
ABSTRACT
Pancreatic ductal adenocarcinoma (PDA) remains a deadly disease that is rarely cured, despite many recent successes with immunotherapy for other malignancies. As the human disease is heavily infiltrated by effector T cells, we postulated that accurately modeling the PDA immune microenvironment would allow us to study mechanisms of immunosuppression that could be overcome for therapeutic benefit. Using viable precision-cut slices from fresh PDA, we developed an organotypic culture system for this purpose. We confirmed that cultured slices maintain their baseline morphology, surface area, and microenvironment after at least 6 d in culture, and demonstrated slice survival by MTT assay and by immunohistochemistry staining with Ki-67 and cleaved-Caspase-3 antibodies. Immune cells, including T cells (CD3+, CD8+, and FOXP3+) and macrophages (CD68+, CD163+ and HLA-DR+), as well as stromal myofibroblasts (αSMA+) were present throughout the culture period. Global profiling of the PDA proteome before and after 6 d slice culture indicated that the majority of the immunological proteins identified remain stable during the culture process. Cytotoxic effects of drug treatment (staurosporine, STS and cycloheximide, CHX) on PDA slices culture confirmed that this system can be used to assess functional response and cell survival following drug treatment in both a treatment time- and dose-dependent manner. Using multicolor immunofluorescence, we stained live slices for both cancer cells (EpCAM+) and immune cells (CD11b+ and CD8+). Finally, we confirmed that autologous CFSE-labeled splenocytes readily migrate into co-cultured tumor slices. Thus, our present study demonstrates the potential to use tumor slice cultures to study the immune microenvironment of PDA.
Introduction
Pancreatic ductal adenocarcinoma (PDA), commonly referred to as pancreatic cancer, is the fourth-leading cause of cancer-related death in the United States, owing to its 5-y survival rate of less than 7%.Citation1 Despite decades of research, PDA, which represents 80% of pancreatic malignancies, is often diagnosed at a late stage and is rapidly fatal.Citation2,3 Among the challenges that we must overcome to improve therapeutics for PDA is accurately modeling its microenvironment, as it is composed of a wide array of cells (including fibroblasts, pancreatic stellate cells, and immune cells), blood vessels, and the extracellular matrix (ECM), in addition to the cancer cells themselves.Citation4
Using an autochthonous murine model of PDA, Clark et al. showed that a leukocyte-predominant infiltrate paralleled disease progression, and that there were many immunosuppressive cell types, such as tumor-associate macrophages (TAM), myeloid derived suppressor cells (MDSC), and regulatory T cells; in contrast, there were very few effector T cells present.Citation5 In contrast, our previous study demonstrated that in human PDA, there exists a more complex mixture of both inflammatory and regulatory immune cells (including effector cells), and that neoadjuvant therapy reduces the number of cells associated with immunosuppression and with worsened survival.Citation6
The recent development of immunotherapy designed to block immune checkpoints, such as the programmed death 1 (PD-1) pathway, has demonstrated dramatic results in melanoma and other malignancies.Citation7,8 However, clinical results in small numbers of patients with advanced PDA treated with inhibitors of the PD-1 and CTLA-4 pathways have been disappointing.Citation9,10 This outcome may be attributed to the distinctive and hostile immune microenvironment of PDA containing elements such as exhausted intratumoral effector T cells, a massive infiltration of immunosuppressive leukocytes,Citation11,12 a strong desmoplastic reaction with multiple cell types, immunosuppressive molecular factors inherent to the ECM,Citation4 and an inflammatory program driven by oncogenes such as Ras.Citation13 Such a complex microenvironment, therefore, demands a similarly complex model system to accurately predict response to therapy.
To faithfully recapitulate the tumor microenvironment of PDA, including the critical elements of the immune system, we developed a model system using slices of fresh tumor samples. Tissue slice culture has recently demonstrated its ability to closely resemble the architecture of original organ or tumor to allow for performance of toxicity studies, drug metabolism studies in liver, lung, kidney, and intestine,Citation14,15 studies of drug susceptibility and mechanisms of resistance in head and neck cancer,Citation16 cell biology studies in both mouseCitation17,18 and human pancreas,Citation19 and gene therapy testing in pancreatic cancer.Citation20 Live antibody-labeled fluorescence imaging was also tested in slice culture tissue, most notably in lung slices (with staining of Lyve-1 and CD3121), as well as in the localization of dendritic cells in the mouse skin, both normal and malignant mouse vasculature,Citation22 and in epithelial DCs in human cornea in vivo.Citation23
On the other hand, there have been few instances in the literature studying the immune response in the slice culture model. Salmon et al. used slice cultures to determine that the ECM structure in human lung tumors restricts the preferential localization and migration of T cells into the stromal zone.Citation24 However, to our knowledge, no reports have thus far shown immune cell survival after slice culture in pancreatic cancer. In this study, we used precision-cut PDA slice cultures to test tissue viability, architecture and morphology preservation (including immune and stromal cell survival), and response to drug treatments. We also used this model in vitro to stain and specifically label both immune and epithelial cells in live pancreatic tumor tissue slices by antibody-labeled fluorescence, as well as to monitor the migration of carboxyfluorescein succinimidyl ester (CFSE) labeled autologous leukocytes through the tumor. Therefore, we have demonstrated that tumor slice cultures have the potential to expand our understanding of immune responses in the PDA microenvironment and aid in the development of novel immunotherapies for PDA.
Results
Slice cultures maintain morphology and surface area for over 1 week
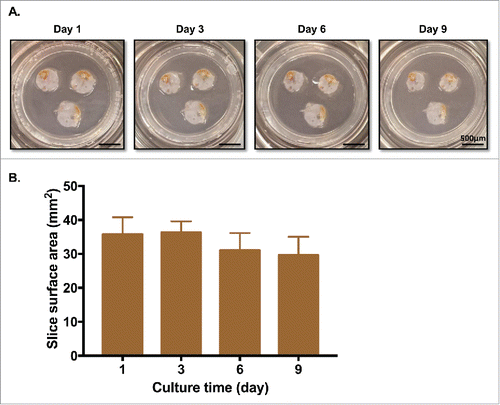
Fresh excess sterile PDA specimens were obtained immediately following surgical resection and pathology evaluation of margins from 13 patients (). Precision-cut sections were prepared and cultured as detailed in the Methods section, and were subjected to a variety of tests (). First, the surface areas of slices of PDA were measured on days 1, 3, 6, and 9 of culture. There was minimal change of surface area through day 9, and gross morphology of the slices remained quite similar to day 1 ().
Table 1. Overview of patient demographics.
Figure 1. The procedure flowchart for tumor slice culture. Human tumor tissue is transported in ice-cold 10% FBS RPMI 1640 media with 1% penicillin and streptomycin from the operating room to the laboratory and cut into 250-μm slices in buffer solution using a vibratome. The slices are transferred to culture medium and then carefully placed on membrane inserts in 6- or 24-well plates to create an air–liquid interface. The slices are cultured in vitro for survival and cytotoxic assays, architectural characterization identification and immunohistochemistry, as well as live immune-fluorescence imaging.
Figure 2. PDA tumor slices maintain morphology and surface area for over a week in culture. (A) PDA slices were cultured for up to 9 d, with fresh media changes performed every 2–3 d. Bar = 500 μm. (N = 3) (B) Surface area (mm2) of each slice was measured by analyzing photographs with Fiji Image J. There were no significant differences in surface area among days 1, 3, 6, and 9. (N = 3) Error bars represent STDEV.
Critical cellular components of the tumor microenvironment survive in slice culture
Next, we determined the health of the slices based on histology and the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In particular, we were interested to see if cells throughout the thickness of the slice remained viable in culture. We vertically embedded the slices in paraffin upon completion of each time point and cut them into 4-µm sections. To evaluate changes in tissue histology over time, H&E staining was performed on PDA slice culture sections. In PDA slices, we found that cell morphology was well-preserved over 9 d throughout the full thickness of the slice ().
Figures 3. Slice cultures preserve the overall tumor microenvironment. (A) Slice tissues maintain their architecture through their entire thickness. PDA slices were harvested on the indicated days, fixed and embedded in paraffin. The slices were cut vertically, stained with H&E, and imaged using brightfield microscopy. Bar = 100 μm. (N = 4) (B) MTT assay showed minimal changes over the culture periods. (N = 3) (C) PDA slices were stained with antibodies to Ki-67 and cleaved-Caspase-3 on days 1 and 6. Bar = 50 μm. (N = 3) (D) Quantification of each marker's expression demonstrated similar levels of cellular proliferation and apoptosis at both time points. (N = 2) Error bars represent STDEV.
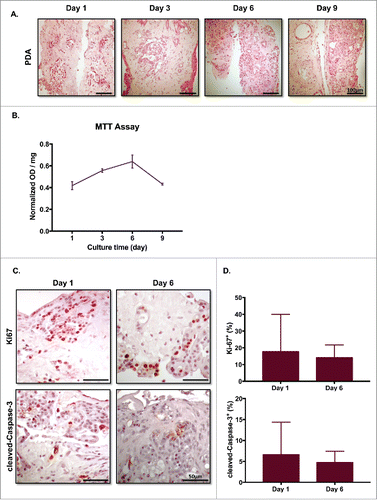
The MTT assay was used to quantify cell metabolism. Consistent with our findings from histology, there were minimal differences in normalized OD readings over the entire culture period from 1 to 9 d ().
To further verify slice culture cellular survival over various periods, we performed IHC for the proliferation marker Ki-67 and the apoptosis marker cleaved-Caspase-3 at days 1 and 6. Cells positive for either Ki-67 or cleaved-Caspase-3 were observed at both time periods across the whole slice vertical thickness (); there were no significant differences between day 1 and day 6 in the percentages of Ki-67+ or cleaved-Caspase-3+ cells ().
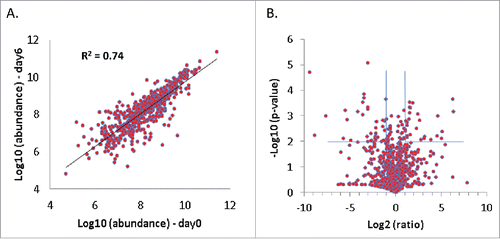
Next, to determine the extent of preservation of protein composition in vitro, PDA tissue slices before being placed in culture were compared with those cultured for 6 d using mass spectrometry-based proteomic analysis. 2,868 proteins were identified using stringent identification criteria, representing proteins from a wide range of cellular components with various molecular functions, including a large number of hydrophilic cytosolic proteins as well as hydrophobic membrane proteins. Among these proteins, more than 700 of them are annotated as immunologically-related proteins by Immunology Database and Analysis Portal (ImmPort) developed by Scheuermann et alCitation25. shows the correlation of expressional abundance of these immunological proteins before and after 6 d of slice culture. Proteins with a p-value ≤ 0.01 and abundance change ≥ 2-fold are defined as differentially expressed proteins (). For the given analytical sensitivity, the proteomic data showed a global similarity in protein profile between the PDA slices before and after culture. The impact on immunological proteins appeared to be minor, as only 63 immunological proteins identified showed differential expression after 6 d of culture (Table S1). Functional clustering analysis indicated that many of these proteins are extracellular proteins that have a functional role in biomolecular binding and are diversely involved in various immunologic processes (Fig. S1). While most of these proteins do not show significant interactions, 14–3–3 domain was enriched among the differentially expressed proteins (Fig. S2), possibly suggesting an influence on 14–3–3 proteins' binding ability to signaling proteins. Similar to the global level, most of the macrophage markers, including HLA-DR proteins, CD163, CD14, Integrin α M, CXCL2, CXCL3, CXCL9, and STAT1, did not show significant changes in abundance after 6 d culture. T-cell markers (e.g., CD3, CD4+, CD8+, and FOXP3) were subject to much lower abundance in the tissue proteome, and were not detected in the global profiling analysis. Overall, over 90% of the immunological proteins identified did not show significant alterations in abundance at global scale, suggesting that the slice culture model largely preserved the immunologically related proteome, consistent with the observation at the overall proteome level.
Figure 4. The majority of identified immunologic proteins remain stable over 6 d of slice culture. Duplicate PDA slices before and after 6 d of culture were analyzed by proteomics and compared. (A) Correlation of abundance of immunologic proteins annotated by ImmPort before and after 6 d of slice culture. (B) Volcano plot of the immunologic proteins before and after 6 d of slice culture. Proteins with a p-value ≤ 0.01 and an abundance change ≥ 2-fold are considered to have significant changes after 6 d in culture (N = 2).
T cells, macrophages, and stromal myofibroblasts survive in slice cultures
Given the challenges inherent in evaluating the effects of immunotherapies on human cancer, we postulated that slice cultures could fill this void. Therefore, we examined the survival of immune and stromal cells in slice culture. We performed IHC at days 1 and 6 of culture for the following markers: markers of T cells, specifically CD3 for all T cells, CD8+ for cytotoxic T cells, and FOXP3 for regulatory T cells (); and markers of macrophages, including CD68 for all macrophages, HLA-DR for classically activated M1 macrophages, and CD163 for alternatively activated M2 macrophages (). The percentage of each immune cell among total cells, as determined by the above staining, was not significantly different between days 1 and 6 of culture ().
Figure 5. Immune and stromal cells survive in slice culture. IHC for (A) T cell markers (CD3, CD8+, and FOXP3) (N = 3) and (B) macrophage markers (CD68, HLA-DR and CD163) was performed on days 1 and 6 of PDA slice cultures (all Bar = 50 μm). (N = 3) (C) Quantitation of cells staining for each marker demonstrates stability of immune cell populations. (N = 3) Error bars represent STDEV. (D) Day 1 and 6 slices were stained with α smooth muscle actin (αSMA) (Bar = 100 μm). (N = 4) The right panels are the expanded views of the region of interest indicated by the black boxes in the left panels.
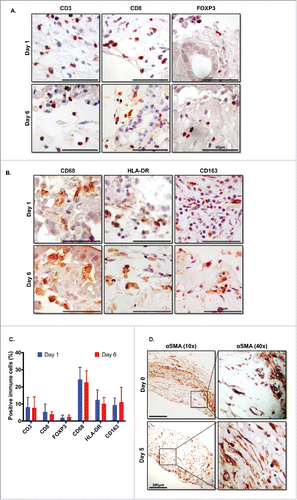
One feature of PDA that distinguishes it from other cancer types is the large amount of stroma that comprises the tumor, occupying up to 80% of the entire cancer.Citation26,27 This fibrous tissue component consists mainly of cancer-associated fibroblasts (CAFs), small blood vessels, inflammatory cells, and ECM.Citation27 As CAFs have been shown to interact with and potentially regulate T cell function in PDA, we set out to confirm the presence of CAFs over the course of the culture period.Citation28 IHC for α-smooth muscle actin (αSMA) was used to identify CAFs. After 6 d in slice culture, the slices continued to stain strongly for αSMA, demonstrating similar expression as in the original tumor before culture (). Further, the morphology of the CAFs within the surrounding ECM remained unchanged in culture.
The cytotoxic effects of drug treatment can be evaluated in slice culture
To investigate functional responses of slice cultures to cytotoxic drugs, we treated the PDA slices with staurosporine (STS), a protein kinase inhibitor commonly used experimentally to induce widespread apoptosis. After 48 h treatment, the slices were fixed and processed to stain with Ki-67 and cleaved-Caspase-3 antibodies. Pre-culture tumor (day 0) was used as an additional comparison to the day 3 negative control. We found that STS treated slices had slightly fewer Ki-67+ cells, but more cleaved-Caspase-3+ cells when compared with day 3 untreated slices and day 0 pre-culture tumor (). We quantified the percentage of Ki-67+ cells or cleaved-Caspase-3+ cells and found that there was a trend toward lower proliferation, but significantly increased apoptosis in PDA slices after 48 h of STS treatment (). We confirmed that STS induced significantly more cell death than untreated control slices at day 3 by comparing the total cell nuclei count (). Consistent with our prior data, there was no significant difference in the percentages of Ki-67+ and cleaved-Caspase-3+ cells between original tumor slices and day 3 untreated cultured slices (). Slices treated with 1-µM STS for 6 and 24 h at day 3 of slice culture had more cells undergoing apoptosis than did untreated slices. Additionally, 24 h of treatment resulted in more apoptotic cells compared with slices treated for 6 h ().
Figure 6. Dose- and time-dependent responses with drug treatment. (A) Effect of staurosporine (STS) on slice cell apoptosis. PDA slices were treated with vehicle or 1 µm STS for 48 h after one day culture. Slices were stained with antibodies Ki67 and cleaved-Caspase-3 at original tumor slice (day 0) and control and 1-μM STS 48 h treatment at day 3 culture. Bar = 50 μm. (N = 3) (B) Graphs show the percentages of Ki-67+ cells and cleaved-Caspase-3+ cells of total counted cells at day 0, day 3 control and 1-μM STS treatment of 48 h. Total cell nuclei numbers were counted for each condition. (N = 3) (C) Time-dependent response with STS treatment. Appearance of cleaved-Caspase-3+ cells following treatment of PDA slices with 1-μM STS for 6 or 24 h. Bar = 50 μm. Error bars represent STDEV. (N = 2) (D) Dose-dependent response with STS and cycloheximide (CHX) treatment. Appearance of cleaved-Caspase-3+ cells following treatment of PDA slices with 2 μM and 10 μM STS or 10 μg/mL and 50 μg/mL CHX for 6 h. Bar = 50 μm. Error bars represent STDEV. p-values are as follows: p < 0.05; **p < 0.005; ***p < 0.001. (N = 2).
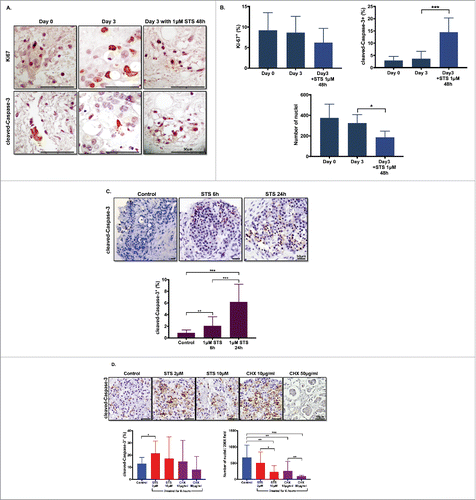
We also treated the slices at day 4 of culture with two different doses of STS (2 and 10 µM) and with another inhibitor of protein biosynthesis, cycloheximide (CHX), at 10 and 50 µg/mL doses for 6 h. Both doses of STS and 10 µg/mL CHX induced higher percentages of cleaved-Caspase-3+ cells than untreated slices, and this difference was significant when comparing untreated slices to 2-µM STS slices. Higher dose of STS (10 µM) and CHX (10 and 50 µg/mL) induced significant cell death with necrosis, as compared with untreated slices. There was a significant difference in total numbers of nuclei between 2 and 10-µM STS, as well as between 10 and 50 µg/mL of CHX; there were lower numbers of cell nuclei in each group compared with untreated slices (). Collectively, these data demonstrated that PDA slice cultures can be used to assess functional response and cell survival to drug treatment in both a treatment time-dependent and dose-dependent manner.
Imaging of cancer cells and immune cells from live slices
The trafficking of immune cells in human cancers affects their immunobiology but this has been difficult to study. This is particularly true in pancreatic cancer, due to the variety of potentially immunosuppressive cell types, such as regulatory T cells, M2 macrophages, and activated stromal myofibroblasts that accompany the effector CD8+ T cell infiltrate. Therefore, we devised methods to track immune cells and their interaction with cancer cells within live slice cultures.
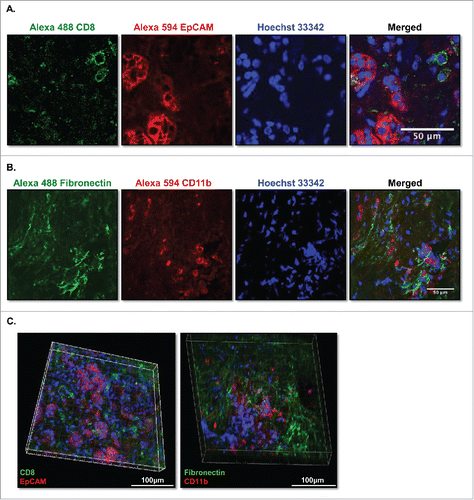
Following incubation of live slices with fluorescently conjugated antibodies, we identified cytotoxic T cells stained with anti-CD8+ antibody and epithelial tumor cells stained with anti-EpCAM antibody in PDA slices (). Double staining of PDA slices with both antibodies allowed us to identify CD8+ cytotoxic T cells (green) in or adjacent to clusters of EpCAM+ carcinoma cells (red) (). Stromal myofibroblasts were stained with fibronectin, and specific staining of macrophages was achieved with anti-CD11b antibody (). Double staining of PDA slices with fibronectin and CD11b allowed us to identify macrophages (red) among stromal myofibroblasts (green) (). To better understand the spatial relationship between cancer cells and immune cells in the tumor microenvironment, we double stained the slices of PDA with Alexa 594 EpCAM, and Alexa 488 CD8 and imaged serial Z stacks with a 2-µm z-step size ( and ). This live slice culture staining technique, which allows for identification and colocalization of a variety of fluorescently labeled cell types, represents a novel tool to study the interactions among multiple cell types in the 3D tumor microenvironment.
Figure 7. Multicolor immunofluorence images of live-stained culture slices. (A) PDA slices were stained with 10 μg/mL of Alexa Fluro 488 CD8 and Alexa Fluor 594 EpCAM (CD326) (N = 2) or (B) 10 μg/mL of Alexa Fluro 488 fibronectin and Alexa Fluor 594 CD11b antibodies for 3 h, followed by Hoechst 33342 nuclear stain for 5 min. Slices were then fixed and imaged by confocal microscopy. Bar = 50 μm. (N = 2) (C) The PDA slices of (A) and (B) were subjected to Z serial stack imaging (2 μm/stack) by confocal microscopy and were visualized in Volume views by Imaris software and Fiji Image J. Left panel, CD8+ (green), EpCAM+ (CD326) (red); right panel, fibronectin+ (green) and CD11b+ (red) and nuclei stain (blue) stained cells. Bar = 100 μm. (N = 2).
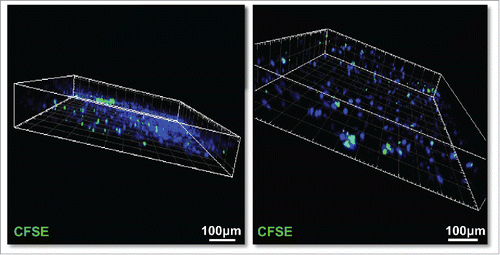
To examine the ability of immune cells to migrate into live slices, we isolated a single-cell suspension of autologous splenocytes or PBMC, labeled them with CFSE, and added them to the top of the slices on day 2 of culture. After 6 additional days of culture, the CFSE-labeled immune cells (green) were found to have migrated throughout the slices (). No positive cells were found in the slices without the addition of CFSE-labeled immune cells (data not show). The presence of adoptively transferred leukocytes in the slice cultures 6 d after addition to the culture system demonstrates the feasibility of using slice cultures as a preclinical model for the evaluation of adoptive cellular immunotherapies.
Figure 8. CFSE-labeled human slpenocytes migrate into live PDA slices. Isolated single-cell suspensions of autologous splenocytes (1×106) from a PDA patient were labeled with 1-μM CFSE and were added to tumor slice cultures on day 2 of culture. After 6 additional days of co-culture, the slices nuclei were stained with Hoechst 33342 (blue), fixed with 4% paraformaldehyde, and imaged by confocal microscopy with Z series stack image capture (2 μm/stack). Bar = 100 μm. (N = 2).
Discussion
Having a robust platform to accurately and reproducibly study the interactions of immune cells with carcinoma and stromal cells in the tumor microenvironment is of paramount importance in the development of effective immunotherapies for pancreatic cancer. In the present study, we have shown that slice cultures from fresh human PDA can survive for approximately one week and consistently maintain the morphology, a major component of the proteome, surface area and microenvironment (including cancer cells, a variety of immune cells, and stromal cells) that was present in the tumor in situ before resection ().
There are several lines of evidence to support our contention that slice cultures allow for precise, short-term modeling of the PDA tumor immune microenvironment. First, consistent with a prior study of head and neck slice cultures, our histology images show that there are no discernable structural changes between days 1 and 9 of culture, and that cell survival is consistent throughout the slice from membrane to air interface ( and ).Citation16 In contrast, Davies et al. studied multiple murine and human tumor types and did find some loco-regional changes induced by culture at the air–liquid interfaces (including more necrotic cells observed at the membrane interface).Citation29 This discrepancy might be attributable to the innate differences in both tissue type and species of origin, as well as small experimental differences in culture tissue, species and culture conditions. Second, our proteomics data confirmed that the PDA slice culture did not trigger major alterations in the immunologically related proteome after 6 d of culture, demonstrating a stable tumor organ culture system (). Third, we show that the major immune cell populations in pancreatic cancer, namely T cells and macrophages, maintain stable levels during ex vivo organotypic culture (); however, we have not yet determined the survival of other lower frequency immune cell populations, such as NK cells, neutrophils, or B cells in this system. Finally, our data demonstrate that PDA slices react in a predictable manner to drug treatment, and have readily measurable time- and dose-dependent functional responses ().
There is a small, but recently growing body of literature describing precision cut slice cultures from different normal and tumor tissues, which describe cellular survival, as well as preservation of the tissue microenvironment,Citation15-17 cellular stress,Citation19 and physiologic behavior.Citation30 However, to our knowledge, the present study is the first to demonstrate the survival of immune cells, cancer cells, and stromal cells in PDA slice cultures. To further enhance our ability to understand cell–cell interactions in PDA, we found that we could use primarily conjugated fluorescent antibodies to stain cancer cells and immune cells in live slices (). Additionally, we show that “adoptive transfer” of fluorescently labeled autologous immune cells is readily achieved in tumor slice cultures, and that these cells can be tracked in three dimensions as they traffic through the slice ().
In addition to the dense immune infiltrate, a well-established distinguishing feature of PDA is the great amount of fibrotic scar tissue that comprises up to 80% of the entire cancer mass.Citation26,27 This so called desmoplasia consists mainly of CAFs, small blood vessels, inflammatory cells, and ECM.Citation27 αSMA expression by activated pancreatic stellate cells appears to be a marker for CAF, and may have prognostic relevance for pancreatic cancer patients; therefore, we felt that confirming stable αSMA expression is critical to any model system of human PDA.Citation31,32 Importantly, our present study demonstrates that there is no difference of αSMA expression in PDA slice cultures between day 1 and 6 in culture.
As the primary goal of our work is to develop slice cultures as a platform for evaluating the effects of immunotherapy, particularly focusing on the development of personalized cancer care, we developed techniques to directly examine live slice cultures. Staining viable slices for cancer cells with fluorescently conjugated antibodies allowed us to acquire three-dimensional images by confocal microscopy. Taking advantage of the high spatial resolution in viable slices and the variety of readily available fluorescently labeled antibodies, we anticipate being able to further study the endogenous 3D interactions of cancer cells, immune cells, stromal cells, and endothelial cells in patient samples. To mimic adoptive transfer, as in the burgeoning field of adoptive T cell therapy, we co-cultured autologous CFSE-labeled splenocytes (or PBMC) and PDA slices for 6 d, and captured the migration of those cells into the slice.
As with any model system, we have identified several important limitations inherent to PDA organotypic slice cultures. First, although we did demonstrate stability of the slice culture microenvironment and the number of immune cells over the span of approximately one week in culture, significantly longer periods of study may be needed to accurately model an antitumor immune response. Second, the potential for intratumoral heterogeneity introduces the possibility that individual slices from geographically distinct regions of a tumor may vary in their response to cytotoxic or immune activating treatments; however, this can be accounted for by carefully planning the distribution of replicate slices for each experimental condition. Third and most importantly, the composition of the immune microenvironment of an organotypic slice in culture is limited to the tumor-infiltrating immune cells. Therefore, testing immunotherapeutics with this system cannot consider the contribution of cells entering the tumor from the circulation after the institution of such therapy. An additional aspect of the human slice culture system is that there is inherent variability among different individual patient tumors. This can be both a limitation and an asset to the system, as it does make cross-tumor comparisons challenging, while simultaneously increasing the validity associated with consistent results from experiments conducted with more than one patient tumor.
In conclusion, our study provides the baseline data and rationale to establish slice culture as an important model to study the tumor immunobiology of pancreatic cancer; we anticipate that further work will eventually make this technique useful for personalized clinical immunotherapy, particularly pertaining to adoptive cellular therapy.
Materials and methods
Ethics statement
All investigations performed in relation to this manuscript were conducted according to the principles expressed in the Declaration of Helsinki. Fresh tumor and spleen (or blood) were procured from patients undergoing pancreatic resection for pancreatic tumors, and who provided prior written-informed consent under a research protocol approved by the Cancer Consortium Institutional Review Board (CC-IRB) at the Fred Hutchinson Cancer Research Center. Tissue blocks were gathered under a separate CC-IRB-approved protocol for which there was a waiver of consent, as the study was considered to be of minimal risk since the tissue and data were collected solely for non-research purposes.
Preparation of tissue slices and process of slice samples
Tumors were collected directly from the operating room at the University of Washington Medical Center, USA and were placed into cold media (RPMI 1640 media with 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin (Gibco) before being transported to the nearby laboratory. Histologic examination of tumor samples was determined by a board certified pathologist (FJ). Slices were prepared and cut in extracellular solution consisting of (mmol/L): 140 NaCl, 5 KCl, 2 NaHCO3, 1 NaH2PO4, 1.2 MgCl2, 1.5 CaCl2, 3 glucose, and 10 HEPES (pH 7.4). Thick tissue slices (250 µm) were prepared under sterile conditions using a Vibratome, Leica VT1200 S (Leica Biosystems Nussloch GmbH, Germany) within 2–6 h after surgery. Slices were then carefully placed on 0.4-µm pore size membrane culture inserts (Millipore Corporation) pre-coated with collagen gel mixture (0.025 N NaOH, 3 mg/mL rat tail collagen type I in PBS buffer) in 6- or 24-well plates containing tumor culture medium. Slice culture medium is composed of RPMI 1640 consisting of 10% fetal bovine serum, 1% GlutaMAX (Gibco), 1.134 g /l NaHCO3, 14.5 mm HEPES, and 20 mg/L l-cystine (all from Sigma) and 1% penicillin and streptomycin. Later, slices were cultivated in a humidified incubator at 5% CO2 and 37 °C in atmospheric oxygen. Medium was changed on the second day and every 2–3 d thereafter. The slices were fixed with 4% paraformaldehyde for at least 1 h at 4°C and embedded in paraffin in vertical orientation for H&E and immunohistochemistry stain or following live antibody stain and in horizontal orientation for the immune migration assay.
Immunohistochemistry
Slides were de-paraffinized in xylene and rehydrated through graded ethanol followed by heat mediated antigen retrieval using 10 mM Sodium Citrate Buffer (pH 6.0). IHC was performed via incubations with one primary antibody followed by host-matched secondary polymer reagent and color substrate. Primary antibodies used were as follows: CD3+, CD8+, HLA-DR, CD68 (A0452, M7103, M0746, M0814 – Dako), CD163, Ki-67, and αSMA (ab189915, ab16667, ab5694 – Abcam), cleaved-Caspase-3 (#9661, Cell Signal Technologies), and FOXP3 (14–4777–82, eBioscience). Secondary reagents were ImmPress Rabbit HRP and Mouse HRP (Vector Laboratories). Color development was performed using Quanto DAB brown (Fisher Scientific). Slides were counterstained with Harris hematoxylin (Sigma) as appropriate, dehydrated and mounted.
Drug treatment
Slices were treated with staurosporine (STS) 1 µM for 6 h and 24 h, 2 µM and 10 µM for 6 h and Cycloheximide (CHX; Sigma) 10 µg/mL and 50 µg/mL for 6 h.
MTT assay
Slices (three slices per group) were weighed dry before being placed in 6-well or 24-well plates with membrane culture inserts in tumor culture medium, followed by MTT assay on various days. Briefly, slices were transfer to 24-well plates and 100 µL of 5 mg/mL MTT was added to each well with 400 µL medium and incubated for 3.5 h. The media was removed, and 750 µL of MTT solvent (4 mM HCl, 0.1% Nondet P-40 in isopropanol) was added to each well, followed by homogenization using the Kontes Glass Duall 20 Grinder (VWR). Optical density was read at 600 nm using an OPTImax tunable microplate reader (Molecular Devices). The OD value was normalized to dry slice weight.
Slice morphology and area measurement
Slice morphology was monitored after culture in different time points. Slice area was quantified manually in Fiji-ImageJ (NIH) by encircling the tissue borders and calculating area (12.1 pixels/mm2).Citation33
Proteomics
Sample preparations
Tissue lysate in RIPA lysis buffer was diluted with PBS buffer. The proteins were reduced by TCEP and alkylated by iodoacetamin (IDA). The proteins were precipitated and collected by a two-step cold acetone precipitation procedure, then resuspended in 50 mM ammonium bicarbonate. Digestion was performed in two steps with trypsin at 1:50 enzyme-to-protein ratio. Half of the trypsin was added and the sample was incubated at 37 °C for 2 h with vortexing at every 30 min. The second half of the trypsin was added and the mixture was incubated at 37 °C for 16 h. The trypsin digestion was terminated by adding 1% of formic acid (v/v).
LC MS/MS analysis
LC MS/MS analysis was performed using an Orbitrap FUSION Tribrid mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled with a nanoACQUITY Ultra Performance LC system (Waters, Milford, MA). 1 µg of sample was loaded first to a C18 trapping column (I.D. 100 µm, 5-µm particle size, 200 Å pore size), and resolved on a 25-cm C18 analytical capillary column (I.D. 75 µm, 5-µm particle size, 120 Å pore size). Solvents A and B were water and acetonitrile with 0.1% formic acid, respectively. A 90 min gradient of 5 to 30 % solvent B was applied to resolve the tryptic peptides. Eluted peptides were analyzed using a data-dependent analysis procedure on the mass spectrometer. MS1 survey scan, from 400 to 1600 m/z, was performed with a resolution of 120 K at m/z 200 with the Orbitrap. Selected precursor ions were isolated on the quadrupole MS with an isolation window of 1.6 m/z. The isolated precursor ions were activated by 28% normalized collision energy with HCD. The fragmented ions were then analyzed with the ion trap operating in tandem with the survey scan analysis.
Data analysis
The database search for peptide and protein identification was performed using Trans-Proteomic Pipeline (TPP, v.4.8.0). The MS raw data files were converted to mzML open format, and searched against UNIPROT human protein database using Comet. The search parameters are as follows: static modifications: 57.0215 at cysteine; differential modifications: 15.9949 at methionine. The peptide sequence assignment was assessed with PeptideProphet. Only peptides identified with a PeptideProphet score ≥ 0.95 were included for protein identification and quantification. Peptide and protein quantification was achieved using Skyline software (v. 3.1). For any given protein, the areas under the curve for all the corresponding peptides were summed to represent the protein quantification.
Antibody-labeled fluorescence live staining and imaging of slice tissue
Live slices were rinsed with PBS twice and incubated with different fluorescent antibody-labeled cocktails in PBS with Alexa Fluor 594 anti-human CD326 (EpCAM), Alexa fluor 594 CD11b (324228, 101254 – BioLegend), Alexa Fluor 488 CD8a, Alexa Fluor fibronectin (53–0008, 53–9869 – eBiosciences) (10 µg/mL each) for 3 h at room temperature (RT). After antibody incubation and washing with PBS, slices were stained with Hoechst 33342 (10 µg/mL) for 10 min. To reduce the autofluorescence in the slices, 0.1% Sudan Black B in 70% ethanol was added to slices (20 min at RT), followed by PBS with 0.02% Tween 20 wash. Then, the slices were fixed with 4% paraformaldehyde. Finally, slices were imaged using a Leica SP8X confocal microscope (Leica Microsystems Inc.). The image process was analyzed by Fiji-ImageJ and Imaris software (Bitplane AG).
Isolation of splenocytes
When the spleen was also resected for clinical care, autologous patient spleen samples were collected directly from the operating room and transported to the laboratory in the same manner as that described for tumor above. To make single-cell suspensions, we minced spleen and incubated at 37°C for 30–45 min in an enzymatic cocktail (DNAse (0.1 mg/mL, Roche), collagenase type IV (1 mg/mL), and hyaluronidase (0.1 mg/mL, Worthington) in RPMI 1640. The suspension was then passed through a 70-µm filter, washed, and red blood cells were lysed with ACK RBC lysis. The resulting single-cell suspension of splenocytes was counted and either used immediately for co-culture with tumor slices or cryopreserved in media+10% DMSO for later analysis.
CFSE-labeled leukocyte migration assay
Single-cell suspensions of splenocytes (or PBMC) were stained with 1 µM CFSE (Invitrogen) in PBS for 9 min at 37°C, combined with 20 mL of 10% FBS RPMI-1640 medium at RT for 2 min, centrifuged, washed, and counted. 2 × 106 CFSE-labeled splenocytes were added to the top of the slices in 24-well membrane culture insert. After 6 d co-culture, slices were fixed with 4% paraformaldehyde and stained the nuclei with DAPI. Finally, slices were visualized with the Leica SP8X confocal microscope with Z stack of 2 µm/step. The image process was analyzed by Fiji-ImageJ and Imaris software.
Quantification of nuclei and statistical analysis
Total cell numbers and cells positive for the proliferative marker Ki-67 and the apoptosis marker cleaved-Caspase-3, as well as immune cells (CD3, CD8+, FOXP3, CD68, CD163, and HLA-DR) were determined by counting the nuclei in stained slides with Fiji-ImageJ plug-in Cell Counter (NIH). Values in graphs represent mean ± STDEV. Differences between groups were determined using parametric two-tailed t-tests (paired or unpaired, as appropriate). Number (N) in figure legends represents number of repetitions of each experiment (i.e., number of individual tumors tested). Number of slices used in each experimental repetition varied from 2 to 3; refer to Table S2 for exact number of slices used from each tumor. p-values ≤ 0.05 were considered to be statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_materials.zip
Download Zip (1.2 MB)Acknowledgments
We acknowledge Dr Ru Chen from Department of Medicine for support and overview of proteomics data and Dr Nathanial Peters for imaging support from the NIH to the UW W. M. Keck Microscopy Center (S10 OD016240), University of Washington.
Funding
This work was supported by generous funding from the Swim Across America (VGP), National Institutes of Health Cancer Center Support Grant P30-CA015704 (ALC) and the University of Washington Department of Surgery Research Reinvestment Fund (VGP).
References
- Cancer Facts & Figures 2016. Available at: http://www.cancer.org/acs/groups/content/@research/documents/…/acspc-047079.pdf.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10-29; PMID:22237781; https://doi.org/10.3322/caac.20138
- Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100,313 patients diagnosed from 1985-1995, using the National Cancer Database. J Am Coll Surg 1999; 189:1-7; PMID:10401733; https://doi.org/10.1016/S1072-7515(99)00075-7
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res 2012; 18:4266-76; PMID:22896693; https://doi.org/10.1158/1078-0432.CCR-11-3114
- Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007; 67:9518-27; PMID:17909062; https://doi.org/10.1158/0008-5472.CAN-07-0175
- Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM, Whiting SH, Yeh MM, Yee C, Riddell SR et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS One 2014; 9:e96565; PMID:24794217; https://doi.org/10.1371/journal.pone.0096565
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; https://doi.org/10.1056/NEJMoa1003466
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320-30; PMID:25399552; https://doi.org/10.1056/NEJMoa1412082
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; https://doi.org/10.1056/NEJMoa1200694
- Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010; 33:828-33; PMID:20842054; https://doi.org/10.1097/CJI.0b013e3181eec14c
- Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer 2013; 108:914-23; PMID:23385730; https://doi.org/10.1038/bjc.2013.32
- Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: Impact on clinical outcome. Nat Rev Cancer 2012; 12:298-306; PMID:22419253; https://doi.org/10.1038/nrc3245
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012; 122:639-53; PMID:22232209; https://doi.org/10.1172/JCI59227
- de Kanter R, Monshouwer M, Meijer DK, Groothuis GM. Precision-cut organ slices as a tool to study toxicity and metabolism of xenobiotics with special reference to non-hepatic tissues. Curr Drug Metab 2002; 3:39-59; PMID:11878310; https://doi.org/10.2174/1389200023338071
- Guyot C, Lepreux S, Combe C, Sarrazy V, Billet F, Balabaud C, Bioulac-Sage P, Desmoulière A. Fibrogenic cell phenotype modifications during remodelling of normal and pathological human liver in cultured slices. Liver Int 2010; 30:1529-40; PMID:20846345; https://doi.org/10.1111/j.1478-3231.2010.02342.x
- Gerlach MM, Merz F, Wichmann G, Kubick C, Wittekind C, Lordick F, Dietz A, Bechmann I. Slice cultures from head and neck squamous cell carcinoma: A novel test system for drug susceptibility and mechanisms of resistance. Br J Cancer 2014; 110:479-88; PMID:24263061; https://doi.org/10.1038/bjc.2013.700
- Marciniak A, Cohrs CM, Tsata V, Chouinard JA, Selck C, Stertmann J, Reichelt S, Rose T, Ehehalt F, Weitz J et al. Using pancreas tissue slices for in situ studies of islet of Langerhans and acinar cell biology. Nat Protoc 2014; 9:2809-22; PMID:25393778; https://doi.org/10.1038/nprot.2014.195
- Marciniak A, Selck C, Friedrich B, Speier S. Mouse pancreas tissue slice culture facilitates long-term studies of exocrine and endocrine cell physiology in situ. PLoS One 2013; 8:e78706; PMID:24223842; https://doi.org/10.1371/journal.pone.0078706
- Rebours V, Albuquerque M, Sauvanet A, Ruszniewski P, Lévy P, Paradis V, Bedossa P, Couvelard A. Hypoxia pathways and cellular stress activate pancreatic stellate cells: Development of an organotypic culture model of thick slices of normal human pancreas. PLoS One 2013; 8:e76229; PMID:24098783; https://doi.org/10.1371/journal.pone.0076229
- van Geer MA, Kuhlmann KF, Bakker CT, ten Kate FJ, Oude Elferink RP, Bosma PJ. Ex-vivo evaluation of gene therapy vectors in human pancreatic (cancer) tissue slices. World J Gastroenterol 2009; 15:1359-66; PMID:19294766; https://doi.org/10.3748/wjg.15.1359
- Thornton EE, Krummel MF, Looney MR. Live imaging of the lung. Current Protocols in Cytometry 60:12.28:12.28.1–12.28.12; PMID:22470155; https://doi.org/10.1002/0471142956.cy1228s60
- Cummings RJ, Mitra S, Lord EM, Foster TH. Antibody-labeled fluorescence imaging of dendritic cell populations in vivo. J Biomed Opt 2008; 13:044041; PMID:19021368; https://doi.org/10.1117/1.2966122
- Mastropasqua L, Nubile M, Lanzini M, Carpineto P, Ciancaglini M, Pannellini T, Di Nicola M, Dua HS. Epithelial dendritic cell distribution in normal and inflamed human cornea: In vivo confocal microscopy study. Am J Ophthalmol 2006; 142:736-44; PMID:17056357; https://doi.org/10.1016/j.ajo.2006.06.057
- Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, Mami-Chouaib F, Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest 2012; 122:899-910; PMID:22293174; https://doi.org/10.1172/JCI45817
- Kong YM, Dahlke C, Xiang Q, Qian Y, Karp D, Scheuermann RH. Toward an ontology-based framework for clinical research databases. J Biomed Inform 2011; 44:48-58; PMID:20460173; https://doi.org/10.1016/j.jbi.2010.05.001
- Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM et al. Stromal biology and therapy in pancreatic cancer. Gut 2011; 60:861-8; PMID:20966025; https://doi.org/10.1136/gut.2010.226092
- Kadaba R, Birke H, Wang J, Hooper S, Andl CD, Di Maggio F, Soylu E, Ghallab M, Bor D, Froeling FE et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J Pathol 2013; 230:107-17; PMID:23359139; https://doi.org/10.1002/path.4172
- De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med 2011; 208:469-78; PMID:21339327; https://doi.org/10.1084/jem.20101876
- Davies EJ, Dong M, Gutekunst M, Närhi K, van Zoggel HJ, Blom S, Nagaraj A, Metsalu T, Oswald E, Erkens-Schulze S et al. Capturing complex tumour biology in vitro: Histological and molecular characterisation of precision cut slices. Sci Rep 2015; 5:17187; PMID:26647838; https://doi.org/10.1038/srep17187
- Kang C, Qiao Y, Li G, Baechle K, Camelliti P, Rentschler S, Efimov IR. Human organotypic cultured cardiac slices: New platform for high throughput preclinical human trials. Sci Rep 2016; 6:28798; PMID:27356882; https://doi.org/10.1038/srep28798
- Fujita H, Ohuchida K, Mizumoto K, Nakata K, Yu J, Kayashima T, Cui L, Manabe T, Ohtsuka T, Tanaka M. alpha-smooth muscle actin expressing stroma promotes an aggressive tumor biology in pancreatic ductal adenocarcinoma. Pancreas; November 2010; 39(8):1254-1262; PMID:20467342; https://doi.org/10.1097/MPA.0b013e3181dbf647
- Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A, Giese NA, Esposito I, Friess H, Kleeff J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol 2008; 6:1155-61; PMID:18639493; https://doi.org/10.1016/j.cgh.2008.05.006
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al. Fiji: An open-source platform for biological-image analysis. Nat Methods 2012; 9:676-82; PMID:22743772; https://doi.org/10.1038/nmeth.2019