiertz EJ, van Eggermond MC, van den Elsen PJ, Taminiau AH, Ottaviano L, Schaefer KL et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: implications for immune recognition. J Pathol 2009; 218:222–31; PMID:19274709; http://dx.doi.org/10.1002/path.2537
- Holmes TD, El Sherbiny YM, Davison A, Clough SL, Blair GE, Cook GP. A human NK cell activation/inhibition threshold allows small changes in the target cell surface phenotype to dramatically alter susceptibility to NK cells. J Immunol 2011; 186:1538–45; PMID:21191066; http://dx.doi.org/10.4049/jimmunol.1000951
- Lang P, Pfeiffer M, Muller I, Schumm M, Ebinger M, Koscielniak E, Feuchtinger T, Foll J, Martin D, Handgretinger R. Haploidentical stem cell transplantation in patients with pediatric solid tumors: preliminary results of a pilot study and analysis of graft versus tumor effects. Klin Padiatr 2006; 218:321–6; PMID:17080334; http://dx.doi.org/10.1055/s-2006-942256
- Perez-Martinez A, de Prada Vicente I, Fernandez L, Gonzalez-Vicent M, Valentin J, Martin R, Maxwell H, Sevilla J, Vicario JL, Diaz MA. Natural killer cells can exert a graft-vs-tumor effect in haploidentical stem cell transplantation for pediatric solid tumors. Exp Hematol 2012; 40:882-91.e1; PMID:22771496; http://dx.doi.org/10.1016/j.exphem.2012.07.004
- Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, Campana D, Juergens H, Pule M, Rossig C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res 2009; 15:4857–66; PMID:19638467; http://dx.doi.org/10.1158/1078-0432.CCR-08-2810
- Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, Peng Y, Mao H, Yi L, Ghoshal K et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014; 28:917–27; PMID:24067492; http://dx.doi.org/10.1038/leu.2013.279
- Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ aggressive B-cell non-Hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res 2015; 3:333–44; PMID:25492700; http://dx.doi.org/10.1158/2326-6066.CIR-14-0114
- Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005; 106:376–83; PMID:15755898; http://dx.doi.org/10.1182/blood-2004-12-4797
- Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, Kiessling R, Blankenstein T, Abken H, Charo J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci USA 2008; 105:17481–6; PMID:18987320; http://dx.doi.org/10.1073/pnas.0804788105
- Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, Bachmann M, Fussel M, Schackert G, Temme A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol 2015; 194:3201–12; PMID:25740942; http://dx.doi.org/10.4049/jimmunol.1400330
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385:517–28; PMID:25319501; http://dx.doi.org/10.1016/S0140-6736(14)61403-3
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507–17; PMID:25317870; http://dx.doi.org/10.1056/NEJMoa1407222
- Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, Titze U, Leuchte K, Landmeier S, Hotfilder M et al. The ganglioside antigen GD2 is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer 2012; 106:1123–33; PMID:22374462; http://dx.doi.org/10.1038/bjc.2012.57
- Fisher JP, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, Anderson J. Effective combination treatment of GD2-expressing neuroblastoma and Ewing's sarcoma using anti-GD2 ch14.18/CHO antibody with Vγ9Vδ2+ γδT cells. Oncoimmunology 2016; 5:e1025194; PMID:26942051; http://dx.doi.org/10.1080/2162402X.2015.1025194
- Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 2015; 75:3505–18; PMID:26330164; http://dx.doi.org/10.1158/0008-5472.CAN-15-0139
- Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, Cooper MA, Fehniger TA. Cytokine activation induces human memory-like NK cells. Blood 2012; 120:4751–60; PMID:22983442; http://dx.doi.org/10.1182/blood-2012-04-419283
- Leuchte K, Altvater B, Hoffschlag S, Potratz J, Meltzer J, Clemens D, Luecke A, Hardes J, Dirksen U, Juergens H et al. Anchorage-independent growth of Ewing sarcoma cells under serum-free conditions is not associated with stem-cell like phenotype and function. Oncol Rep 2014; 32:845–52; PMID:24927333; http://dx.doi.org/10.3892/or.2014.3269
- Mueller SK, Altvater B, Chen C, Kailayangiri S, Ahlmann M, Dirksen U, Juergens H, Rossig C. Zoledronic acid negatively affects the expansion of in vitro activated human NK cells and their cytolytic interactions with Ewing sarcoma cells. Oncol Rep 2013; 29:2348–54; PMID:23525469; http://dx.doi.org/10.3892/or.2013.2350
- Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther 2005; 12:933–41; PMID:15979412; http://dx.doi.org/10.1016/j.ymthe.2005.04.016
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA 2009; 106:3360–5; PMID:19211796; http://dx.doi.org/10.1073/pnas.0813101106
- Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 "super-stimulation". Mol Ther 2013; 21:2268–77; PMID:23985696
- Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 2014; 20:4262–73; PMID:24919573; http://dx.doi.org/10.1158/1078-0432.CCR-13-2627
- John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res 2013; 19:5636–46; PMID:23873688; http://dx.doi.org/10.1158/1078-0432.CCR-13-0458
- Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, Ritz J. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015; 4:e1008824; PMID:26155422; http://dx.doi.org/10.1080/2162402X.2015.1008824
- Balsamo M, Vermi W, Parodi M, Pietra G, Manzini C, Queirolo P, Lonardi S, Augugliaro R, Moretta A, Facchetti F et al. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur J Immunol 2012; 42:1833–42; PMID:22585684; http://dx.doi.org/10.1002/eji.201142179
- Lila N, Rouas-Freiss N, Dausset J, Carpentier A, Carosella ED. Soluble HLA-G protein secreted by allo-specific CD4+ T cells suppresses the allo-proliferative response: a CD4+ T cell regulatory mechanism. Proc Natl Acad Sci USA 2001; 98:12150–5; PMID:11572934; http://dx.doi.org/10.1073/pnas.201407398
- Rajagopalan S, Long EO. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med 1999; 189:1093–100; PMID:10190900; http://dx.doi.org/10.1084/jem.189.7.1093
- Riteau B, Rouas-Freiss N, Menier C, Paul P, Dausset J, Carosella ED. HLA-G2, -G3, and -G4 isoforms expressed as nonmature cell surface glycoproteins inhibit NK and antigen-specific CTL cytolysis. J Immunol 2001; 166:5018–26; PMID:11290782; http://dx.doi.org/10.4049/jimmunol.166.8.5018
- Rouas-Freiss N, Goncalves RM, Menier C, Dausset J, Carosella ED. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc Natl Acad Sci USA 1997; 94:11520–5; PMID:9326642; http://dx.doi.org/10.1073/pnas.94.21.11520
- Wiendl H, Mitsdoerffer M, Schneider D, Melms A, Lochmuller H, Hohlfeld R, Weller M. Muscle fibres and cultured muscle cells express the B7.1/2-related inducible co-stimulatory molecule, ICOSL: implications for the pathogenesis of inflammatory myopathies. Brain : a journal of neurology 2003; 126:1026–35; PMID:12690043; http://dx.doi.org/10.1093/brain/awg114
- Ibrahim EC, Guerra N, Lacombe MJ, Angevin E, Chouaib S, Carosella ED, Caignard A, Paul P. Tumor-specific up-regulation of the nonclassical class I HLA-G antigen expression in renal carcinoma. Cancer Res 2001; 61:6838–45; PMID:11559559
- Polakova K, Bandzuchova E, Sabty FA, Mistrik M, Demitrovicova L, Russ G. Activation of HLA-G expression by 5-aza-2 - deoxycytidine in malignant hematopoetic cells isolated from leukemia patients. Neoplasma 2009; 56:514–20; PMID:19728760; http://dx.doi.org/10.4149/neo_2009_06_514
- Urosevic M, Kurrer MO, Kamarashev J, Mueller B, Weder W, Burg G, Stahel RA, Dummer R, Trojan A. Human leukocyte antigen G up-regulation in lung cancer associates with high-grade histology, human leukocyte antigen class I loss and interleukin-10 production. Am J Pathol 2001; 159:817–24; PMID:11549573; http://dx.doi.org/10.1016/S0002-9440(10)61756-7
- Wiendl H, Mitsdoerffer M, Hofmeister V, Wischhusen J, Bornemann A, Meyermann R, Weiss EH, Melms A, Weller M. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol 2002; 168:4772–80; PMID:11971028; http://dx.doi.org/10.4049/jimmunol.168.9.4772
- Paul P, Rouas-Freiss N, Khalil-Daher I, Moreau P, Riteau B, Le Gal FA, Avril MF, Dausset J, Guillet JG, Carosella ED. HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci USA 1998; 95:4510–5; PMID:9539768; http://dx.doi.org/10.1073/pnas.95.8.4510
- Liu KJ, Wang CJ, Chang CJ, Hu HI, Hsu PJ, Wu YC, Bai CH, Sytwu HK, Yen BL. Surface expression of HLA-G is involved in mediating immunomodulatory effects of placenta-derived multipotent cells (PDMCs) towards natural killer lymphocytes. Cell Transplant 2011; 20:1721–30; PMID:21669042; http://dx.doi.org/10.3727/096368911X580590
- Rizzo R, Lanzoni G, Stignani M, Campioni D, Alviano F, Ricci F, Tazzari PL, Melchiorri L, Scalinci SZ, Cuneo A et al. A simple method for identifying bone marrow mesenchymal stromal cells with a high immunosuppressive potential. Cytotherapy 2011; 13:523–7; PMID:21171826; http://dx.doi.org/10.3109/14653249.2010.542460
- Riggi N, Cironi L, Provero P, Suva ML, Kaloulis K, Garcia-Echeverria C, Hoffmann F, Trumpp A, Stamenkovic I. Development of Ewing's sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res 2005; 65:11459–68; PMID:16357154; http://dx.doi.org/10.1158/0008-5472.CAN-05-1696
- Morandi F, Levreri I, Bocca P, Galleni B, Raffaghello L, Ferrone S, Prigione I, Pistoia V. Human neuroblastoma cells trigger an immunosuppressive program in monocytes by stimulating soluble HLA-G release. Cancer Res 2007; 67:6433–41; PMID:17616704; http://dx.doi.org/10.1158/0008-5472.CAN-06-4588
- Beatty GL, O`Hara MH, Nelson AM, McHGarvey M, Torigian DA, Lacey SF, Melenhorst JJ, Levine BL, Plesa G, Chew A et al. Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. J Clin Oncol 2015; 33:149s; http://dx.doi.org/10.1200/JCO.2014.55.5409
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu EL, Gee AP, Mei Z et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14:1264–70; PMID:18978797; http://dx.doi.org/10.1038/nm.1882
- Ottaviano L, Schaefer KL, Gajewski M, Huckenbeck W, Baldus S, Rogel U, Mackintosh C, de Alava E, Myklebost O, Kresse SH et al. Molecular characterization of commonly used cell lines for bone tumor research: a trans-European EuroBoNet effort. Genes Chromosom Cancer 2010; 49:40–51; PMID:19787792; http://dx.doi.org/10.1002/gcc.20717
- Hanenberg H, Xiao XL, Dilloo D, Hashino K, Kato I, Williams DA. Colocalization of retrovirus and target cells on specific fibronectin fragments increases genetic transduction of mammalian cells. Nat Med 1996; 2:876–82; PMID:8705856
- Corson TW, Samuels BC, Wenzel AA, Geary AJ, Riley AA, McCarthy BP, Hanenberg H, Bailey BJ, Rogers PI, Pollok KE et al. Multimodality imaging methods for assessing retinoblastoma orthotopic xenograft growth and development. PLoS One 2014; 9:e99036; PMID:24901248; http://dx.doi.org/10.1371/journal.pone.0099036
- Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of GD2-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer 2001; 94:228–36; PMID:11668503; http://dx.doi.org/10.1002/ijc.1457