
ABSTRACT
Optimal approaches to differentiate tumor antigen-specific cytotoxic T lymphocytes (CTLs) from pluripotent stem cells (PSCs) remain elusive. In the current study, we showed that combination of in vitro priming through Notch ligands and in vivo development facilitated the generation of tumor Ag-specific CTLs that effectively inhibited tumor growth. We co-cultured the murine induced PSCs (iPSCs) genetically modified with tyrosinase-related protein 2 (TRP2)-specific T cell receptors with OP9 cell line expressing both Notch ligands Delta-like 1 and 4 (OP9-DL1/DL4) for a week before adoptively transferred into recipient C67BL/6 mice. Three weeks later, B16 melanoma cells were inoculated subcutaneously, and the antitumor activity of the iPSC-derived T cells was assessed. We observed the development of the TRP2-specific iPSC-CD8+ T cells that responded to Ag stimulation and infiltrated into melanoma tissues, significantly inhibited the tumor growth, and improved the survival of the tumor-bearing mice. Thus, this approach may provide a novel effective strategy to treatment of malignant tumors.
Introduction
Recent studies demonstrate that immunotherapy based on adoptive cell transfer (ACT) of T cells can induce tumor regression in patients with metastatic cancersCitation1 or eliminate pathogens in viral infection.Citation2 T cells can be genetically engineered by a variety of techniques to express major histocompatibility complex (MHC)-restricted T cell receptor (TCR) or -unrestricted chimeric Ag receptor (CAR), with high affinity and specificity for tumor-associated antigens (Ags). Genetic engineering approaches can be used to effectively target any cancer tissues because T cells can be engineered to target Ags expressed by the transformed and non-transformed cells within the tumor mass.Citation3
Naive or central memory T cell-derived effector cytotoxic T lymphocytes (CTLs) are used in the ACT-based therapies due to their high proliferative potential, less prone to apoptosis than terminally differentiated cells, and strong ability to respond to homeostatic cytokines such as IL-7 and IL-15. Nevertheless, the difficulties involved in obtaining sufficient cells from patients limit the utility of the ACT-based therapies. To date, stem cells are considered good sources to generate a large amount of naive Ag-specific T cells. Induced pluripotent stem cells (iPSCs) can be easily generated from patients' somatic cells by transduction of various transcription factors, and they exhibit characteristics identical to those of embryonic stem cells (ESCs).Citation4,5 This approach provides promise for generating patient- or disease-specific PSCs.Citation6,7 A number of genetic methods as well as protein-based approaches have been developed to produce iPSCs with potentially reduced risks, including reduced immunogenicity.Citation8,9 Our previous studies indicated that in vivo programming of Ag-specific iPSC-T cells can be used for cell-based therapies of cancers and autoimmune disorders.Citation10-13
Reports by several groups including our own have shown successful development of stem cell-derived Ag-specific T lymphocytes for immunotherapy.Citation10,14,15 Nevertheless, there are concerns with the previous approaches, i.e., they may result in non-specific differentiation and prolonged period of in vitro and/or in vivo development. In this study, we co-cultured murine iPSCs genetically modified with tyrosinase related protein 2 (TRP2; an enzyme highly expressed in cutaneous melanoma and melanocytes)-specific TCR with OP9 cell line expressing Notch ligands for a week before adoptively transferred into recipient mice. TRP2 protein is a single-pass type I membrane protein belonging to the melanocyte differentiation Ags, and has been considered as a target for melanoma immunotherapy. We showed that the combinative in vitro and in vivo development of TRP2-specific iPSC-CD8+ T cells remarkably infiltrated into the melanoma tissues, significantly inhibited the tumor growth and improved the survival of tumor-bearing mice. Thus, this novel approach to generating naive single-type tumor Ag-specific PSC-CD8+ T cells may have great potential to be adapted for cancer immunotherapy.
Materials and methods
Cell lines and mice
The mouse iPS-MEF-Ng-20D-17 cell line was obtained from RIKEN Cell Bank.Citation10 Expressions of Oct3/4, Sox2, Klf4, and c-Myc were confirmed by RT-PCR, and expression of GFP was confirmed by flow cytometry. OP9 cells and B16-F10 melanoma cells were obtained from ATCC (Manassas, Virginia). OP9 cell line expressing either GFP or murine DL1 (OP9-DL1) was a kind gift from Dr Juan Carlos Zúñiga-Pflücker at the University of Toronto. OP9 cells that overexpress DL4 (OP9-DL4) or both (OP9-DL1/DL4) ligands were generated by retrovirus-mediated gene introduction and enriched by fluorescent activated cell sorting (FACS). C57BL/6 mice (4–6-week-old) were purchased from The Jackson Laboratory (Bar Harbor, ME). All experiments were performed in compliance with the regulations of the Institutional Committee of Animal Use and Care of The Pennsylvania State University College of Medicine, and in accordance with guidelines by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Cell culture
iPSCs were maintained on feeder layers of irradiated SNL76/7 cells as described previously.Citation10
Retroviral transduction
Retroviral transduction was performed as described previously.Citation10 Expression of DsRed was determined by flow cytometry gating on GFP+ cells. DsRed+ GFP+ cells were purified by cell sorting using a MoFlo high performance cell sorter (Dako Cytomation, Fort Collins, CO).
PCR assay
Genomic DNA from different cells were extracted using the DNeasy Blood & Tissue DNA isolation Kit (Qiagen, Valencia, CA). DNA assay to evaluate the incorporated TRP2 TCR was performed using the Qiagen PCR mastermix kit (Qiagen, Valencia, CA). The primers used for detecting integrated TRP2 TCR are: forward: 5′–ATTTTAGATCTCCACCATGCTGATTCTAAGCCTGTTG–3′; reverse: 5′– TAAGAATTCTCAGGAATTTTTTTTCTTGAC−3′.
Western blot
Western blot was performed as described previously.Citation10
Antibodies
PE- or APC-conjugated anti-mouse IL-2 (JES6–5H4) and IFNγ (XMG1.2) antibodies were obtained from BD PharMingen (San Diego, CA). FITC-, PE-, PE/Cy7-, or PerCP-, PE/Cy5-, APC/Cy7-, Pacific Blue-, PerCP/Cy5.5- or APC-conjugated anti-mouse TCRVβ11, CD4+, CD8+, CD25, CD69, CD44, CD19, CD62L, CD127, CD11b, and CD117 antibodies were obtained from either BioLegend (San Diego, CA) or BD PharMingen (San Diego, CA). Delta antibody (C-20) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Adoptive cell transfer
In vitro primed TRP2-iPSC-derived DsRed+ progenitor cells (3 × 106) and control cells were washed and re-suspended in cold PBS before i.v. injection into mice through the tail vein. Four-six weeks later, CD8+ T cell development in the lymph nodes and spleen was determined by flow cytometry.
Flow cytometric analysis
Expressions of GFP, CD117, CD25, CD69, CD44, CD4+, CD8+, and other markers were analyzed by flow cytometry after gating on DsRed+ cells or other markers such as GFP expression. iPSC-derived TRP2-specific cells recovered from the adoptively transferred mice were evaluated by surface markers CD4, CD8, and TCRVβ11.
TCR profiling
The specific TCR Vα or Vβ of in vitro generated SP CD3+CD8+ T cells were determined using the TCRExpress™ mouse TCR Vα and Vβ repertoire screening kits (BioMed Immunotech, FL). RNA was purified from iPSC-T cells and used for one step PCR amplification and TCR gel analysis.
Ex vivo peptide stimulation assay
iPSC-derived TRP2-specific cells recovered from the adoptively transferred mice were evaluated for their antigen reactivity by ex vivo stimulation assay. Briefly, CD8+ cells were negatively selected from pooled lymph nodes and spleens using MACS separation column (Miltenyi Biotec, San Diego, CA), and then pulsed with irradiated splenocytes loaded with TRP2180–188 peptide (GenScript, Piscataway, NJ). T cell activation was assessed by the synthesis of IL-2 and IFNγ using intracellular cytokine staining.
Murine melanoma model
Four weeks after adoptive transfer of in vitro primed cells, 5 × 105 B16-F10 melanoma cells were subcutaneously inoculated into the right flank of C57/BL6 mice. Tumor sizes were measured by a caliper and tumor volumes were calculated as: V = long diameter × short diameter2 × 0.52.Citation12
Histology and immunofluorescence
For H&E staining, tumor tissues were fixed by 10% neutral formalin solution (VWR, West Chester, PA), and the fixed samples were prepared and stained as described.Citation10 For immunofluorescent microscopy, tissues were frozen in cryovials on dry ice immediately after resection. Cryosectioning and immunofluorescent staining were performed as described.Citation10 FITC-labeled anti-TCRVβ11 antibody was used to detect the tumor infiltrating TRP2-iPSC derived CTLs.
Statistical analysis
One-way ANOVA analysis was used for analysis of difference between the groups using GraphPad Prism (GraphPad Software, San Diego, CA), and the significance was set at 5%.
Results and discussion
Generation of TRP2 TCR gene-transduced iPSCs (TRP2-iPSCs)
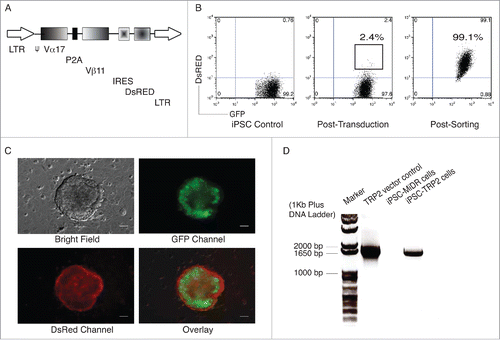
The retroviral vector MiDRCitation10 that contains TRP2-specific TCR Vα17 and Vβ11 genes was used in this study (). Following the transduction, DsRed expression was visualized by fluorescent microscopy (). Although the transduction efficiency was slight, we were able to sort for DsRed+ GFP+ cells (). Moreover, we confirmed the gene integration by the PCR amplification of the full length TRP2-specific TCR genes of isolated gemonic DNA from TRP2-iPSCs (). We also generated the OP9 cell line that co-expresses DL1 and DL4 ligands (OP9-DL1/DL4). OP9-DL1/DL4 cells highly expressed both of DL1 and DL4 ligands on the cell surface (Fig. S1), and this cell line induces stem cell differentiation toward CD8+ T cell in vitro.Citation16
Figure 1. Genetic modification of iPSCs with TRP2-specific TCR. Mouse iPSCs were transduced with the MIDR vector or TRP2-specific TCR (MiDR-TCR). (A) Schematic representation of the retroviral construct. (B) GFP+ iPSCs (left) were transduced with the retroviral construct MiDR-TCR, and GFP+ DsRed+ iPSCs (middle) were analyzed by flow cytometry and sorted by a high-speed cell sorter (right). (C) TCR-transduced iPSCs were visualized by fluorescence microscopy (scale bars: 50 μm). (D) GFP+ DsRed+ iPSCs were sorted and DNA were analyzed for TCR (Vα17 and Vβ11) gene expression by PCR. The forward primer is ATTTTAGATCTCCACCATGCTGATTCTAAGCCTGTTG and the reverse primer is TAAGAATTCTCAGGAATTTTTTTTCTTGAC. Data are representative of three identical experiments.
In vitro differentiation of TRP2-specific iPSC-CD8+ T cells
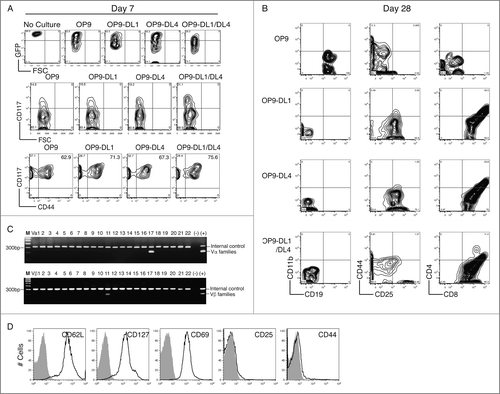
On Day 7, TRP2-iPSCs started to differentiate, as evidenced by a decreased GFP expression in comparison to the undifferentiated cells. A higher percentage of CD117+ cells were identified in the GFPlo TRP2-iPSCs co-cultured with OP9-DL1/DL4 cells (31.1%). CD44 expression was also detected in the CD117+ population with a higher ratio in the OP9-DL1/DL4 feeder cell group (75.6%) (), which is typical of hematopoietic differentiation. On Day 12, no significant expression of CD19, CD11b, CD25, CD44, CD8+, and CD4 were detected in all of the OP9 groups with or without stimulation with Notch ligands. CD44 expression was observed in all of the OP9 groups that expressed Notch ligands. By contrast, TRP2-iPSCs on OP9 cells showed a notably decreased CD44 expression (Fig. S2A). On Day 18, TRP2-iPSCs with Notch ligand stimulation differentiated toward early T lineage in the context of expressions of CD25, CD44, and CD8+; however, without Notch ligand stimulation, TRP2-iPSCs differentiated into B lineage, as evidenced by CD19 expression (Fig. S2B). TRP2-iPSCs stimulated by both DL1 and DL4 had a higher percentage of CD8+ single positive (SP) (63.1%) and CD4+CD8+ double positive (DP) (11.8%) cells than those stimulated with either DL1 or DL4 alone. In the DL1/DL4 stimulation group, there were more cells found in the double negative stage 3 (DN3) as characterized as CD44−CD25+ (17.6%). After 28 d of co-culture in the presence of both DL1 and DL4, only a small fraction of TRP2-iPSCs was CD4+CD8+ DP (11.4%) and the majority of them was CD8+ SP (85.3%) and CD25−CD44− (; the bottom panel). TCR analysis demonstrated the gene expression of TCR Vα 17 and Vβ11 only in the CD8+ SP and CD25−CD44− cells (). In addition, CD8+ TCRVβ11+ SP cells showed high expressions of CD62L and CD127 but not CD25 (), indicating that the TRP2-specific iPSC-CD8+ T cells have a naive phenotype. These results clearly demonstrate that the TRP2-specific iPSC-CD8+ T cells are naive single-type T cells.
Figure 2. In vitro differentiation of TRP2-specific iPSC-CD8+ (T)cells. GFP+ DsRed+ iPSCs were co-cultured with OP9, OP9-DL1, OP9-DL4, or OP9-DL1/DL4 cells for various periods of time, and the cell surface markers were analyzed by flow cytometry. (A) Expression of GFP, CD117, CD117, and CD44 on day 7. (B) Expression of CD19 with CD11b, CD25 with CD44, and CD8+ with CD4+ on day 28. (C) TCR profiles of SP CD8+ cells on day 28. (D) Expression of CD62L, CD127, CD69, CD25, and CD44 on SP CD8+ TCRVβ11+ cells on day 28. The data shown are representative of three identical experiments.
In vitro priming of TRP2-iPSCs
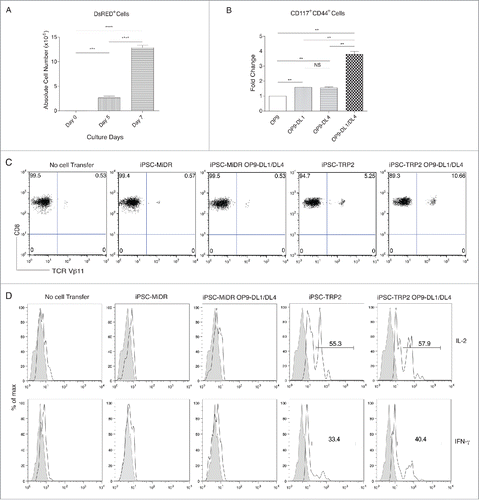
TRP2-iPSCs were co-cultured with OP9, OP9-DL1, OP9-DL4, or OP9-DL1/DL4 cells for 7 d before adoptively transferred into host animals. shows that the numbers of DsRED+ cells on various days in the group of OP9-DL1/DL4 expanded drastically: from 5 × 104 on day 0 to 2.67 × 106 on day 5 and to 1.29 × 107 on day 7. The percentage of CD117+CD44+ DP cells in the OP9-DL1/DL4 group is 3.79 times higher than the OP9 control group (). These results indicate that short-term priming by OP9-DL1/DL4 promotes a robust early T lineage differentiation.
Figure 3. Development of TRP2-specific iPSC-CD8+ (T)cells by a combination approach of in vitro priming and in vivo differentiation. TRP2-iPSCs were co-cultured with OP9, OP9-DL1, OP9-DL4, or OP9-DL1/DL4 cells for 7 days, and then adoptively transferred into host animals for 3-week in vivo differentiation. (A) Cell numbers of DsRED+ populations on various days following co-culture of TRP2-iPSCs with OP9-DL1/DL4 cells for 7 d. Data shown represent mean ( ±SD) counts from three experiments. ***p < 0.0001. (B) Fold changes of numbers of CD117+CD44+ cells co-cultured with OP9, OP9-DL1, OP9-DL4, or OP9-DL1/DL4 cells for 7 d. Data shown represent mean ( ±SD) counts from three experiments. **p < 0.001). (C) Three weeks after in vivo differentiation, CD8+TCRVβ11+ cells from the pooled lymph nodes and spleen of mice in different groups were analyzed by flow cytometry, after gating on CD8+ T cell population. Data shown are representative of three identical experiments. (D) Three weeks after in vivo differentiation, the pooled lymph nodes and spleen of mice in different groups were stimulated with TRP2180–188 peptide and analyzed by intracellular cytokine staining (dark lines; shaded areas indicate isotype controls), after gating on CD8+ T cells. Data shown are representative of three identical experiments.
In vivo differentiation of primed TRP2-iPSCs
TRP2-iPSCs were co-cultured with OP9, OP9-DL1, OP9-DL4, or OP9-DL1/DL4 cells for 7 days, and then adoptively transferred into host animals. Three weeks later, a larger population of CD8+TCRVβ11+ DP cells were identified in mice receiving the primed TRP2-iPSCs (10.66%) as compared with the non-primed cells (5.25%) or negative controls (). Furthermore, CD8+ T cells from mice receiving the primed TRP2-iPSCs responded to TRP2180–188 peptide stimulation and produced IL-2 and IFNγ (). In addition, in mice that received primed TRP2-iPSCs and inoculated with melanoma cells, the local draining lymph nodes and spleens had reactive responses and presented larger sizes (Fig. S3). These results show that the functional TRP2-specific iPSC-CD8+ T cells can be generated by a combinative in vitro and in vivo development.
Effective tumor suppression by adoptive transfer of the primed TRP2-iPSCs
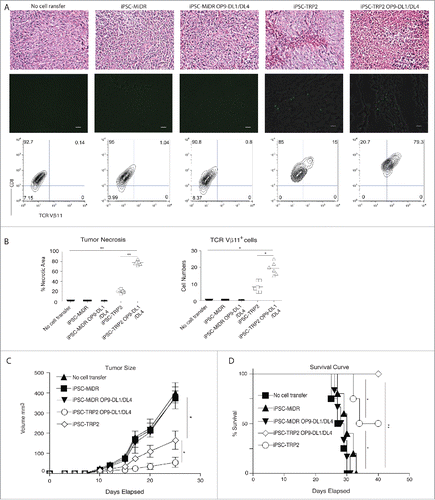
To determine the potential usefulness and benefits of this approach in cancer treatment, we next determine the effects of adoptive transfer of the primed TRP2-iPSCs on tumor development and growth. TRP2-iPSCs were co-cultured with OP9, OP9-DL1, OP9-DL4, or OP9-DL1/DL4 cells for 7 d and then adoptively transferred into host animals. Three weeks later, mice were inoculated s.c. with B16 melanoma cells in the flank region. From day 21 to 22 after tumor challenge, tumor tissues were examined for tumor-reactive T cell infiltration. In the group of the primed TRP2-iPSCs, tumors had larger areas of necrotic tissue and hypocellularity (75% of total area) as compared with that in the control groups. A significant population of the TRP2-specific CD8+TCRVβ11+ T cells was detected in the melanoma tissues, suggesting a high infiltration rate of those cells (79.3%) ( and ). Notably, the tumor mass from mice receiving the primed TRP2-iPSCs was substantially smaller than that of other groups. Furthermore, the mice receiving the primed TRP2-iPSCs showed a delayed pattern of tumor growth as compared with other control groups (Fig. S4). Mice that received the primed TRP2-iPSCs also survived longer: the mice in control groups generally started to succumb from day 28 following melanoma challenge; in contrast, all the mice that received the primed TRP2-iPSCs remained alive during a 40-d observation ().
Figure 4. Effective suppression of tumor growth by adoptive transfer of primed TRP2-iPSCs. TRP2-specific iPSC-CD8+ T cells were generated as described in . Three weeks after in vivo differentiation, mice were inoculated s.c. with B16 melanoma cells in the flank region. (A) On day 21 and 22 after tumor challenge, tumor tissues were examined for tumor-reactive T cell infiltration. Necrotic tissue and hypocellularity by HE staining (up panel), immunofluorescent staining (middle panel; TRP2-specific TCRVβ11+ CD8+ T cells in green infiltrated in tumor tissues in black) and by flow cytometry (low panel) (scale bars: 20 μm). Data shown are representative of three identical experiments. (B) Quantitation of tumor necrosis and cell infiltration of CD8+ TCRVβ11+ DP population in tumor tissue. Data represent mean ( ± SD) counts from six individual mice. *p < 0.05; **p < 0.001. (C) Tumor growth. Data represent mean ( ± SD) tumor volume from six individual mice. One-way ANOVA test was used for statistical analyses between two groups (*p < 0.05). (D) Kaplan–Meier survival curves (n = 6). *p < 0.05; **p < 0.001, One-way ANOVA with Newman–Keuls Multiple comparison test.
The TRP2-specific iPSC-CD8+ T cells are identified after adoptive transfer of non-primed TRP2-iPSCs (in vivo programming), and these iPSC-CD8+ T cells are functional in controlling tumor growth in the melanoma challenged mice. However, the primed TRP2-iPSCs show a better profile in expansion and tumor interaction. As compared with the mice receiving no cell transfer or iPSCs transduced with the vector control (iPSC-MiDR), the mice treated with either the non-primed or primed TRP2-iPSCs were well protected, and the primed TRP2-iPSCs had an increased efficiency, which might be partly due to the large number of mature TRP2-specific CD8+ T cells in circulation. Re-challenge studies should further prove that these cells are long-living and exert protective effects on tumor-bearing animals. Although the TRP2-specific iPSC-CD8+ T cells can suppress melanoma growth in murine models, the translational potential of the strategy reported here needs further investigation. As the development of iPSCs into the fully differentiated T cells would take 6–8 weeks, the current iPSC technology would not be practical in clinic, because cancer patients might not have the time left for the T cell maturation, and tumor Ag-specific iPSC-CTLs may be negatively selected during T-cell development in patients due to the presence of specific tumor Ag. The goal of TCR transduction and a one-week in vitro differentiation is to escape the negative selection and generate tumor Ag-specific T progenitors, which also reduces the risk of the transferred cells possibly (a) differentiating into other cell types in vivo and initiate lymphoma or leukemia or (b) becoming tolerized/dysfunctional. Additionally, the efficacy of developing mature CTLs can be improved by administration of agonist anti-Notch antibodies and/or supplement of cytokines such as Flt3L and IL-7, which might be considered as possible approach to enhancing this development. Notably, the efficacy of ACT therapy has been evaluated in the tumor-bearing mice receiving the tumor-specific CD8+ T cells at various stages of differentiation. These studies demonstrate that administration of naive and early effector T cells in combination with a lymphodepleting pretreatment regimen, γc cytokine administration, and vaccination, can result in eradication of established tumors.Citation17-19 It is thus anticipated that combined treatments such as sublethal irradiation before ACT, plus cytokine administration (IL-2 or IL-15) and anti-PD1/PD-L1 therapy, should improve effectiveness of the iPSC-CD8+ T cell-based therapies.Citation20 Based on the results of this report, this strategy warrant further exploration of its benefits in treatment of human cancer.
Overall, a current barrier to progress in the field is the lack of an efficient approach for generating large numbers of naive Ag-specific T cells that could be used for cell-based therapies. Here, we use PSC-T cells to address this limitation as a means of deriving large numbers of persistent Ag-specific PSC-CTLs for ACT. Development of such an approach likely provide an important step toward personalized immunotherapies for cancer treatment. In addition, such an approach helps illuminate signaling mechanisms in T cell differentiation. The project may advance basic and translational research to move the field toward the clinical use of stem cell-derived products such as therapeutic Ag-specific T cells.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_materials.pdf
Download PDF (348.2 KB)Acknowledgments
The authors thank Dr Arthur A. Hurwitz from National Cancer Institute for providing TRP2-specific TCR genes.
Funding
This project was funded, in part, by grants from National Institute of Health Grant R01AI121180, R21AI109239, and R21AI128325, and the American Diabetes Association (1–16-IBS-281) to J.S.
References
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62-8; PMID:25838374; https://doi.org/10.1126/science.aaa4967
- Antsiferova O, Muller A, Ramer PC, Chijioke O, Chatterjee B, Raykova A, Planas R, Sospedra M, Shumilov A, Tsai MH et al. Adoptive transfer of EBV specific CD8+ T cell clones can transiently control EBV infection in humanized mice. PLoS Pathog 2014; 10:e1004333; PMID:25165855; https://doi.org/10.1371/journal.ppat.1004333
- Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 2016; 22:26-36; PMID:26735408; https://doi.org/10.1038/nm.4015
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 2008; 26:101-6; PMID:18059259; https://doi.org/10.1038/nbt1374
- Kim JB, Sebastiano V, Wu G, Arauzo-Bravo MJ, Sasse P, Gentile L, Ko K, Ruau D, Ehrich M, van den Boom D et al. Oct4-induced pluripotency in adult neural stem cells. Cell 2009; 136:411-9; PMID:19203577; https://doi.org/10.1016/j.cell.2009.01.023
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009; 136:964-77; PMID:19269371; https://doi.org/10.1016/j.cell.2009.02.013
- Raya A, Rodriguez-Piza I, Guenechea G, Vassena R, Navarro S, Barrero MJ, Consiglio A, Castellà M, Río P, Sleep E et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 2009; 460:53-9; PMID:19483674; https://doi.org/10.1038/nature08129
- Li Z, Yang CS, Nakashima K, Rana TM. Small RNA-mediated regulation of iPS cell generation. EMBO J 2011; 30:823-34; PMID:21285944; https://doi.org/10.1038/emboj.2011.2
- Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature 2011; 474:212-5; PMID:21572395; https://doi.org/10.1038/nature10135
- Lei F, Zhao B, Haque R, Xiong X, Budgeon L, Christensen ND, Wu Y, Song J. In vivo programming of tumor antigen-specific T lymphocytes from pluripotent stem cells to promote cancer immunosurveillance. Cancer Res 2011; 71:4742-7; PMID:21628492; https://doi.org/10.1158/0008-5472.CAN-11-0359
- Haque R, Lei F, Xiong X, Bian Y, Zhao B, Wu Y, Song J. Programming of regulatory T cells from pluripotent stem cells and prevention of autoimmunity. J Immunol 2012; 189:1228-36; PMID:22732595; https://doi.org/10.4049/jimmunol.1200633
- Haque M, Song J, Fino K, Sandhu P, Wang Y, Ni B, Fang D, Song J. Melanoma immunotherapy in mice using genetically engineered pluripotent stem cells. Cell Transplant 2016; 25:811-27; PMID:26777320; https://doi.org/10.3727/096368916X690467
- Haque M, Song J, Fino K, Sandhu P, Song X, Lei F, Zheng S, Ni B, Fang D, Song J. Stem cell-derived tissue-associated regulatory T cells ameliorate the development of autoimmunity. Sci Rep 2016; 6:20588; PMID:26846186; https://doi.org/10.1038/srep20588
- Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol 2013; 31:928-33; PMID:23934177; https://doi.org/10.1038/nbt.2678
- Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, Nakayama-Hosoya K, Iriguchi S, Uemura Y, Shimizu T et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013; 12:114-26; PMID:23290140; https://doi.org/10.1016/j.stem.2012.11.002
- Haque R, Song J, Haque M, Lei F, Sandhu P, Ni B, Zheng S, Fang D, Yang JM, Song J. c-Myc-induced survivin is essential for promoting the notch-dependent T Cell differentiation from hematopoietic stem cells. Genes (Basel) 2017; 8:pii: E97; https://doi.org/10.3390/genes8030097
- Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013; 24:589-602; PMID:24209604; https://doi.org/10.1016/j.ccr.2013.09.014
- Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ. Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res 2013; 73:1547-58; PMID:23436794; https://doi.org/10.1158/0008-5472.CAN-12-3343
- Palucka AK, Coussens LM. The basis of oncoimmunology. Cell 2016; 164:1233-47; PMID:26967289; https://doi.org/10.1016/j.cell.2016.01.049
- Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016; 13:394; PMID:27118494; https://doi.org/10.1038/nrclinonc.2016.65