
ABSTRACT
Human pancreatic cancer does not respond to immune check point blockade immunotherapy. One key feature of pancreatic cancer is the association between its progression and chronic inflammation. Emerging evidence supports a key role for the JAK-STAT pathway in pancreatic cancer inflammation. We aimed at testing the hypothesis that sustained JAK-STAT signaling suppresses cytotoxic T lymphocyte (CTL) activation to counteract anti-PD-1 immunotherapy-induced CTL activity in pancreatic cancer. We show that human pancreatic carcinomas express high level of PD-L1 and exhibit low level of CTL infiltration. JAK-STAT inhibitor Ruxolitinib selectively inhibits STAT1 and STAT3 activation and increases CTL infiltration to induce a Tc1/Th1 immune response in the tumor microenvironment in an orthotopic pancreatic cancer mouse model. Ruxilitinib-mediated tumor suppressive efficacy diminishes in T-cell-deficient mice. Pancreatic tumor grows significantly faster in IFNγ-deficient mice. However, neutralizing IFNγ does not alter tumor growth but diminishes Ruxolitinib-induced tumor suppression in vivo, indicating that lymphocytes and IFNγ are essential for Ruxolitinib-induced host antitumor immune response. Both type I and type II interferons upregulate PD-L1 expression through the JAK-STAT signaling pathway in mouse pancreatic tumor cells. Tumor cells respond to activated T cells by activating STAT3. The inhibition of STAT3 downregulates immune suppressive cytokines production by tumor cells, resulting in increased T cell activation and effector function. Consequently, Ruxolitinib significantly improves the efficacy of anti-PD-1 immunotherapy. Our data demonstrate that Ruxolitinib is effective in the inhibition of systemic inflammation in the tumor microenvironment and therefore upregulates CTL infiltration and activation to overcome pancreatic cancer resistance to anti-PD-1 immunotherapy.
Introduction
Although relatively rare, pancreatic cancer is almost a death sentence for patients diagnosed with this disease, as there are few effective treatments. The current standard first-line therapy for patients with metastatic pancreatic cancer includes FOLFIRINOX (oxaliplatin, irinotecan, fluorouracil, and leucovorin), germcitabine, or combined germcitabine and paclitaxel.Citation1-3 These therapies are often highly toxic and thus intolerant in certain patients.Citation3 Furthermore, it is inevitable that some patients develop chemoresistance to these first-line therapies. In addition, there are currently few effective second-line therapies for human pancreatic cancer.Citation4 Anti-PD-1/PD-L1 antibody-based immune check point blockade immunotherapy has shown durable efficacy in many types of human cancers.Citation5-7 However, pancreatic cancer stands out as one of the few human cancer types that show no response to anti-PD-1/PD-L1 immunotherapy.Citation6 The immunological mechanism underlying this non-response in pancreatic cancer is unknown.Citation8-10
Patients with pancreatic cancer typically have a shorter survival time if they show signs of systemic inflammation in the tumor microenvironment.Citation11,12 Emerging clinical data suggest that chronic systemic inflammatory response is associated with poor outcomes in pancreatic cancer patients.Citation13 An elevated systemic inflammation is independently associated with lower overall survival after pancreaticoduodenectomy in pancreatic cancer patients. Additionally, in an adjuvant therapy subgroup of pancreatic patients, an elevated systemic inflammation remains independently associated with reduced overall survival.Citation14 The JAK/STAT signaling pathways are essential for immune responses of the host immune system and for interactions between host immune cells and non-immune cells.Citation15-19 Both type I (i.e., IFNα and IFNβ) and type II (i.e., IFNγ) IFNs are potent activators of the JAK-STAT signaling pathways and play essential roles in host cancer immune surveillance.Citation19-24 The JAK/STAT signaling transduction transits and also terminates quickly under normal physiologic conditions.Citation25 However, JAK mutations and aberrant or chronic JAK-STAT signaling often occurs in human inflammatory disease and malignancies.Citation15,17,26-29 Accordingly, the inhibition of JAK signaling has been shown to be effective in the suppression of lymphomas and leukemia.Citation30-33 On the other hand, sustained JAK/STAT signaling occurs under certain pathological conditions and often causes chronic inflammation and inflammation-mediated cancer progression.Citation34 For example, the IFNγ-STAT1 signaling pathway is essential for host cancer immune surveillance.Citation21 However, sustained IFNγ-STAT1 signaling leads to chronic inflammationCitation35 and inflammation-mediated cancer progression.Citation36 Similarly, proinflammatory cytokines and resultant STAT3 activation promote tumor initiation and progression.Citation37-40 Furthermore, STAT3 is essential for pancreatic ductal adenocarcinoma progression in mouse models that harbor constitutive active KRAS, which is the oncogenic driver of human pancreatic ductal adenocarcinoma.Citation38,41,42 In addition, STAT3 may decrease the immunogenicity of cancer cells via cell-autonomous pathways and induce an immunosuppressive tumor microenvironment.Citation43-45 Consistent with the critical roles of JAK/STAT signaling pathways in chronic inflammation and inflammation-mediated solid tumor progression, JAK-STAT inhibitors have shown promising efficacy in the suppression of non-haematopoietic tumors including pancreatic cancer.Citation46-51 These studies demonstrated that the chronic JAK-STAT pathways promote pancreatic cancer development and lead us to the hypothesis that the JAK-STAT pathway-mediated inflammation might counteract anti-PD-1 immunotherapy-induced antitumor immune response. We report here that Ruxolitinib therapy increases cytotoxic T lymphocyte (CTL) infiltration and activation in the tumor microenvironment to effectively enhance the efficacy of anti-PD-1 immunotherapy against orthotopic pancreatic cancer in vivo. Our data suggest that Ruxolitinib is potentially an effective adjunct agent that can be further developed to overcome cancer cell resistance to immune check point blockade immunotherapy in pancreatic cancer patients.
Results
Inhibition of the JAK-STAT signaling pathway decreases pancreatic tumor growth in vivo
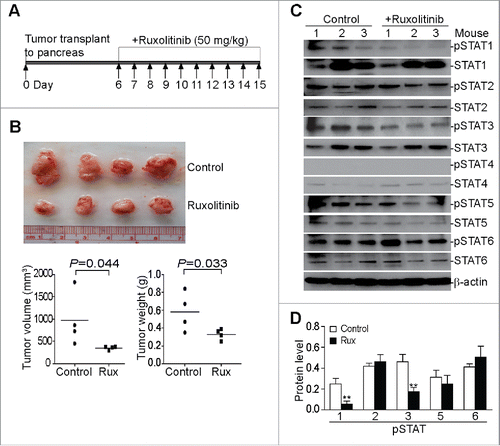
Mouse pancreatic tumor cells PANC02-H7 were injected to the pancreas of wt mice. The orthotopic pancreatic tumors grow fast in the syngeneic mice and the oral administration of Ruxolitinib significantly suppressed tumor growth ( and ). The analysis of the tumor tissues for the six known STAT proteins revealed that Ruxolitinib inhibits STAT1 and STAT3 phosphorylation in vivo ( and ), whereas the phosphorylation of STAT2, STAT5, and STAT6 was not significantly altered ( and ). The STAT4 protein level is low in all tumor tissues analyzed and no STAT4 phosphorylation was detected in these tumor tissues ( and ).
Figure 1. Ruxolitinib inhibits STAT1 and STAT3 activation to suppress pancreatic tumor growth in vivo. (A) Scheme of the orthotopic PANC02-H7 pancreatic tumor mouse model and Ruxolitinib (Rux) therapy. PANC02-H7 cells were injected into pancreas of mice. Tumor-bearing mice were treated with Ruxolitinib (50 mg/kg body weight) daily starting on day 6 for 10 d. (B) The orthotopic tumors were dissected from control (n = 4) and Ruxolitinib-treated (n = 4) tumor-bearing mice 15 d after tumor transplant. Shown are the images of the dissected tumors. Bottom panel: tumors were measured using a digital caliber. The tumor volume was calculated by the formula of length × width2/2 (left panel). Tumor weights of the control and treatment groups are presented at the right. (C) Tumor tissues were homogenized in total protein lysis buffer and analyzed by Western blotting using the indicated antibodies. β-actin was used as normalization control. (D) The protein band intensities of pSTAT1, pSTAT2, pSTAT3, pSTAT5, and pSTAT6 as shown in (C) were quantified using NIH image J and normalized as the ratios of each over the intensities of β-actin. Column: Mean of three mice; Bar: SD. **p < 0.01.
To determine whether Ruxolitinib suppresses pancreatic tumor growth through the inhibition of tumor cell proliferation, PANC02-H7 cells were cultured in the presence of Ruxolitinib. Analysis of cell cycle indicates that Ruxolitinib does not alter pancreatic cell cycle progression (Fig. S1A). The analysis of cellular proliferation shows that Ruxolitinib does not inhibit pancreatic tumor cell proliferation at dose as high as 1,000 nM (Fig. S1B). Therefore, Ruxolitinib suppresses pancreatic tumor growth through a mechanism that is independent of tumor cell proliferation.
Ruxolitinib-mediated suppression of pancreatic tumor growth in vivo depends on T cells
The JAK/STAT signaling pathway plays a key role in immune cell activation and differentiation.Citation52 T lymphocytes are essential for host cancer immune surveillance.Citation20,21 We then sought to determine whether T cells are involved in the Ruxolitinib-mediated tumor growth suppression in vivo. We made use of Rag1 KO mice. The rationale is that if Ruxolitinib enhances T cell-mediated tumor suppression, then the efficacy of Ruxolitinib in inhibition of tumor growth should diminish in T cell-deficient mice. PANC02-H7 cells were injected into the pancreas of Rag1 KO mice. Tumor-bearing mice were then treated with Ruxolitinib. The analysis of the orthotopic tumors indicates that Ruxolitinib exhibits no tumor suppressive efficacy in T-cell-deficient mice. Both tumor size and weight were not significantly different between control and Ruxolitinib-treated groups (). Therefore, we conclude that Ruxolitinib suppresses pancreatic tumor growth through a T-cell-dependent mechanism in vivo.
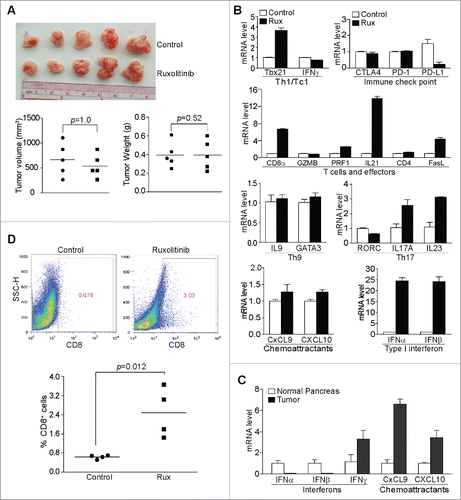
Figure 2. Ruxolitinib-mediated tumor suppression depends on host T cells in vivo. (A) PANC02-H7 cells were transplanted to the pancreas of Rag1 KO mice. Five days later, the tumor-bearing mice were treated with solvent (control, n = 5) or Ruxolitinib (n = 5) daily for 10 d. The orthotopic tumors were dissected from tumor-bearing mice 15 d after tumor transplant. Shown are images of the dissected tumors. Tumors were measured using a digital caliber. The tumor volume was calculated by the formula of length × width2/2 and presented at the left panel. Tumor weights of the control and treatment group are presented at the right panel. (B) Tumor tissues from control (n = 4) and Ruxolitinib-treated (n = 4) tumor-bearing mice were dissected 15 d after tumor transplant as in (A) and analyzed by real-time PCR to determine the levels of Th1/Tc1 cell markers, immune checkpoint molecules, T cells and T cell effector molecules, Th9, Th17 cell markers, T cell chemoattractants and type I interferons using the indicated gene-specific PCR primers. Column: Mean; Bar: SD. (C) RNAs were isolated from normal pancreas (n = 5) and orthotopic pancreatic tumor tissues (n = 5) and analyzed by real-time PCR for interferons and T cell chemoattractants using the indicated gene-specific PCR primers. (D) Tumor tissues from control (n = 4) and Ruxolitinib-treated (n = 4) tumor-bearing mice were dissected 15 d after tumor transplant as in A to be prepared for single cells. The cells were stained with fluorescent-conjugated anti-mouse CD8α mAb and analyzed by flow cytometry. Top panels show percentage of CD8+ cells in the tumor tissues of one representative mouse of the control and the ruxolitinib-treated tumor-bearing mice, respectively. Bottom panel: quantification of % CD8+ T cells in the tumor tissues.
Inhibition of the JAK-STAT signaling pathways increase CTL activation and infiltration in the tumor microenvironment
To further determine the effects of Ruxolitinib on T cell function in the tumor microenvironment, we analyzed tumor tissues for the expression levels of immune cell signature genes in the tumor microenvironment. Ruxolitinib treatment increased the expression level of Tbx21/T-bet, a marker for Th1/Tc1 cells, by 3.7-folds (). Among the three immune check point genes, Ruxolitinib decreased the PD-L1 expression level by 6.5-folds (). The CD8α expression level increased by 6.7-folds in Ruxolitinib-treated tumors as compared with untreated tumors, suggesting increased tumor-infiltration of CD8+ T cells. The IL21 expression level increased by 13.9-folds. CTL effectors perforin (PRF1) and FasL expression levels increased by 2.6- and 4.4-folds, respectively, in the treated tumor tissues (), indicating upregulated CTL cell activation in the tumor microenvironment.
The JAK-STAT signaling pathways regulate both Th9 and Th17 cell differentiations and these two subsets of T cells plays opposing roles in host cancer immune surveillance.Citation53 The analysis of the expression levels of IL9 and GATA3, markers for Th9 T cells, indicates that the Th9 cell level was not affected by Ruxolitinib therapy (). Among the three Th17 cell markers analyzed, IL17A and IL23 level increased (). With no change in the IFNγ level in the tumor tissues, Ruxolitinib therapy did not dramatically change the levels of IFNγ-activated T cell chemoattractants CXCL9 and CXCL10 (). Interestingly enough, the expression levels of IFNα and IFNβ increased approximately by 24.4- and 24.1-folds, respectively, in Ruxolitinib-treated tumor tissues as compared with the untreated tumors ().
Next, we compared normal pancreas and the orthotopic pancreatic tumor tissues for expression levels of IFNs and IFNγ-activated T cell chemoattractants. IFNα and IFNβ are actually much lower in the tumor tissues than in normal pancreas. However, levels of IFNγ, CXCL9, and CXCL10 are 3.3-, 3.6-, and 3.4-folds higher, respectively, in the tumor tissues than in the normal pancreas ().
To determine whether the CD8α protein level in the tumor tissue is also upregulated after Ruxolitinib treatment, we stained the total tumor cells with a CD8α-specific antibody and analyzed CD8+ T cell levels in the tumor tissues. Ruxolitinib treatment significantly increased CD8+ T cell infiltration in the tumor microenvironment (). In contrast to the inhibition of PD-L1 expression and increased CTL activation and infiltration in the tumor microenvironment (), Ruxolitinib treatment did not significantly alter the levels of general myeloid-derived suppressor cells (MDSCs)(CD11b+Gr1+) (Fig. S2A). The analysis of the sunsets of MDSCs indicates that the level of M-MDSC (CD11b+Ly6G−Ly6Chi) also did not change in the tumor-bearing mice after Ruxolitinib treat-ment (Fig. S2B). Very low level of PMN-MDSCs (CD11b+Ly6G+Ly6Clo) exists in this orthotopic pancreatic tumor mouse model and Ruxolitinib treatment did not change PMN-MDSC level (Fig. S2C).
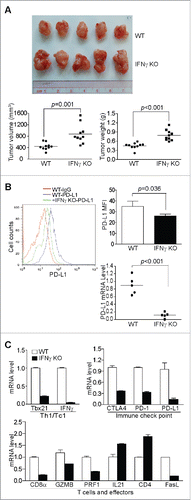
Figure 3. IFNγ is essential for suppression of pancreatic cancer growth in vivo. (A) Tumor cells were transplanted to pancreas of WT (n = 5) and IFNγ KO (n = 5) mice. The orthotopic tumors were dissected from tumor-bearing mice 15 d after tumor transplant. Shown are images of the dissected tumors from one of the two experiments. Tumors from WT (n = 10) and IFNγ KO (n = 10) mice from two experiments were weighed and measured and presented at the bottom panel. (B) Tumor tissues from WT (n = 5) and IFNγ KO (n = 5) mice from one of the two experiments were digested to make single cells and stained with fluorescent dye-conjugated anti-mouse PD-L1 mAb. Shown are the overlay of representative plots of tumor cell PD-L1 protein staining from one of five tumor-bearing mice. The mean fluorescence intensity of PD-L1 protein was quantified and presented at the top right panel. Column: Mean; Bar: SD. Bottom right panel: RNA was prepared from the tumor tissues and analyzed by real-time PCR for PD-L1 mRNA level. Each dot represents the relative PD-L1 mRNA level from the tumor of one tumor-bearing mouse. (C) RNA was isolated from tumor tissues of tumor-bearing WT (n = 5) and IFNγ KO (n = 5) mice as shown in (A) and analyzed by real-time PCR to determine the levels of Th1/Tc1 cell markers, immune checkpoint molecules, T cells, and T cell effector molecules using the indicated gene-specific PCR primers. Column: Mean; Bar: SD.
IFNγ is essential for host cancer immune surveillance against pancreatic cancer growth in vivo
In addition to lymphocytes, IFNγ is also essential for host cancer immune surveillance.Citation20,21 STAT1 is a direct target of IFNγ and Ruxolitinib inhibits STAT1 activation (). To determine the relative contributions of the IFNγ-STAT1 signaling pathway in Ruxolitinib-mediated PD-L1 repression and tumor suppression, PANC02-H7 cells were orthotopically transplanted to IFNγ KO and age-matched WT mice and analyzed for their growth in vivo. It is clear that pancreatic tumor cells grow significantly faster in IFNγ KO mice than in wt mice (). As expected, IFNγ deficiency decreased the PD-L1 protein level in tumor cells (). Interestingly, IFNγ deficiency in the host also downregulated the mRNA levels of CTLA4, PD-1, and PD-L1 by 2.7-, 3.3-, and 7.1-folds, respectively, in the tumor microenvironment (). The mRNA levels of CD8α, granzyme B, perforin, and FasL were 4-, 1.4-, 2.5-, and 5-folds lower, respectively, in tumor tissues grown in IFNγ KO mice than in wt mice (). These observations validate the concept that IFNγ is essential for host cancer immune surveillance.Citation20,21
IFNγ neutralization represses PD-L1 expression in tumor cells but exhibits no effects on pancreatic tumor growth in vivo
We next sought to determine whether decreasing IFNγ signaling, not completely ablating IFNγ function, can suppress pancreatic tumor growth as Ruxolitinib did. The tumor-bearing mice were treated with an anti-IFNγ mAb daily for 10 d. However, IFNγ neutralization mAb therapy did not significantly decrease tumor growth in vivo (). As expected, IFNγ neutralization mAb therapy significantly decreased the PD-L1 expression level in tumor cells in the tumor microenvironment ().
Figure 4. Ruxolitinib-exerted tumor suppression depends on host IFNγ in vivo. (A) PANC02-H7 cells were orthotopically transplanted to WT mice. The tumor-bearing mice were treated daily from day 6 with IgG isotype control (n = 5,200 μg/mouse) or anti-mouse IFNγ mAb (n = 5,200 μg/mouse) for 10 d. The orthotopic tumors were then dissected from tumor-bearing mice. Shown are the images of the dissected tumors. Tumor volume and weight were quantified and are presented at the bottom. (B) Tumor tissues were digested to make single cells and stained with anti-mouse PD-L1 mAb. Shown is the overlay of representative plots of tumor cell PD-L1 protein staining from one of five tumor-bearing mice of the IgG control and anti-IFNγ mAb treatment groups. The PD-L1 protein MFI was quantified and presented at the top right panel. Column: Mean; Bar: SD. Bottom right panel: RNA was prepared from the tumor tissues and analyzed by the real-time PCR for PD-L1 mRNA level. Each dot represents the relative PD-L1 mRNA level from tumor of one tumor-bearing mouse. (C) RNA was isolated from tumor tissues of tumor-bearing IgG control (n = 5) and anti-IFNγ mAb-treated (n = 5) mice. The gene expression level was analyzed by real-time PCR to determine the levels of Th1/Tc1 cells, immune checkpoint molecules, T cells, and T cell effector molecules using the indicated gene-specific PCR primers. (D) PANC02-H7 cells were orthotopically transplanted to wt mice. The tumor-bearing mice were treated daily from day 6 with IgG isotype control (n = 5), Ruxolitinib (n = 5), anti-mouse IFNγ mAb (n = 5), or Ruxolitinib and anti-mouse IFNγ mAb for 10 d. The orthotopic tumors were then dissected from tumor-bearing mice. Shown are the images of the dissected tumors. Tumor volume and weight were quantified and presented at the right panels.
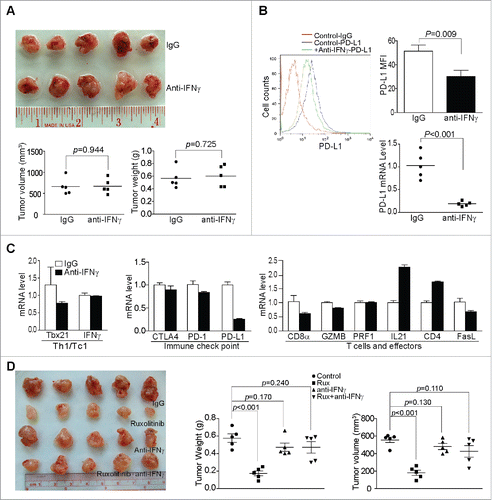
The analysis of immune cell signature gene expression profiles indicates that Th1/Tc1 signature gene and CTL signature genes are downregulated by IFNγ neutralization mAb therapy (). As in Ruxolitinib-treated tumors and tumors from IFNγ KO mice, IL21 is 2.3-folds higher in the IFNγ mAb-treated tumors () as compared with WT mice. IFNγ mAb therapy also increased CD4+ T cells expression in the tumor tissues by 1.8-folds (). These observations suggest that although the complete loss of IFNγ expression leads to immune deficiency and increased tumor growth, decreasing IFNγ function with neutralizating mAb does not cause significant immune deficiency and tumor growth promotion.
To determine whether IFNγ plays a role in Ruxolitinib-exerted tumor suppression, the tumor-bearing mice were treated with Ruxolitinib, IFNγ neutralizing mAb or both Ruxolitinib and IFNγ neutralization mAb. As observed above, Ruxolitinib therapy significantly suppresses the established tumor growth as measured by both the tumor size and tumor weight (). However, IFNγ-neutralizing mAb diminished Ruxolitionib function in the suppression of tumor growth (). These observations indicate that the low level of IFNγ is sufficient for host cancer immune surveillance and for Ruxolitinib-induced tumor suppression.
Both type I and Type II interferons regulate PD-L1 expression through the JAK-STAT pathway in pancreatic tumor cells
Tumor cells respond to IFNγ signaling to upregulate PD-L1.Citation54 Our above observations indicate that IFNγ upregulates PD-L1 expression in the tumor tissues (). We next sought to further determine the role of Ruxolitinib in the regulation of PD-L1 expression in pancreatic tumor cells. The analysis of tumor cells from control and Ruxolitinib-treated tumors revealed that Ruxolitinib therapy significantly downregulated the PD-L1 protein level on the tumor cell surface (). The analysis of tumor cell PD-L1 mRNA level indicates that Ruxolitinib also represses PD-L1 transcription in the orthotopic pancreatic tumors in vivo (). To determine whether Ruxolitinib specifically inhibits IFNγ-induced PD-L1 expression in pancreatic tumor cells, PANC02-H7 cells were treated with IFNγ in the absence or presence of Ruxolitinib. Flow cytometry analysis revealed that IFNγ upregulates PD-L1 expression and Ruxolitinib effectively diminishes IFNγ-induced PD-L1 expression in pancreatic tumor cells in vitro ().
Figure 5. Inhibition of the JAK-STAT signaling pathway decreases tumor cell PD-L1 expression. (A) Tumor tissues from control and Ruxolitinib-treated tumor-bearing mice were digested to make single cells. Cells were stained with anti-mouse PD-L1 mAb and analyzed by flow cytometry. The top panel shows the overlay of tumor cell PD-L1 staining from one of control and Ruxolitinib-treated tumor-bearing mice. Control-IgG: IgG isotype staining of tumor cells from a control mouse; Control-PD-L1: Anti-PD-L1 mAb staining of tumor cells from a control mouse; +Rux-PD-L1: anti-PD-L1 mAb staining of tumor cells from a Ruxolitinib-treated (+Rux) mouse. The mean fluorescence intensity (MFI) of PD-L1 protein was quantified and presented in the bottom left panel. Column: Mean; Bar: SD. Bottom right panel: RNA was prepared from the tumor tissues of control (n = 4) and Ruxolitinib-treated (n = 4) mice and analyzed by real-time PCR for the PD-L1 mRNA level. Each dot represents the relative PD-L1 mRNA level from tumor of one tumor-bearing mouse. (B) PANC02-H7 cells were cultured in the presence of recombinant IFNγ (100 U/mL) or IFNγ + Ruxolitinib (Rux, 100 nM) for approximately 24 h. Cells were then stained with anti-PD-L1 mAb and analyzed by flow cytometry. The top panel shows the overlay of PD-L1 staining from the control and the indicated treatment groups of tumor cells. The PD-L1 MFI is shown at the bottom left panel. The tumor cells were also analyzed by real-time RT-PCR for the PD-L1 mRNA level (bottom right panel). (C) Tumor cells were treated with recombinant IFNα (100 U/mL) or IFNα and Ruxolitinib (100 nM) for approximately 24 h. Cells were stained with anti-PD-L1 mAb and analyzed by flow cytometry. Left panel shows the overlay of PD-L1 protein staining of the control and the indicated treatment groups of tumor cells. Right panel shows the quantification of PD-L1 protein MFI. (D) Tumor cells were treated with recombinant IFNβ (100 U/mL) or IFNβ and Ruxolitinib (100 nM) for approximately 24 h. Cells were stained with anti-PD-L1 mAb and analyzed by flow cytometry. Left panel shows the overlay of PD-L1 protein staining of the control and indicated treatment groups of tumor cells. Right panel shows PD-L1 protein MFI. (E) Tumor cells were cultured in the presence of IFNγ or IFNγ with Fludarabine (Fluda) at the indicated concentrations for approximately 24 h. Cells were then stained with anti-PD-L1 mAb and analyzed by flow cytometry. Left panel shows the overlay of PD-L1 protein staining of the control and the indicated treatment groups of tumor cells. Right panel shows PD-L1 protein MFI.
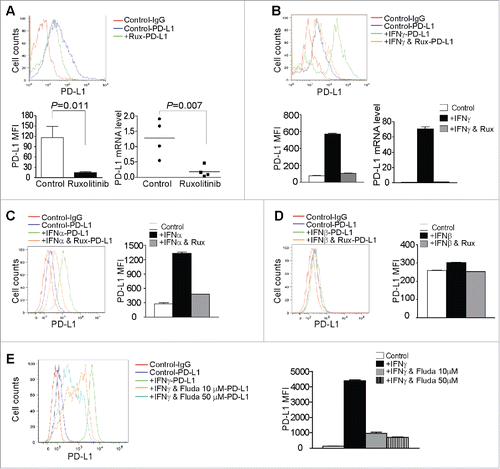
Ruxolitinib increases IFNα and IFNβ expression levels in the tumor tissues (). We next tried to determine whether IFNα and IFNβ regulate PD-L1 expression in pancreatic cancer cells. IFNα exhibited potent activity in upregulating PD-L1 expression in pancreatic tumor cells in vitro and Ruxolitinib also inhibits IFNα-induced PD-L1 expression in the tumor cells (). In contrast, IFNβ shows minimal activity in upregulating PD-L1 expression in pancreatic tumor cells (). To determine whether PD-L1 upregulation by IFNs is through STAT1 activation, tumor cells were treated with IFNγ with or without the pSTAT1 inhibitor Fludarabine and analyzed for the PD-L1 protein level. It is clear that the inhibition of STAT1 activation blocks the IFNγ induction of PD-L1 (). Taken together, our data indicate that PD-L1 is activated by both type I and type II IFNs through the JAK-STAT1 pathway, and Ruxolitinib is effective in downregulating PD-L1 expression in vivo in pancreatic tumor cells.
Ruxolitinib inhibits STAT3 activation in tumor cells to reverse tumor-mediated immune suppression to enhance T cell activation
Ruxolitinib inhibits the activation of both STAT1 and STAT3 in the tumor microenvironment (). It is known that STAT3 promotes T cell survival under physiologic conditions, Citation55 and the JAK/STAT signaling pathway is essential for immune cell activation and differentiation.Citation52 Indeed, it has been reported that Ruxolitinib inhibits JAK/STAT signaling to decrease effector T cell activation and proliferation.Citation56,57 To determine whether Ruxolitinib inhibits STAT3 activation in T cells in the tumor microenvironment, tumor-infiltrating CD8+ T cells were isolated from the tumor tissues of control and Ruxolitinib-treated tumor-bearing mice. Western blotting analysis indicates that STAT3 activation is inhibited in CD8+ CTLs by Ruxolitinib in the tumor microenvironment (Fig. S3). Indeed, Ruxolitinib inhibited T cell proliferation in vitro (Fig. S4) and T cell effector expression in vitro (Fig. S5).
These observations are in direct contrast to the above observations that Ruxolitinib therapy increases CTL activation and infiltration in the tumor microenvironment under pathological conditions (), and suggest that the tumor cell-T cell interactions in the tumor microenvironment may dictate Ruxolitinib functions under pathological conditions. To elucidate the biochemical and molecular mechanisms underlying Ruxolitinib-mediated CTL activation, we use both T-cell and tumor-cell-conditioned medium to model T-cell-tumor cell interactions in the tumor microenvironment. We first used the activated T-cell-conditioned medium to culture tumor cells in the absence or presence of a pSTAT3-selective inhibitor STATTIC (). The rationale is that activated T cells produce cytokines to activate STAT3 and activated STAT3 upregulates the transcription of immune suppressive cytokines in tumor cells. Indeed, activated T cell-conditioned medium increased STAT3 activation in tumor cells (). The analysis of tumor cells revealed that several immune suppressive cytokines, including IL6, IL10, and GM-CSF, are downregulated by STATTIC (). These observations suggest that tumor cells respond to T cell activation by activating STAT3 to produce immune suppressive cytokines.
Figure 6. Inhibition of STAT3 activation decreases immune suppressive cytokines in tumor cells to enhance T cell activation. (A) Scheme of modeling functions of STAT3 in T cell and tumor cell interactions in vitro. (B) Tumor cells as treated in (A) were analyzed by Western blotting for STAT3 activation. The protein band intensities were quantified using NIH image J. The pSTAT3 protein level was normalized as the ratio over the intensity of STAT3. Column: Mean; Bar: SD. (C) Tumor cells were either cultured in fresh medium, or T cell-conditioned medium in the absence or presence of pSTAT3 inhibitor STATTIC for 21 h, and then analyzed by qPCR for the indicated cytokines. β-actin was used as internal normalization control. (D) Scheme of modeling STAT3 functions in the effect of T cell-conditioned tumor cells on T cell activation and effector expression. Purified CD3+ T cells were stimulated for 3 d in anti-CD3/CD28-coated plates. Culture supernatant was collected and used to culture tumor cells in the absence or presence of pSTAT3 inhibitor (STATTIC) for 6 h. The medium was then replaced with fresh medium to remove pSTAT3 inhibitor (STATTIC) and the tumor cells were cultured for approximately 15 h. Tumor culture supernatant was then collected and used to culture T cells in anti-CD3/CD28-coated plates. T cells were analyzed after stimulation for 6 h. (E) T cells as treated in (D) were collected and analyzed by qPCR for the expression levels of effectors. The expression level of each effector of unstimulated cells (0 h) was arbitrarily set as 1. β-actin was used as internal control for qPCR. (F) Purified CD3+ T cells were cultured under the conditions as indicated for 3 d and analyzed for proliferation by 3H thymidine incorporation assay and T-bet expression by qPCR. Column: Mean; Bar: SD.
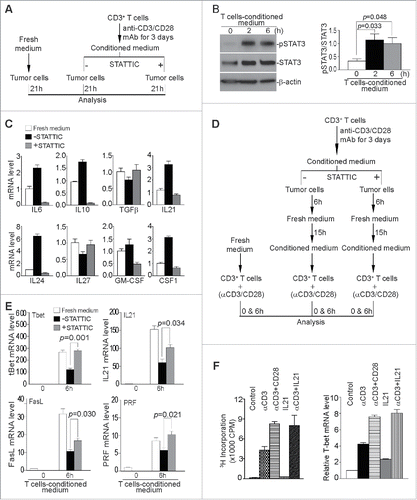
We then used activated T cell-conditioned medium to condition tumor cells in the absence or presence of STATTIC. Culture supernatants from these conditioned tumor cells were then used to culture T cells under activation conditions (). The rationale is that the inhibition of pSTAT3 decreases tumor cell production of immune suppressive cytokines to reverse tumor-induced suppression of T cell activation. We used four immune effector molecules as indicators of T cell activation. The analysis of T cells indicated that tumor cells represses T-bet, IL21, FasL, and perforin expression, and inhibition of STAT3 activation in tumor cells reversed tumor-mediated repression of expression of T-bet, IL21, FasL, and perforin ().
IL21 is known to suppress tumor growth and promote CTL activity Citation58 and is activated by Ruxolitinib in the tumor microenvironment (). We then hypothesized that IL21 may induces T cell activation and effector expression. To test this hypothesis, T cells were cultured in the presence of IL21 or IL21 plus anti-CD3. Anti-CD3 and anti-CD28 were used as positive control for T cell activation and proliferation. IL21 alone exhibited no stimulatory function in T cell activation and minimal effect on T-bet expression (). However, IL21 exhibited potent co-stimulatory function in enhancing anti-CD3-induced T cell proliferation and T-bet expression (). Therefore, IL21 mimics CD28 and functions as a T cell co-stimulatory cytokine. Taken together, our data suggest that tumor cells counteract activated T cells by STAT3-dependent suppressive cytokine production and Ruxolitinib inhibits STAT3 signaling in tumor cells to reverse immune suppression to activate CTLs in the tumor microenvironment, resulting in the increased secretion of IL21 that acts as a co-stimulatory factor to compensate loss of STAT3 function in T cells to activate CTLs in the tumor microenvironment.
Human pancreatic cancer exhibits high PD-L1 expression and low CTL infiltration in the tumor microenvironment
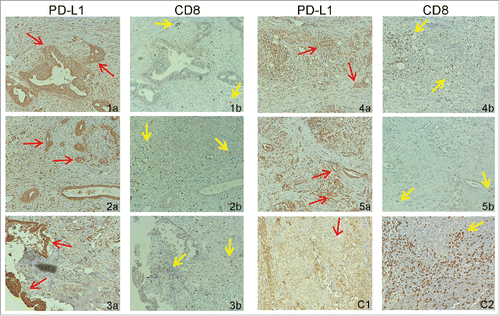
To determine the PD-L1 protein level in human pancreatic carcinoma tissues, we made use of a recently developed and FDA approved highly specific and sensitive PD-L1 mAb Citation59 to analyze the PD-L1 protein level by immunohistochemical method in human pancreatic tumor specimens. Tumor specimen information is included in Table S3. Adrenal tumor tissue was used as a positive control for PD-L1 protein (). Among the five tumor tissue specimens analyzed, high PD-L1 protein levels were observed in all five tumor specimens (). We also analyzed CD8+ T cell infiltration in these five tumor tissues. Human tonsil tissue was used as a positive control for CD8+ T cells (). CD8+ T cell levels were lower in three of the five tumor specimens (). Two tumor tissues exhibit the medium level of CD8+ T cell infiltration in certain tumor regions ().
Figure 7. PD-L1 protein and tumor-infiltrating CD8+ T cell levels in human pancreatic carcinoma. Tumor tissue specimens from five human pancreatic cancer patients were stained with antibodies that are specific for human PD-L1 (1a–5a) and human CD8+ (1b–5b), respectively. Brown color indicates PD-L1 protein and tumor-infiltrating CD8+ T cells staining. The tissues were counterstained with hematoxylin. Each image represents representative image of one patient. Red arrows indicate tumor cells and yellow arrows point to tumor-infiltrating CD8+ T cells. (C1) Human Adrenal tumor tissue was used as a positive control for human PD-L1-specific antibody. (C2) Human tonsil tissue was used as a positive control for human CD8+-specific antibody.
Inhibition of JAK-STAT pathway increases the efficacy of anti-PD-1 immunotherapy to suppress pancreatic cancer growth
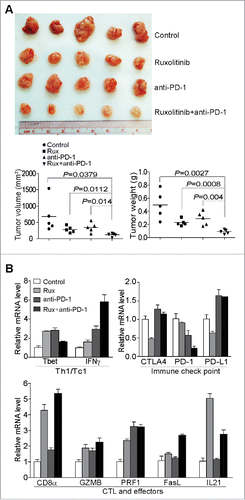
Anti-PD-1 check point blockade immunotherapy suppresses PD-L1-induced T cell inhibition to increase antitumor immune response.Citation8,60 If there are few tumor-infiltrating CTLs, the anti-PD-1 immunotherapy is unlikely to be effective. Our above observation that Ruxolitinib treatment increases CTL infiltration and activation in the tumor microenvironment suggest that Ruxolitinib might be effective in enhancing anti-PD-1 immunotherapy efficacy. To test this hypothesis, we used the PANC02-H7 orthotopic tumor model and treated tumor-bearing mice with Ruxolitinib and anti-PD-1 mAb, either alone or in combination. The analysis of tumor volume and tumor weight indicate that combined Ruxolitinib and anti-PD-1 immunotherapy exhibit significantly greater efficacy than either Ruxolitinib or anti-PD-1 mAb alone (). Furthermore, the combined therapy resulted in higher IFNγ, CD8+ T cell and FasL levels in the tumor microenvironment (). As expected, Ruxolitinib treatment decreases the PD-L1 level in the tumor tissues (). However, combined Ruxolitinib and anti-PD-1 treatment increases the PD-L1 level in the tumor tissues, which is correlated with the increased IFNγ level in the tumor tissue (), suggesting that increased IFNγ might upregulate PD-L1 expression in the tumor microenvironment and offset Ruxolitinib-mediated PD-L1 downregulation. Nevertheless, our observations indicate that Ruxolitinib can increase CTL infiltration and activation in the tumor microenvironment to improve the efficacy of anti-PD-1 mAb immunotherapy to effectively suppress pancreatic tumor growth in vivo.
Figure 8. Ruxolitinib increases the efficacy of anti-PD-1 mAb immunotherapy to suppress pancreatic tumor growth in vivo. (A) PANC02-H7 cells were injected into pancreas to establish orthotopic pancreatic tumors. The tumor-bearing mice were treated with solvent (Control, n = 5), Ruxilitinib (50 mg/kg body weight, n = 5) daily, anti-PD-1 mAb (200 μg/mouse, n = 5) every 2 d, and Ruxolitinib + anti-PD-1 mAb (n = 5) for 10 d. Shown are the images of the dissected tumors. Right panel: tumors were measured using a digital caliber. The tumor volume was calculated by the volume of length × width2/2 (left panel). Tumor weights of the control and treatment groups are presented at the right. (B) RNAs were isolated from tumor tissues of the four group mice as in (A) (n = 5 for each group). The RNA samples from the five mice of each group were pooled and the expression levels of the indicated genes were analyzed by real-time PCR using gene-specific PCR primers. Column: Mean; Bar: SD.
Discussion
Ruxolitinib is a potent and selective JAK1 and JAK2 inhibitor, with IC50 of 5.9 nM and 5.7 nM, respectively.Citation61 Among the six STATs of the JAK-STAT signaling pathways, we observed that Ruxolitinib selectively inhibits the phosphorylation of STAT1 and STAT3 in pancreatic tumor tissues in tumor-bearing mice. STAT1 is the key mediator of IFNγ signaling pathway,Citation62 and IFNγ signaling pathway is essential for host cancer immune surveillance.Citation20,21 Consistent with the essential role of IFNγ in host cancer immune surveillance and cancer suppression,Citation20,21 we observed that pancreatic tumor grows significantly faster in IFNγ KO mice than in wt mice. Thus, complete loss of IFNγ signaling causes immune deficiency and tumor growth promotion. In pancreatic tumor tissue, the IFNγ level is higher as compared with normal pancreas. Therefore, the IFNγ level might be constantly elevated in the tumor microenvironment. It is known that elevated and sustained IFNγ signaling often lead to chronic inflammation and inflammation-mediated tumor development.Citation36 Therefore, neutralizing IFNγ mAb therapy may reduce IFNγ to a low but still physiologically relevant level, which may explain why neutralizing IFNγ mAb treatment does not cause immune deficiency. However, in the presence of both Ruxolitinib and IFNγ neutralizing mAb, Ruxolitinib may inhibit IFNγ signaling more effectively since the level of IFNγ has been decreased by IFNγ-neutralizing mAb, and thus diminishes the function of the low level IFNγ in the tumor microenvironment. Our data therefore indicated that a low level of IFNγ is essential for Ruxolitinib function in the suppression of pancreatic tumor growth in vivo.
IL21 is a cytokine with pleiotropic functions in the regulation of T cell differentiation and function.Citation58,63 In this study, we observed that the IL21 expression level is higher in the tumor microenvironment in IFNγ KO mice and after IFNγ-neutralization mAb immunotherapy of pancreatic tumor-bearing mice. CD4+ T cell level is also higher in tumors in IFNγ KO mice and after IFNγ-neutralization mAb immunotherapy. It is thus reasonable to assume Ruxolitinib suppresses the chronic IFNγ signaling to increase CD4+ T cell infiltration in the tumor to upregulate IL21 production in the tumor microenvironment. IL21 then acts as a co-stimulatory signal to enhance tumor antigen-stimulated CD8+ T cell activation and effector function to suppress tumor growth. Ruxolitinib therapy does not cause a dramatic change in the IFNγ level but induces a significant increase in IL21 in the tumor microenvironment. The mechanism underlying the disconnection between IL21 and IFNγ in Ruxolitinib-treated tumor tissues remains to be elucidated.
On the other hand, one of the signatures of antitumor immune response by T cells is secretion of IFNγ.20,21 Indeed, the IFNγ level is higher in the tumor tissue as compared with the normal pancreas. However, tumor cells can respond to IFNγ to upregulate PD-L1.Citation64,65 As expected, we observed that Ruxolitinib can inhibit the IFNγ-STAT1 signaling to repress PD-L1 expression in tumor cells.Citation65 In addition to the type II IFNγ, we show here that the type I IFNα and IFNβ, particularly IFNα, can also upregulate PD-L1 in pancreatic tumor cells through the JAK-STAT signaling pathway. It is known that IFNα/β play a key role in the regulation of CTL response, and the intratumoral levels of type I IFNs correlate with favorable disease outcomes in several cohorts of cancer patients.Citation19,24 The roles of IFNα and IFNβ in Ruxolitinib-exerted suppression of pancreatic cancer growth remain to be determined.
kRAS is the oncogenic driver of human pancreatic ductal adenocarcinoma and STAT3 is essential for the progression of kRAS-driven pancreatic ductal adenocarcinoma.Citation37,38,41,42 We demonstrated that tumor cells respond to activated T cells to activate STAT3. It is known that activated STAT3 then upregulates immune suppressive cytokines and mediators.Citation39,40,45,66,67 It is also known that STAT3 suppresses type I interferon and the loss of STAT3 leads to increased Type I interferon and CTL infiltration,Citation45 which may explain the significant increase of IFNα and IFNβ in Ruxolitinib-treated tumor tissues. Therefore, tumor cells may counterattack T-cell-mediated antitumor immune response by responding to T-cell-secreted cytokines to activate STAT3. Activated STAT3 then augments the expression of immune suppressive cytokines, including IL10 and IL6, to inhibit T cell activation and function. Therefore, Ruxolitinib suppresses pancreatic tumor growth at least in part through inhibiting STAT3 activation in tumor cells to reverse STAT3-mediated immune repression and an immunosuppressive tumor microenvironment,Citation43-45 which might also contribute to increased CTL infiltration and activation in the tumor microenvironment.
In addition to promoting pancreatic cancer progression, STAT3 is also essential for T cell survival under physiologic conditions.Citation55 The JAK/STAT signaling pathway is essential for immune cell activation and differentiation.Citation52 Ruxolitinib has been shown to inhibit JAK/STAT signaling to decrease effector T cell activation and proliferation.Citation56,57 We observed here that although Ruxolitinib inhibits STAT3 activation in tumor-infiltrating CTLs in vivo and downregulates T cell activation and proliferation in vitro, Ruxolitinib therapy actually increases CTL activation in the tumor microenvironment in vivo. It is also possible that Ruxolitinib suppresses STAT3 activation in the tumor cells to reverse tumor cell-induced immune suppression, which overweighs the STAT3-mediated T cell survival and results in an overall increase in CTL activation and infiltration in the tumor microenvironment.
Decreased CTLs is linked to increased immune checkpoint markers, such as PD-L1, in mouse models of EGFR-driven lung cancer.Citation68,69 In this study, we observed that PD-L1 is abundantly expressed in human pancreatic carcinoma cells. Furthermore, CTL infiltration in human pancreatic cancer cells is low. Ruxolitinib effectively suppresses tumor cell PD-L1 activation as well as enhances CTL infiltration and activation in the tumor microenvironment. Furthermore, the efficacy of Ruxolitinib diminished in Rag1 KO mice. These observations further indicate that Ruxolitinib functions through enhancing antitumor T cell response.
Based on these observations, we propose a model to outline Ruxolitinib functions in inhibition of pancreatic tumor growth in vivo. In this model, the host immune system recognizes antigens of the growing tumor and activates T cells. T cell-mediated tumor responses include secretion of cytokines that induce STAT1 and STAT3 activation in tumor cells. Tumor cells respond to IFN signaling by upregulating PD-L1 as an adaptive mechanism to counterattack the antitumor T cells. STAT3 activation induces the expression and production of immune suppressive cytokine (i.e., IL6 and IL10) in tumor cells as another counter attack mechanism to repress antitumor T cells in the tumor microenvironment. The inhibition of STAT1 activation represses IFNγ-induced PD-L1 upregulation in tumor cells, whereas the inhibition of STAT3 activation decreases the tumor cell production of immune suppressive cytokines. Therefore, the inhibition of STAT1 and STAT3 activation coordinately reverse tumor cell-mediated T cell suppression, resulting in increased T cell infiltration and activation in the tumor microenvironment.
Our observations that human pancreatic carcinomas lack CTL infiltration suggest that the limited CTL level in the tumor microenvironment might be one of the limiting factors for pancreatic cancer nonresponsiveness to anti-PD-1 immunotherapy. The effectiveness of Ruxolitinib in inducing CTL tumor infiltration and activation thus lead to the notion that Ruxolitinib should be effective in overcoming pancreatic cancer resistance to anti-PD-1 immunotherapy. Indeed, Ruxolitinib therapy significantly enhanced the efficacy of anti-PD-1 immunotherapy in an orthotopic pancreatic cancer mouse model. Ruxolitinib has been tested in human pancreatic cancer patients and found to have certain direct antitumor activity.Citation51 Our data suggest that Ruxolitinib might be more effective if it is used as an adjunct agent to suppress chronic inflammation and increase CTL infiltration, instead of a monotherapeutic agent, to overcome human pancreatic cancer resistance to anti-PD-1 immunotherapy.
Methods
Tumor cells and specimens
PANC02-H7 cells were kindly provided by Dr. Min Li (University of Oklahoma Health Sciences Center).Citation70 Human pancreatic cancer specimens were collected from the Augusta University Medical Center from consented patients under approved protocols. The tumor tissue specimens were analyzed by board-certified pathologists.
Orthotopic mouse pancreatic tumor model
WT C57BL/6, IFNγ KO, and Rag1 KO mice were obtained from the Jackson Laboratory. Mouse was continuously anesthetized under 2% isoflurane in oxygen flow. A small abdominal incision at the right side near the spleen was made and the pancreas was pulled out with a sterile forcep. Tumor cells (1 × 104 cells in 20 μL saline) were injected into the pancreas using a sterile tuberculin syringe. The abdomen was closed with wound clips. Tumor-bearing mice was treated with Ruxolitinib (50 mg/kg body weight) 5 d after tumor transplant daily for 10 d by oral gavage. All mouse studies are performed according to protocols approved by Augusta University Animal Care and Use Committee.
Reagents
Ruxolitinib was obtained from LC laboratories (Woburn, MA). pSTAT3 inhibitor STATTIC was obtained from Santa Cruz Biotech (Dallas, TX). Anti-CD3 mAb, anti-CD28 mAb, and recombinant IL21 protein were obtained from Biolegends (San Diego, CA). Anti-IFNγ and anti-PD-1 (clone RMP1–14) mAb were obtained from Bio X cell Inc. (West Lebanon, NH).
Cell surface protein analysis
Tumor cells were stained with fluorescent dye-conjugated anti-mouse PD-L1 mAbs (Biolegend) and analyzed by flow cytometry. Spleen and blood were collected from tumor-bearing mice and stained with anti-mouse CD11b, anti-mouse Gr1, anti-mouse Ly6G, and anti-mouse Ly6C antibodies (Biolegend). Tumor tissues were collected from tumor-bearing mice and digested with collagenase solution (Collagenase 1 mg/mL, Hyaluronidase 0.1 mg/mL, and DNase I 30 U/mL) to make single cells. The tumor cell mixtures were stained with fluorescent-conjugated anti-mouse CD11b, anti-mouse Gr1, anti-mouse Ly6G, anti-mouse Ly6C, and anti-mouse CD8+ mAbs (Biolegend), and analyzed by flow cytometry.
Isolation of tumor-infiltrating CTLs
Tumor cell mixture as prepared above was incubated with Dynabeads mouse CD8+ (lyt-2) (Invitrogen Dynal AS. Norway) at 4°C for 30 min. The beads were washed three times with PBS and the bead-bound cells were lysed in total cellular protein lysis buffer (20 mM HEPES pH7.4, 20 mM NaCl, 10% glycerol, and 1% Triton X-100) plus protease and phosphatase inhibitor cocktails (Calbiochem, Darmstadt, Germany).
Western blotting analysis
Western blotting analysis was performed as described previously.Citation21 Antibodies are listed in Table S1.
Gene expression analysis
Fresh normal pancreas and PANC02-H7 tumor tissues were homogenized in Trizol (Life Technologies) to isolate total RNA. Cultured tumor cells were lyzed directly in Trizol to isolate total RNA. cDNA was synthesized from total RNA and used for the analysis of gene expression using gene-specific primers (Table S2) in the StepOne Plus Real-Time PCR System (Applied Biosystems).
Immunohistochemistry
Human pancreatic carcinoma tissues were deparaffinized and rehydrated, followed by treatment with the Universal HIER antigen retrieval reagent (Abcam, Cat# ab208572) according to the manufacturer's instructions. The slides were then blocked with goat serum, rinsed, and probed with anti-human PD-L1 Rab mAb (Abcam, clone 28–8). The tissues were probed with the rabbit-specific IHC polymer detection kit (Abcam, Cat# ab209101) according to the manufacturer's instructions. The stained tissues were counterstained with hematoxylin (Richard-Allan Scientific, Kalamazoo, MI).
Cell viability assays
Cell viability assays were performed using the MTT cell proliferation assay kit (ATCC, Manassas, VA) according to the manufacturer's instructions.
In vitro T cell activation and culture of tumor cells in T cell-conditioned medium
Lymph nodes and spleens were collected from C57BL/6 mice. Single cell suspension was prepared from lymph nodes and used to purify CD3+ T cells using the MojoSort mouse CD3 T cell isolation kit (Biolegend) according to the manufacturer's instructions. For T cell activation, 24-well culture plate was coated with anti-mouse CD3 and anti-mouse CD28 MAbs (1 μg/well in 150 μL PBS) overnight. The purified T cells were then seeded in the coated plate at a density of 5 × 105 cells/well in RPMI medium plus 10% FBS. T cell culture supernatants were collected 3 d later, diluted with 1/4 fresh medium, and used to culture tumor cells,
3H incorporation and T cell proliferation assay
Total 96-well plates were coated with anti-mouse CD3 and anti-mouse CD28 MAbs. Purified CD3+ T cells were then seeded at a density of 1 × 105 cells/well in 100 mL RPMI medium plus 10% FBS. 3H-thymidine was added to each well and cultured for another 6 h. Cells were harvested to a filter using the TOMTEC cell harvester. 3H incorporation was counted in a PerKinElmer 1450 LSC and Luminescence counter.
Statistical analysis
Statistical analysis was performed using ANOVA and Student's t test. A p < 0.05 was taken as statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
C. L. and A.T. contributed in the development of methodology and acquisition of data, N.Sa. in the analysis and interpretation of data, and C.L., N.Si., and K.L. in conception and design and writing manuscript.
KONI_A_1291106_s02.pdf
Download PDF (564.6 KB)Acknowledgments
We thank Dr. Roni Bollag for analyzing the human pancreatic tumor specimens and Ms. Sameera Qureshi for collecting the human pancreatic tumor specimens. We also thank Dr. Kimya Jones at the Georgia Esoteric and Molecular Laboratory in assistance for immunohistochemical staining of tumor specimens.
References
- Seufferlein T, Bachet JB, Van Cutsem E, Rougier P. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012; 23 Suppl 7:vii33-vii40; PMID:22997452; http://dx.doi.org/10.1093/annonc/mds224
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013; 369:1691-1703; PMID:24131140; http://dx.doi.org/10.1056/NEJMoa1304369
- Ho MY, Kennecke HF, Renouf DJ, Cheung WY, Lim HJ, Gill S. Defining eligibility of FOLFIRINOX for first-line metastatic pancreatic adenocarcinoma (MPC) in the province of British Columbia: A population-based retrospective study. J Clin Oncol 30, 2012 (suppl; abstr e14588); PMID:26165420; http://dx.doi.org/10.1097/COC.0000000000000205
- Rahma OE, Duffy A, Liewehr DJ, Steinberg SM, Greten TF. Second-line treatment in advanced pancreatic cancer: A comprehensive analysis of published clinical trials. Ann Oncol 2013; 24:1972-1979; PMID:23670093; http://dx.doi.org/10.1093/annonc/mdt166
- Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J Clin Invest 2015; 125:3384-3391; PMID:26325035; http://dx.doi.org/10.1172/JCI80011
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-2465; PMID:22658128; http://dx.doi.org/10.1056/NEJMoa1200694
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-2454; PMID:22658127; http://dx.doi.org/10.1056/NEJMoa1200690
- Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res 2015; 3:399-3411; PMID:25678581; http://dx.doi.org/10.1158/2326-6066.CIR-14-0215
- Hutcheson J, Balaji U, Porembka MR, Wachsmann MB, McCue PA, Knudsen ES, Witkiewicz AK. Immunologic and metabolic features of pancreatic ductal adenocarcinoma define prognostic subtypes of disease. Clin Cancer Res 2016; 22:3606-3617; PMID:26858311; http://dx.doi.org/10.1158/1078-0432.CCR-15-1883
- Luheshi NM, Coates-Ulrichsen J, Harper J, Mullins S, Sulikowski MG, Martin P, Brown L, Lewis A, Davies G, Morrow M et al. Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget 2016; 7:18508-18520; PMID:26918344; http://dx.doi.org/10.18632/oncotarget.7610
- Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013; 13:759-771; PMID:24154716; http://dx.doi.org/10.1038/nrc3611
- Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol 2014; 15:e493-e503; PMID:25281468; http://dx.doi.org/10.1016/S1470-2045(14)70263-3
- Haruki K, Shiba H, Shirai Y, Horiuchi T, Iwase R, Fujiwara Y, Furukawa K, Misawa T, Yanaga K. The C-reactive protein to albumin ratio predicts long-term outcomes in patients with pancreatic cancer after pancreatic resection. World J Surg 2016; 40(9):2254-2260; PMID:26956901; http://dx.doi.org/10.1007/s00268-016-3491-4
- Jamieson NB, Denley SM, Logue J, MacKenzie DJ, Foulis AK, Dickson EJ, Imrie CW, Carter R, McKay CJ, McMillan DC. A prospective comparison of the prognostic value of tumor- and patient-related factors in patients undergoing potentially curative surgery for pancreatic ductal adenocarcinoma. Ann Surg Oncol 2011; 18:2318-2328; PMID:21267785; http://dx.doi.org/10.1245/s10434-011-1560-3
- Villarino AV, Kanno Y, Ferdinand JR, O'Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol 2015; 194:21-27; PMID:25527793; http://dx.doi.org/10.4049/jimmunol.1401867
- Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciume G, Laurence A, Sun HW, Brooks SR, Vahedi G, Shih HY et al. Asymmetric action of STAT transcription factors drives transcriptional outputs and cytokine specificity. Immunity 2015; 42:877-889; PMID:25992861; http://dx.doi.org/10.1016/j.immuni.2015.04.014
- Schwartz DM, Bonelli M, Gadina M, O'Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol 2016; 12:25-36; PMID:26633291; http://dx.doi.org/10.1038/nrrheum.2015.167
- Wan CK, Andraski AB, Spolski R, Li P, Kazemian M, Oh J, Samsel L, Swanson PA 2nd, McGavern DB, Sampaio EP et al. Opposing roles of STAT1 and STAT3 in IL-21 function in CD4+ T cells. Proc Natl Acad Sci U S A 2015; 112:9394-9399; PMID:26170288; http://dx.doi.org/10.1073/pnas.1511711112
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol 2015; 15:405-414; PMID:26027717; http://dx.doi.org/10.1038/nri3845
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001; 410:1107-1111; PMID:11323675; http://dx.doi.org/10.1038/35074122
- Bardhan K, Paschall AV, Yang D, Chen MR, Simon PS, Bhutia YD, Martin PM, Thangaraju M, Browning DD, Ganapathy V et al. IFNgamma induces DNA methylation-silenced GPR109A expression via pSTAT1/p300 and H3K18 acetylation in colon cancer. Cancer Immunol Res 2015; 3(7):795-805; PMID:25735954; http://dx.doi.org/10.1158/2326-6066.CIR-14-0164
- Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: Implications for cancer therapy. Nat Rev Cancer 2016; 16:131-144; PMID:26911188; http://dx.doi.org/10.1038/nrc.2016.14
- Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol 2014; 41:156-173; PMID:24787290; http://dx.doi.org/10.1053/j.seminoncol.2014.02.002
- Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, Mueller YM, Fraietta JA, Wherry EJ, Turner M et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol 2013; 14:593-602; PMID:23603793; http://dx.doi.org/10.1038/ni.2576
- Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, Marque F, Régnier CH, De Pover A, Ryckelynck H et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov 2012; 2:512-523; PMID:22684457; http://dx.doi.org/10.1158/2159-8290.CD-11-0324
- Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015; 27:516-532; PMID:25873174; http://dx.doi.org/10.1016/j.ccell.2015.03.006
- Losdyck E, Hornakova T, Springuel L, Degryse S, Gielen O, Cools J, Constantinescu SN, Flex E, Tartaglia M, Renauld JC et al. Distinct acute lymphoblastic leukemia (ALL)-associated Janus kinase 3 (JAK3) mutants exhibit different cytokine-receptor requirements and JAK inhibitor specificities. J Biol Chem 2015; 290:29022-29034; PMID:26446793; http://dx.doi.org/10.1074/jbc.M115.670224
- Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: Mutations, inhibitors, and resistance. Clin Cancer Res 2013; 19:1933-1940; PMID:23406773; http://dx.doi.org/10.1158/1078-0432.CCR-12-0284
- Gadina M, Schwartz DM, O'Shea JJ. Editorial: decernotinib: A next-generation jakinib. Arthritis Rheumatol 2016; 68:31-34; PMID:26479275; http://dx.doi.org/10.1002/art.39463
- Ju W, Zhang M, Wilson KM, Petrus MN, Bamford RN, Zhang X, Guha R, Ferrer M, Thomas CJ, Waldmann TA. Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or Navitoclax in a murine model of human Hodgkin's lymphoma. Proc Natl Acad Sci U S A 2016; 113:1624-1629; PMID:26811457; http://dx.doi.org/10.1073/pnas.1524668113
- Maude SL, Dolai S, Delgado-Martin C, Vincent T, Robbins A, Selvanathan A, Ryan T, Hall J, Wood AC, Tasian SK et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015; 125:1759-1767; PMID:25645356; http://dx.doi.org/10.1182/blood-2014-06-580480
- Silvennoinen O, Hubbard SR. Targeting the inactive conformation of JAK2 in hematological malignancies. Cancer Cell 2015; 28:1-2; PMID:26175407; http://dx.doi.org/10.1016/j.ccell.2015.06.010
- Zhang M, Mathews Griner LA, Ju W, Duveau DY, Guha R, Petrus MN, Wen B, Maeda M, Shinn P, Ferrer M et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Proc Natl Acad Sci U S A 2015; 112:12480-12485; PMID:26396258; http://dx.doi.org/10.1073/pnas.1516208112
- Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: A rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 2012; 22:796-811; PMID:23238015; http://dx.doi.org/10.1016/j.ccr.2012.10.023
- Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, Piwnica-Worms D, DiPersio JF. IFNgamma R signaling mediates alloreactive T-cell trafficking and GVHD. Blood 2012; 120:4093-4103; PMID:22972985; http://dx.doi.org/10.1182/blood-2012-01-403196
- Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, Minoda Y, Sanada T, Yoshioka T, Mimata H et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med 2006; 203:1391-1397; PMID:16717119; http://dx.doi.org/10.1084/jem.20060436
- Wormann SM, Diakopoulos KN, Lesina M, Algul H. The immune network in pancreatic cancer development and progression. Oncogene 2014; 33:2956-2967; PMID:23851493; http://dx.doi.org/10.1038/onc.2013.257
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011; 19:456-469; PMID:21481788; http://dx.doi.org/10.1016/j.ccr.2011.03.009
- Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007; 7:41-51; PMID:17186030; http://dx.doi.org/10.1038/nri1995
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer 2009; 9:798-809; PMID:19851315; http://dx.doi.org/10.1038/nrc2734
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res 2011; 71:5020-5029; PMID:21586612; http://dx.doi.org/10.1158/0008-5472.CAN-11-0908
- Fukuda A, Wang SC, Morris JPt, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011; 19:441-455; PMID:21481787; http://dx.doi.org/10.1016/j.ccr.2011.03.002
- Kroemer G, Galluzzi L, Zitvogel L. STAT3 inhibition for cancer therapy: Cell-autonomous effects only? Oncoimmunology 2016; 5:e1126063; PMID:27467938; http://dx.doi.org/10.1080/2162402X.2015.1126063
- Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, Zhou T, Schmidt J, Jo M et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med 2015; 7:314ra185; PMID:26582900; http://dx.doi.org/10.1126/scitranslmed.aac5272
- Yang H, Yamazaki T, Pietrocola F, Zhou H, Zitvogel L, Ma Y, Kroemer G. STAT3 inhibition enhances the therapeutic efficacy of immunogenic chemotherapy by stimulating Type 1 interferon production by cancer cells. Cancer Res 2015; 75:3812-3822; PMID:26208907; http://dx.doi.org/10.1158/0008-5472.CAN-15-1122
- Gore J, Craven KE, Wilson JL, Cote GA, Cheng M, Nguyen HV, Cramer HM, Sherman S, Korc M. TCGA data and patient-derived orthotopic xenografts highlight pancreatic cancer-associated angiogenesis. Oncotarget 2015; 6:7504-7521; PMID:25762644; http://dx.doi.org/10.18632/oncotarget.3233
- Craven KE, Gore J, Wilson JL, Korc M. Angiogenic gene signature in human pancreatic cancer correlates with TGF-beta and inflammatory transcriptomes. Oncotarget 2016; 7:323-341; PMID:26586478; http://dx.doi.org/10.18632/oncotarget.6345
- Patel MR, Jacobson BA, Ji Y, Drees J, Tang S, Xiong K, Wang H, Prigge JE, Dash AS, Kratzke AK et al. Vesicular stomatitis virus expressing interferon-beta is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget 2015; 6:33165-33177; PMID:26431376; http://dx.doi.org/10.18632/oncotarget.5320
- Yang S, Luo C, Gu Q, Xu Q, Wang G, Sun H, Qian Z, Tan Y, Qin Y, Shen Y et al. Activating JAK1 mutation may predict the sensitivity of JAK-STAT inhibition in hepatocellular carcinoma. Oncotarget 2015; 7(5):5461-5469; PMID:26701727; http://dx.doi.org/10.18632/oncotarget.6684
- An HJ, Choi EK, Kim JS, Hong SW, Moon JH, Shin JS, Ha SH, Kim KP, Hong YS, Lee JL et al. INCB018424 induces apoptotic cell death through the suppression of pJAK1 in human colon cancer cells. Neoplasma 2014; 61:56-62; PMID:24195509; http://dx.doi.org/10.4149/neo_2014_009
- Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM 3rd, Nemunaitis JJ, Stella PJ, Pipas JM, Wainberg ZA et al. Randomized, double-blind, phase II study of Ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol 2015; 33:4039-4047; PMID:26351344; http://dx.doi.org/10.1200/JCO.2015.61.4578
- Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev 2009; 228:273-287; PMID:19290934; http://dx.doi.org/10.1111/j.1600-065X.2008.00754.x
- Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, Jetten AM, Khoury SJ, Fuhlbrigge RC, Kuchroo VK et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med 2012; 18:1248-1253; PMID:22772464; http://dx.doi.org/10.1038/nm.2856
- Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer 2015; 112:1501-1509; PMID:25867264; http://dx.doi.org/10.1038/bjc.2015.101
- Yu CR, Dambuza IM, Lee YJ, Frank GM, Egwuagu CE. STAT3 regulates proliferation and survival of CD8+ T cells: Enhances effector responses to HSV-1 infection, and inhibits IL-10+ regulatory CD8+ T cells in autoimmune uveitis. Mediators Inflamm 2013; 2013:359674; PMID:24204098; http://dx.doi.org/10.1155/2013/359674
- Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, Verbeek M, Fischer J, Otten V, Schmickl M et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014; 123:3832-3842; PMID:24711661; http://dx.doi.org/10.1182/blood-2013-12-543736
- Heine A, Held SA, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, Wolf D, Brossart P. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013; 122:1192-1202; PMID:23770777; http://dx.doi.org/10.1182/blood-2013-03-484642
- Spolski R, Leonard WJ. Interleukin-21: A double-edged sword with therapeutic potential. Nat Rev Drug Discov 2014; 13:379-395; PMID:24751819; http://dx.doi.org/10.1038/nrd4296
- Phillips T, Simmons P, Inzunza HD, Cogswell J, Novotny J, Jr., Taylor C, Zhang X. Development of an automated PD-L1 immunohistochemistry (IHC) assay for non-small cell lung cancer. Appl Immunohistochem Mol Morphol 2015; 23:541-549; PMID:26317305; http://dx.doi.org/10.1097/PAI.0000000000000256
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005; 65:1089-1096; PMID:15705911
- Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol 2010; 184:5298-5307; PMID:20363976; http://dx.doi.org/10.4049/jimmunol.0902819
- Stark GR, Darnell JE, Jr. The JAK-STAT pathway at twenty. Immunity 2012; 36:503-514; PMID:22520844; http://dx.doi.org/10.1016/j.immuni.2012.03.013
- Sutherland AP, Joller N, Michaud M, Liu SM, Kuchroo VK, Grusby MJ. IL-21 promotes CD8+ CTL activity via the transcription factor T-bet. J Immunol 2013; 190:3977-3984; PMID:23479229; http://dx.doi.org/10.4049/jimmunol.1201730
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013; 5:200ra116; PMID:23986400; http://dx.doi.org/10.1126/scitranslmed.3006504
- Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, Ritz J. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015; 4:e1008824; PMID:26155422; http://dx.doi.org/10.1080/2162402X.2015.1008824
- Herbeuval JP, Lelievre E, Lambert C, Dy M, Genin C. Recruitment of STAT3 for production of IL-10 by colon carcinoma cells induced by macrophage-derived IL-6. J Immunol 2004; 172:4630-4636; PMID:15034082; http://dx.doi.org/10.4049/jimmunol.172.7.4630
- Kida H, Ihara S, Kumanogoh A. Involvement of STAT3 in immune evasion during lung tumorigenesis. Oncoimmunology 2013; 2:e22653; PMID:23482587; http://dx.doi.org/10.4161/onci.22653
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 2013; 3:1355-1363; PMID:24078774; http://dx.doi.org/10.1158/2159-8290.CD-13-0310
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016; 7:10501; PMID:26883990; http://dx.doi.org/10.1038/ncomms10501
- Wang Y, Zhang Y, Yang J, Ni X, Liu S, Li Z, Hodges SE, Fisher WE, Brunicardi FC, Gibbs RA et al. Genomic sequencing of key genes in mouse pancreatic cancer cells. Curr Mol Med 2012; 12:331-341; PMID:22208613; http://dx.doi.org/10.2174/156652412799218868