
ABSTRACT
Simultaneous reestablishment of p53/p19Arf and interferon-β pathways in melanoma cells culminates in a cell death process that displays features of necroptosis along with the release of immunogenic cell death molecules and unleashes an antitumor immune response mediated by natural killer cells, neutrophils as well as CD4+ and CD8+ T lymphocytes.
Introduction
During the multistep process of tumorigenesis, cancer cells must acquire several new characteristics, including the capability to resist intrinsic cell death mechanisms and to escape from immune surveillance in order to establish a highly immune suppressive microenvironment.Citation1 Such characteristics are well documented in human melanoma, that retains wild-type p53 in 90% of cases, although inactivated due to elevated levels of MDM2, seen in 56% of cases,Citation2 or loss of p14ARF, which occurs in approximately 50% of cases.Citation3 Interestingly, the Arf (CDKN2A) and interferon-β (IFN-β) genes are both located on human chromosome 9p21 and their frequent loss points to their importance in preventing tumorigenesis. For melanoma, hemizygotic deletion of 9p21 was observed in 12 of 14 cases,Citation4 which may reflect loss of CDKN2A or IFN-β alone or in combination.Citation5 Thus, the remediation of Arf and IFN-β would not only reinstate the expression of some of these genes, but complement the activity of endogenous p53 and promote an antitumor immune response.
Following this line of thought, we have explored the p53/Arf and IFN-β pathways, developing a set of unique adenoviral vectors that use a p53 responsive promoter and encode the p19Arf tumor suppressor protein (p53 functional partner) and IFN-β (immunomodulatory cytokine).Citation6 As discussed here, the combined gene transfer of these factors to tumors harboring wild-type p53 creates an interplay between (i) transgene control, (ii) p53/Arf apototic stimulus and (iii) an IFN-β antiviral inflammatory context, that together unleashes immunogenic cell death (ICD) as well as a systemic innate and adaptive immune attack against cancer cells at primary and distant sites.
Evidence for cooperation between the p53/Arf and interferon-β pathways
The canonical antitumor mechanism of p14ARF (p19Arf in the mouse) involves its induction by several forms of oncogenic stress and its association with MDM2, thus inhibiting the degradation of p53.Citation7 In this way, ARF enables p53 activity, such as triggering growth arrest or apoptosis. In addition, ARF has p53-independent functions, such as induction of cell cycle arrest, senescence, apoptosis and regulation of autophagy.Citation8
IFN-β, as a member of the type I IFN family, is a cytokine initially known for its antiviral effects, but also exerts an important role in antitumor immune responses.Citation9 Upon binding to IFNAR1/2 transmembrane receptors, IFN-β promotes the transcription of hundreds of IFN-stimulated genes (ISGs), for example, Interleukin-1β (IL-1β), IFN-γ, and pro-apoptotic proteins, like FAS and TRAIL.Citation10 It can also induce the expression of major histocompatibility complex (MHC) class I molecules, maturation of dendritic cells and increase cytotoxic activity of natural killer (NK) cells and CD8+ T lymphocytes.Citation11
Moreover, it has been observed that the p53/Arf and type I IFN pathways intersect and even cooperate. For example, the presence of p53 has been shown to boost the antiviral response when cells were exposed to recombinant type I IFN.Citation12,Citation13 The TP53 promoter contains an IFN-stimulated response elements (ISRE) and p53 transcription is activated by IFN-α/β, while p53 protein is stabilized upon cellular exposure to type I IFN.Citation12 The transcription of IRF-5 and IRF-9, which are ISGs, are also known to be activated by p53, contributing to anti-viral defense.Citation14,Citation15 However, it has also been suggested that cell death in response to IFN-α/β is actually dependent on ARF, not p53.Citation16 While these examples show interplay between the p53/Arf and type I IFN pathways, cooperation between them has not been exploited as a therapeutic target in cancer.
Harnessing adenoviral mediated gene transfer of p19Arf and interferon-β
To successfully mediate the transfer of the Arf and IFN-β cDNAs into tumor cells, our group developed an adenoviral vector where transgene expression is controlled by a p53-responsive promoter, called PGTx-β (or simply PG) , which expresses the transgene 5–7 times more than a traditional constitutive promoter derived from cytomegalovirus.Citation17 Interestingly, when PG promotes the expression of p53, an autoregulatory, positive feedback mechanism is established, providing exceptionally high levels of p53 expression and superior tumor cell killing.Citation18 This positive feedback also occurs when PG promotes the expression of p19Arf. In this scenario, p19Arf was shown to stabilize endogenous p53 in B16 (mouse melanoma) and C6 (rat glioma) cells, both of which harbor endogenous wild-type p53. Interestingly, transfer of p53 to B16 cells did not show an antitumor response, in contrast to p19Arf that significantly affected cell viability.Citation19
As demonstrated in a later study, the response to p19Arf was enhanced when combined with IFN-β. In fact, only combined gene transfer, but not the individual treatment, with p19Arf and IFN-β (p19Arf+IFN-β) resulted in massive cell death of B16 cells in vitro, as well as in a model of in situ gene therapy, where adenoviral vectors were injected directly into subcutaneous (s.c) tumors and significantly inhibited tumor progression and prolonged survival.Citation6 Although this data looked promising, a key question regarding the combination of p19Arf with IFN-β was left unanswered: since IFN-β is already known to promote immune activation, could the addition of p19Arf provide an immunotherapeutic benefit? The hypothesis was that, if the p19Arf+IFN-β gene transfer could provide additional immunomodulatory stimulus, it would come from danger associated molecules released through the death process.
In light of this question, we explored a prophylactic cancer vaccine model in which B16 cells were transduced ex vivo with the p19Arf+IFN-β combination and, while at the beginning of the cell death process, inoculated (s.c) in the left flank (vaccine site) of immune competent syngeneic C57BL/6 mice. Seven days after the last vaccine, fresh untreated B16 cells were inoculated in the contralateral flank (tumor challenge site).Citation20 As expected, mice immunized with p19Arf+IFN-β treated cells showed a dramatic decrease in challenge tumor progression, when compared with control groups. Furthermore, we found that this antitumor protection was maintained even when the challenge was performed 73 days after vaccination, indicating the generation of memory response. Also, tumors displayed a high infiltration of hematopoietic CD45+ cells, and were being rejected in a CD4+ and CD8+ T-cell-dependent manner, thus proving the immunological nature of this response.Citation20
Unexpectedly, in the vaccine site, we observed that tumors grew out in a much delayed fashion when mice received three vaccine inoculations with p19Arf+IFN-β treated cells, suggesting that B16 clones with tumorigenic potential remained after treatment and were being kept under immune control, until eventually escaping to later establish the tumor. Seeking to understand this, cells treated with p19Arf and IFN-β alone or combined were inoculated in C57BL/6 (immune competent), nude (athymic mice that lack mature T lymphocytes) or NOD-SCID (lacking both T and NK cells) mice, without further tumor challenge. Surprisingly, tumor formation was completely abrogated only in hosts with functional NK cells that were inoculated with p19Arf+IFN-β treated cells. In accordance with this, immunosuppression with dexamethasone, an inhibitor of NK cell cytotoxic functions,Citation21 recapitulated this result and RT-qPCR analysis of in vitro treated B16 cells revealed upregulation of IL-15, ULBP1 NK cell activating receptor, KILLER/DR5 and FAS/APO-1 death receptors, therefore, supporting the notion that these cytotoxic innate immune cells were being activated by the p19Arf+IFN-β treated B16 cells and mediated immune rejection at the vaccine site.Citation20
Superior immune stimulus of the combined gene therapy, in comparison with IFN-β treatment, could not be detected when evaluating the magnitude of the T cell response, as popliteal lymphocytes secreted similar levels of Th1 related cytokines (IFNγ, IL-2 and tumor necrosis factor-α), indicating no evident advantage for the addition of p19Arf. Interestingly, when a therapeutic vaccine was used, (i.e. vaccination was performed after tumor challenge), we observed a greater reduction in tumor progression in the p19Arf+IFN-β group, when compared with the treatment with IFN-β alone.Citation20
Additional evidence for the benefit of the combined treatment was obtained from a second immunization model, in which adenoviral vectors were directly injected into established s.c. LLC1 tumors (lewis lung carcinoma, wt p53) and, after four rounds of treatment, a contralateral tumor challenge was performed with naïve LLC1 tumor cells. The hypothesis was that immunization in an established primary tumor, where immunosuppression is at its prime, would be more challenging for an effector immune response to be orchestrated and thus, could reveal if the p19Arf+IFN-β actually provides an immune advantage over IFN-β single treatment. Indeed, even though primary tumors from both groups presented similar growth impairment, tumors from the p19Arf+IFN-β group showed remarkably reduced progression in the secondary site.Citation22 Here, just as seen in the therapeutic vaccine model, the combination provided superior immunological protection.
Seeking to better understand the mechanisms behind immune stimulation triggered by the p19Arf+IFN-β combination, we performed microarray analysis of in situ treated LLC1 tumors. Differentially expressed genes revealed a strong immune response and chemotaxic signature with the involvement of neutrophils and CD8+ T cells, as evidenced by the upregulation of CCL3, CXCL3, IL-1α and IL-1β. Also, immunohistochemical quantification of CD11b+ Ly6G+ neutrophils showed an increase exclusively in the p19Arf+IFN-β group. Moreover, antibody-mediated granulocyte depletion throughout the treatment regime could greatly impair the inhibition of tumor growth caused by p19Arf+IFN-β gene transfer, demonstrating the critical role that this population plays in our model.Citation22 Importantly, this result does not exclude the involvement of CD8+ T lymphocytes or NK cells.
Next, we evaluated the molecular mechanisms of death induced by p19Arf and IFN-β alone or in combination. One key observation was that cell death in response to p19Arf+IFN-β is only possible in the context of adenoviral gene transfer, since the substitution of both p19Arf and IFN-β gene transfer for pharmacological approaches, like Nutlin-3 (a compound that binds to Mdm2 and releases p53), recombinant IFN-β (IFN-R) or Poly (I:C) (a toll-like receptor 3 agonist and inducer of endogenous IFN-β expression), did not cause high levels of cell death. Interestingly, when only one gene was substituted by a pharmacological mimetic, but the other was still transferred by the adenoviral vector, high levels of cell death were detected, confirming the importance of the adenovirus-mediated gene transfer in cell death induction. Indeed, the expression of genes related to an antiviral response (Nlrc5, Dram1, Isg15 and Chop) was detected only after cells were treated with p19Arf and IFN-β together.Citation23
We next analyzed apoptosis markers and detected exposure of Annexin-V and Bax expression mainly after p19Arf treatment. The addition of IFN-β seemed to modify the cell death pathway induced by p19Arf, since increased expression of Rip3 and Tnfrsf1a in cells treated with the p19Arf+IFN-β combination was observed. In fact, the treatment of B16 cells with Z-VAD-FMK, a pan-caspase inhibitor, before gene therapy did not inhibit cell death, but increased its levels, indicating again an alternative mechanism of cell death. The increase in Rip3 and Tnfrsf1 expression points to the occurrence of necroptosis, which was recently associated with the activation of ICD and is proposed to be a molecular mechanism of cell death that provides both antigenicity and adjuvancy stimuli to dendritic cells.Citation24 In fact, detection of all the three bona fide markers of ICD (exposure of Calreticulin, secretion of ATP and release of HMGB1) were only observed after p19Arf+IFN-β combined treatment, possibly explaining the immunomodulatory superiority of the combination.Citation23
In support to these findings, we evaluated the gene signature induced by each treatment. Global gene expression analysis revealed that all treatments (p19Arf, IFN-β or the combination of both) inhibited genes related to cell cycle progression, while only p19Arf+IFN-β induced expression of genes related to the p53 signaling pathway and apoptosis, as well as immune response, response to virus and antigen processing.Citation23
The immunity cycle initiated by combined gene transfer of p19Arf and interferon-β to cancer cells
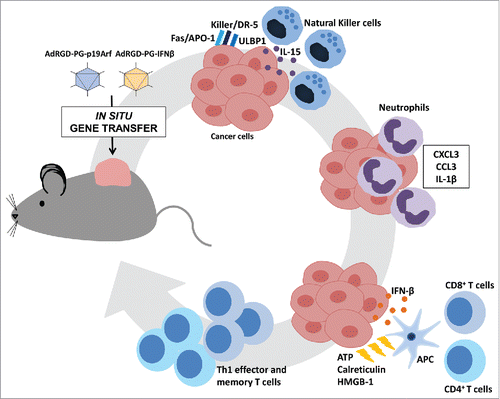
Based on the data presented above, we propose the following hypothesis (): first, at the immunization site either by injection of ex vivo treated cells or in situ gene therapy, the reactivation of the p53/Arf pro-apoptotic pathway in the presence of an antiviral and immunogenic context, that were elicited by the adenoviral vector and IFN-β signaling, starts the cell death process along with the upregulation of IL-15, ULBP1, FAS/APO1, and KILLER/DR5 genes, consequently providing positive stimulus to NK cells to recognize and kill treated tumor cells. Second, the cell death process itself and the upregulation of CCL3, CXCL3 and IL-1β in the tumor mass participates in the recruitment of CD11b+ Ly6G+ neutrophils that may function in the reshaping the tissue after injury, favor the infiltration of CD8+ T cells, directly kill tumor cells or even inhibit angiogenesis.Citation25 The exact function of neutrophils in our model remains to be elucidated. Third, secretion of high levels of IFN-β together with the release of calreticulin, ATP and HMGB1, should provoke profound modulatory effects on tumor associated APCs to achieve an optimal maturation state and, upon encounter with naïve T lymphocytes, induce a Th1 cytotoxic immune response, which was evidenced by the secretion of IFNγ and TNF-α. We speculate that the main importance of these primed tumor-specific CD4+ and CD8+ T cells is not at the primary tumor site, but at the distant challenge tumor site, that mimics metastatic foci, since an increase of adaptive immune negative regulators, such as PD-L1, have been found to be induced at the primary site due to the IFN-β inflammatory response.Citation22,Citation26 Finally, establishment of immunological memory has the potential to attack malignant cells that could eventually relapse after therapy, providing long lasting immune protection.
Figure 1. Proposed mechanism for the immunotherapeutic cycle induced upon combined gene transfer of p19Arf and interferon-β. In situ injection of both AdRGDPG-p19Arf and AdRGDPG-IFN-β adenoviral vectors into the tumor mass results in the reestablishment of p53/Arf pro-apoptotic and interferon-β pathways and culminates in a cell death process that: (i) upregulates Interleukin-15, ULBP1 and KILLER/DR5 to stimulate natural killer cells, (ii) recruits CD11b+ Ly6G+ neutrophils by the up regulation of CCL3, CXCL3 and Interleukin-1β, (iii) and together with the release of calreticulin, ATP and HMGB1 induces a Th1 cytotoxic immune response and establishment of immunological memory.
So far, we believe that we have gathered enough functional evidence to characterize the adenovirus mediated gene transfer of p19Arf together with IFN-β as a novel cancer immunotherapy strategy. To the best of our knowledge, no other gene transfer strategy using a replication deficient viral vector has been shown to induce ICD, a molecular mechanism already reported with chemotherapy (e.g., doxorubicin), radiotherapy and even with oncolytic replication-competent vectors, such as the Newcastle disease virus.Citation27,Citation28
We also have successfully identified critical players of our therapy: NK cells, CD4+ and CD8+ T lymphocytes, neutrophils and ICD molecules. However, the kinetics, location and precise mechanisms by which all these components cooperate needs to be understood in greater detail. Indeed, it has been reported that, in response to IL-15, NK cells can assume a helper phenotype and influence T cell priming toward a Th1 response.Citation29 Neutrophils can also modulate T cell immunity on several levels, including recruitment, activation and effector function.Citation25 Most importantly, since the p19Arf+IFN-β combination provides both ICD and IFN-β stimulus, it will be interesting to investigate how the DC compartment is being affected. As recently demonstrated, DC vaccines based on ICD provoke a T cell-driven rejection of high-grade gliomasCitation30 and may represent a therapeutic opportunity that can be further explored. Nevertheless, as type I IFNs have been shown to regulate PD-L1 expression on macrophages,Citation26 association of p19Arf+IFN-β with PD-1/PD-L1 checkpoint blockade immunotherapy is expected to provide great therapeutic benefit. Even though much remains to be investigated, potential therapeutic applications of our p19Arf+IFN-β approach deserve further development.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
We are grateful for the São Paulo Research Foundation for grants (BES) 13/25167–5 and 15/26580–9; and fellowships (RFVM) 13/09474–5, (AH) 11/10656–5.
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011; 144(5):646-74; PMID:21376230; https://doi.org/10.1016/j.cell.2011.02.013
- Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutant 2003; 21(3):217-28; PMID:12619107; https://doi.org/10.1002/humu.10179
- Sharpless E, Chin L. The INK4a/ARF locus and melanoma. Oncogene 2003; 22(20):3092-8; PMID:12789286; https://doi.org/10.1038/sj.onc.1206461
- Fountain JW, Karayiorgou M, Ernstoff MS, Kirkwood JM, Vlock DR, Titus-Ernstoff L, Bouchard B, Vijayasaradhi S, Houghton AN, Lahti J et al. Homozygous deletions within human chromosome band 9p21 in melanoma. Proc Natl Acad Sci USA 1992; 89(21):10557-61; PMID:1438246; https://doi.org/10.1073/pnas.89.21.10557
- Coleman A, Fountain JW, Nobori T, Olopade OI, Robertson G, Housman DE, Lugo TG. Distinct deletions of chromosome 9p associated with melanoma versus glioma, lung cancer, and leukemia. Cancer Res 1994; 54(2):344-8; PMID:8275465; www.cancerres.aacrjournals.org/content/54/2/344.long
- Merkel CA, Medrano RF, Barauna VG, Strauss BE. Combined p19Arf and interferon-beta gene transfer enhances cell death of B16 melanoma in vitro and in vivo. Cancer Gene Ther 2013; 20(5):317-25; PMID:23618951; https://doi.org/10.1038/cgt.2013.23
- Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res 2011; 71(18):5998-6009; PMID:21764762; https://doi.org/10.1158/0008-5472.CAN-10-3211
- Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev 2000; 14(18):2358-65; PMID:10995391; https://doi.org/10.1101/gad.827300
- Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu Rev Biochem 1987; 56:727-77; PMID:2441659; https://doi.org/10.1146/annurev.biochem.56.1.727 10.1146/annurev.bi.56.070187.003455
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. Interferome v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res 2013; 41(Database issue):D1040-6; PMID:23203888; https://doi.org/10.1093/nar/gks1215
- González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol 2012; 12(2):125-35; PMID:22222875; https://doi.org/10.1038/nri3133
- Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003; 424(6948):516-23; PMID:12872134; https://doi.org/10.1038/nature01850
- Zhu Z, Yang Y, Wei J, Shao D, Shi Z, Li B, Liu K, Qiu Y, Zheng H, Ma Z. Type I interferon-mediated immune response against influenza A virus is attenuated in the absence of p53. Biochem Biophys Res Commun 2014; 454(1):189-95; PMID:25450379; https://doi.org/10.1016/j.bbrc.2014.10.067
- Hu G, Mancl ME, Barnes BJ. Signaling through IFN regulatory factor-5 sensitizes p53-deficient tumors to DNA damage-induced apoptosis and cell death. Cancer Res 2005; 65(16):7403-12; PMID:16103093; https://doi.org/10.1158/0008-5472.CAN-05-0583
- Mori T, Anazawa Y, Iiizumi M, Fukuda S, Nakamura Y, Arakawa H. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 2002; 21(18):2914-8; PMID:11973653; https://doi.org/10.1038/sj.onc.1205459
- Sandoval R, Xue J, Pilkinton M, Salvi D, Kiyokawa H, Colamonici OR. Different requirements for the cytostatic and apoptotic effects of type I interferons. Induction of apoptosis requires ARF but not p53 in osteosarcoma cell lines. J Biol Chem 2004; 279(31):32275-80; PMID:15169789; https://doi.org/10.1074/jbc.M313830200
- Bajgelman MC, Strauss BE. Development of an adenoviral vector with robust expression driven by p53. Virology 2008; 371(1):8-13; PMID:18076963; https://doi.org/10.1016/j.virol.2007.11.015
- Tamura RE, da Silva Soares RB, Costanzi-Strauss E, Strauss BE. Autoregulated expression of p53 from an adenoviral vector confers superior tumor inhibition in a model of prostate carcinoma gene therapy. Cancer Biol Ther 2016; 17(12):1221-30; PMID:27646031; https://doi.org/10.1080/15384047.2016.1235655
- Merkel CA, da Silva Soares RB, de Carvalho AC, Zanatta DB, Bajgelman MC, Fratini P, Costanzi-Strauss E, Strauss BE. Activation of endogenous p53 by combined p19Arf gene transfer and nutlin-3 drug treatment modalities in the murine cell lines B16 and C6. BMC Cancer 2010; 10:316; PMID:20569441; https://doi.org/10.1186/1471-2407-10-316
- Medrano RF, Catani JP, Ribeiro AH, Tomaz SL, Merkel CA, Costanzi-Strauss E, Strauss BE. Vaccination using melanoma cells treated with p19arf and interferon beta gene transfer in a mouse model: A novel combination for cancer immunotherapy. Cancer Immunol Immunother 2016; 65(4):371-82; PMID:26887933; https://doi.org/10.1007/s00262-016-1807-8
- Keil D, Luebke RW, Pruett SB. Quantifying the relationship between multiple immunological parameters and host resistance: Probing the limits of reductionism. J Immunol 2001; 167(8):4543-52; PMID:11591782; https://doi.org/10.4049/jimmunol.167.8.4543
- Catani JP, Medrano RF, Hunger A, Del Valle P, Adjemian S, Zanatta DB, Kroemer G, Costanzi-Strauss E, Strauss BE. Intratumoral immunization by p19Arf and interferon-β gene transfer in a heterotopic mouse model of lung carcinoma. Transl Oncol 2016; 9(6):565-74; PMID:27916291; https://doi.org/10.1016/j.tranon.2016.09.011
- Hunger A, Medrano RF, Zanatta DB, Del Valle PR, Merkel CA, Salles TA, Ferrari DG, Furuya TK, Bustos SO, de Freitas Saito R et al. Reestablishment of p53/Arf and interferon-β pathways mediated by a novel adenoviral vector potentiates antiviral response and immunogenic cell death. Cell Death Discov 2017; 3:17017; PMID:28386458; https://doi.org/10.1038/cddiscovery.2017.17
- Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, Pietrocola F, Vacchelli E, Souquere S, Sauvat A et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016; 5(6):e1149673; PMID:27471616; https://doi.org/10.1080/2162402X.2016.1149673
- Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011; 11(8):519-31; PMID:21785456; https://doi.org/10.1038/nri3024
- Shaabani N, Duhan V, Khairnar V, Gassa A, Ferrer-Tur R, Häussinger D, Recher M, Zelinskyy G, Liu J, Dittmer U et al. CD169(+) macrophages regulate PD-L1 expression via type I interferon and thereby prevent severe immunopathology after LCMV infection. Cell Death Dis 2016; 7(11):e2446; PMID:27809306; https://doi.org/10.1038/cddis.2016.350
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017; 17(2):97-111; PMID:27748397; https://doi.org/10.1038/nri.2016.107
- Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, De Vleeschouwer S, Agostinis P, Graf N, Van Gool SW. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer 2015; 136(5):E313-25; PMID:25208916; https://doi.org/10.1002/ijc.29202
- Pampena MB, Levy EM. Natural killer cells as helper cells in dendritic cell cancer vaccines. Front Immunol 2015; 6:13; PMID:25674087; https://doi.org/10.3389/fimmu.2015.00013
- Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, Agostinis P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 2016; 8(328):328ra27; PMID:26936504; https://doi.org/10.1126/scitranslmed.aae0105