
ABSTRACT
A reduced immune surveillance due to immune deficiency or treatment with immunosuppressive drugs is associated with a higher risk to develop aggressive Non-Hodgkin's lymphoma (NHL). Nevertheless, NHL also develops in immunocompetent patients indicating an escape from the immune system. T cell function in advanced aggressive lymphoma is not well characterized and the molecular mechanisms how malignant B cells influence T cell function are ill-defined. We therefore studied T cell function in Eμ-myc transgenic mice that develop an aggressive B cell lymphoma with some similarities to human Burkitt-lymphoma (BL). In advanced lymphoma, the number of T cells was severely reduced and the remaining CD4+ and CD8+ T cells lost the capacity to produce effector cytokines and expand upon re-stimulation. T cells in lymphoma-bearing mice were characterized by the expression of the immune inhibitory molecules programmed death (PD)-1, 2B4 and lymphocyte activation protein (LAG)-3. The proto-oncogene c-Myc not only drives cell proliferation and disease progression but also induces apoptosis of the malignant cells. We found that apoptotic lymphoma cells release purine metabolites that inhibit T cell function. Taken together, our data document that the characteristic high cell turnover and apoptotic rate in aggressive NHL induce a severe T cell dysfunction mediated by several immune-inhibitory mechanisms including ligation of inhibitory ligands and purine metabolites. Blocking a single mechanism only partially restored T cell function and did not increase survival of lymphoma mice.
Introduction
Malignant transformation of mature B cells and the development of Non-Hodgkin's lymphoma (NHL) require genetic alterations of oncogenic drivers. During an immune response, germinal center (GC) B cells are at risk of acquiring mutations in important oncogenes during the process of hyper-mutation and affinity maturation.Citation1 Nevertheless, NHL is a rare disease. This may be due to an efficient immune control of malignant B cells at early stages. Indeed, the frequency of high-grade NHL is substantially increased in immunodeficient patients.Citation2,3 Close to 100% of post-transplant NHL are associated with Epstein-Barr virus (EBV) infection. In contrast, only 30–60% of NHL in patients with congenital immunodeficiency syndromes are EBV-associated.Citation4 EBV-derived antigen latent membrane protein 1 (LMP1) expressed in B cells is a strong lymphoma-associated antigen that activates specific cytotoxic T cells.Citation5 However, immune surveillance of NHL is not limited to EBV antigens. Recently, Afshar-Sterle et al. demonstrated that T cells prevent spontaneous lymphoma development by Fas ligand–mediated cytotoxicity.Citation6 Additional effector mechanisms such as perforin-, granzyme- and cytokine-mediated cytotoxicity contribute to the immune control of lymphoma cells.Citation7,8 Besides T cells, NK cells are important for the immune control of B cell lymphomas.Citation9 The fact that high-grade lymphomas develop in immunocompetent patients implies that malignant B cells escape immune surveillance. Documented immune escape mechanisms in NHL include downregulation of major histocompatibility class I and II (MHC I and II) and costimulatory molecules that render lymphoma cells unrecognizable for effector T cells.Citation10-12 Alternatively, the malignant B cells escape immune control by impairing T cell function, a process that is well established in solid tumors.Citation13 In contrast, the mechanisms that lead to T cell dysfunction in advanced lymphoma are poorly defined.
In this study, we used Eµ-myc transgenic mice that constitutively express the Myc oncogene under the control of the potent Igh enhancer (Eµ).Citation14 These mice develop aggressive B cell lymphoma with some similarities to human Burkitt-lymphoma (BL), i.e. lymphoblastic morphology and rapidly progressive lymphadenopathies.Citation14,15 However, human BL typically arise from germinal center (GC) B cells with somatically mutated Ig variable regions whereas MYC-driven lymphomas in Eμ-myc transgenic mice typically arise from pre-B or naïve B cells.Citation16
Mice with advanced lymphoma had severely reduced T cell numbers and an impaired T cell function. T cells expressed the co-inhibitory molecules programmed death 1 (PD-1), 2B4 and lymphocyte-activation gene 3 (LAG-3).Citation17-19 Blocking the programmed death ligand 1 (PD-L1)/PD-1 interaction in vitro partially restored T cell expansion but did not improve immune control of the lymphoma in vivo. Furthermore, malignant B cells undergoing apoptosis released metabolites that additionally induced T cell dysfunction. An unbiased screen of molecules secreted by malignant B cells by mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy revealed lactic acid and purine metabolites as additional components inducing T cell dysfunction. Blocking PD-1 signaling and blocking the effect of ATP and its degradation improved T cell function in vitro and partially in vivo. However, neither blocking PD-1 signaling nor ATP-degradation prolonged survival of lymphoma-bearing Eµ-myc mice. Our experiments indicate that in aggressive lymphoma, purine degradation products released by apoptotic lymphoma cells induce a severe T cell dysfunction. Thus, blocking one single immune-inhibitory pathway may not be sufficient to improve the outcome of aggressive lymphomas which are characterized by high cell turnover and apoptosis.
Results
T cells in lymphoma-bearing mice are reduced in numbers and express T cell inhibitory molecules
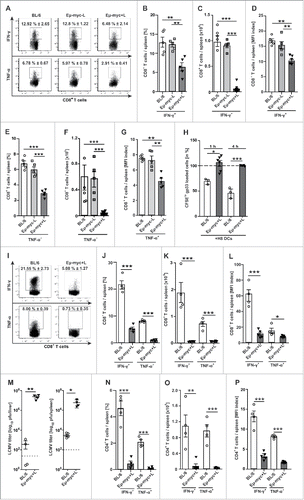
First, we characterized the phenotype and function of T cells in the Eµ-myc lymphoma model.Citation20 Eμ-myc mice developed a c-myc-oncogene-driven aggressive B cell lymphoma with enlarged lymph nodes (LN) and splenomegaly between 6 weeks and 12 months after birth (Fig. S1A, B). Malignant B cells were highly abundant in lymphoid organs whereas CD4+ and CD8+ T cells were significantly reduced in numbers in lymphoma-bearing Eµ-Myc (Eµ-myc+L) mice when compared with naïve BL/6 or non-lymphoma-bearing Eµ-Myc mice (Eµ-myc-L; , ). Phenotypically, CD4+ and CD8+ T cells were characterized by increased expression of the T cell inhibitory molecules PD-1, 2B4 and LAG-3 in blood, spleen and LN (, and data not shown). In Eµ-myc+L mice approximately 40% of all CD8+ T cells and 29% of all CD4+ T cells expressed inhibitory receptors, with a substantial fraction of T cells expressing multiple receptors (). Since continuous T cell receptor stimulation has been shown to lead to the expression of T cell inhibitory receptors,Citation21 we analyzed if non-lymphoma specific cytotoxic CD8+ T lymphocytes (CTLs) similarly express PD-1, 2B4 and LAG-3. To this end, we crossed Eµ-myc mice to lymphocytic choriomeningitis virus (LCMV) gp33 TCR transgenic p14 mice (Eµ-myc x p14). In Eµ-myc x p14 mice, T cells are of known non-lymphoma antigen specificity. Nevertheless, up to 16% of p14 (Vα2+Vβ8.1+CD45.1+) CTLs from lymphoma-bearing Eµ-myc x p14 (Eµ-myc x p14+L) mice expressed one or more inhibitory receptors (). However, the expression of inhibitory receptors on p14 T cells () was substantially lower than on total CD8+ T cells in Eμ-myc mice (), indicating that the expression of inhibitory receptors is driven by T cell receptor-dependent and independent mechanisms. We next analyzed whether the expression of T cell inhibitory receptors correlates with lymphoma load. Indeed, the expression of PD-1, 2B4 and LAG-3 on CD8+ and on CD4+ T cells significantly correlated with the number of B cells in spleen (). The expression of T cell inhibitory markers has been associated with T cell dysfunction, exhaustion and cell death.Citation21,22 In accordance with these studies, 3[H]-TdR uptake of CD8+ () and CD4+ T cells () isolated from spleen of Eµ-myc+L mice was significantly reduced. In addition, a higher fraction of CD8+ and CD4+ T cells in spleens of Eµ-myc+L mice underwent apoptosis (, ). Therefore, CD4+ and CD8+ T cells in BL-like disease undergo apoptosis and are reduced in numbers whereas the remaining T cells are dysfunctional.
Figure 1. Phenotype and function of CD8+ and CD4+ T cells in lymphoma-bearing Eμ-myc mice. (A) Frequencies and (B) absolute numbers of CD8+ and CD4+ T cells in spleens from BL/6 mice, non-lymphoma-bearing (Eµ-myc-L) and lymphoma-bearing Eµ-myc mice (Eµ-myc+L) (n = 7–10 mice/group). (C) Expression and mean fluorescence intensities (MFI) of indicated inhibitory receptors on CD8+ T cells from spleens of BL/6 mice, Eµ-myc-L and Eµ-myc+L mice determined by flow-cytometry (black: staining, gray: isotype, MFI: MFI staining – MFI isotype). One representative FACS plot out of 7–12 is shown. (D) Frequency of co-expression of PD-1, 2B4 and LAG-3 on splenic CD8+ and CD4+ T cells from BL/6 (blue), Eµ-myc-L (green) and Eµ-myc+L (red) mice (n = 5–7 mice/group). (E) Eµ-myc mice were crossed to lymphocytic choriomeningitis virus (LCMV) gp33 TCR transgenic p14 mice (Eµ-myc x p14) and frequency of co-expression of inhibitory receptors on p14pos T cells was analyzed in non-lymphoma bearing Eµ-myc x p14–L (blue) and lymphoma bearing Eµ-myc x p14+L (orange) mice. (n = 2–3 mice/ group). (F) Frequency of PD-1, 2B4, and LAG-3 expressing CD8+ (upper panel) and CD4+ T cells (lower panel) were determined and correlation with absolute B cell numbers was analyzed (n = 6 mice/group). Data are depicted as linear regression curves, calculated using Ozone correlations (Graph Pad Prism5). The “goodness of fit” is displayed as the coefficient of determination ( = r2) (perfect fit: r2 = 1) and significance is displayed as p values (p < 0.05, p < 0.01, p < 0.0001). The correlation coefficient r was calculated using Pearson correlation (perfect correlation: r = 1). (G) CD8+ and (H) CD4+ T cells were isolated from spleens of BL/6, Eµ-myc-L and Eµ-myc+L (n = 25–22 mice/group for CD8+; n = 3–9 mice/group for CD4+) and 3[H]-TdR incorporation was assessed after stimulation with plate-bound αCD3 monoclonal antibody (mAb) for 72 h. (I) Annexin-V staining of splenic CD8+ or CD4+ T cells, from BL/6 and Eµ-myc+L mice directly ex vivo (black: staining, gray: isotype). One representative plot out of 4 is shown. (J) Frequency of Annexin-V+ CD8+ and CD4+ T cells from BL/6 and Eµ-myc+L mice (n = 4 mice/group). Data are displayed as mean ± SEM. Statistics: Student's t test. *p < 0.05, ***p < 0.0001. See also Supplementary Figure S1.
![Figure 1. Phenotype and function of CD8+ and CD4+ T cells in lymphoma-bearing Eμ-myc mice. (A) Frequencies and (B) absolute numbers of CD8+ and CD4+ T cells in spleens from BL/6 mice, non-lymphoma-bearing (Eµ-myc-L) and lymphoma-bearing Eµ-myc mice (Eµ-myc+L) (n = 7–10 mice/group). (C) Expression and mean fluorescence intensities (MFI) of indicated inhibitory receptors on CD8+ T cells from spleens of BL/6 mice, Eµ-myc-L and Eµ-myc+L mice determined by flow-cytometry (black: staining, gray: isotype, MFI: MFI staining – MFI isotype). One representative FACS plot out of 7–12 is shown. (D) Frequency of co-expression of PD-1, 2B4 and LAG-3 on splenic CD8+ and CD4+ T cells from BL/6 (blue), Eµ-myc-L (green) and Eµ-myc+L (red) mice (n = 5–7 mice/group). (E) Eµ-myc mice were crossed to lymphocytic choriomeningitis virus (LCMV) gp33 TCR transgenic p14 mice (Eµ-myc x p14) and frequency of co-expression of inhibitory receptors on p14pos T cells was analyzed in non-lymphoma bearing Eµ-myc x p14–L (blue) and lymphoma bearing Eµ-myc x p14+L (orange) mice. (n = 2–3 mice/ group). (F) Frequency of PD-1, 2B4, and LAG-3 expressing CD8+ (upper panel) and CD4+ T cells (lower panel) were determined and correlation with absolute B cell numbers was analyzed (n = 6 mice/group). Data are depicted as linear regression curves, calculated using Ozone correlations (Graph Pad Prism5). The “goodness of fit” is displayed as the coefficient of determination ( = r2) (perfect fit: r2 = 1) and significance is displayed as p values (p < 0.05, p < 0.01, p < 0.0001). The correlation coefficient r was calculated using Pearson correlation (perfect correlation: r = 1). (G) CD8+ and (H) CD4+ T cells were isolated from spleens of BL/6, Eµ-myc-L and Eµ-myc+L (n = 25–22 mice/group for CD8+; n = 3–9 mice/group for CD4+) and 3[H]-TdR incorporation was assessed after stimulation with plate-bound αCD3 monoclonal antibody (mAb) for 72 h. (I) Annexin-V staining of splenic CD8+ or CD4+ T cells, from BL/6 and Eµ-myc+L mice directly ex vivo (black: staining, gray: isotype). One representative plot out of 4 is shown. (J) Frequency of Annexin-V+ CD8+ and CD4+ T cells from BL/6 and Eµ-myc+L mice (n = 4 mice/group). Data are displayed as mean ± SEM. Statistics: Student's t test. *p < 0.05, ***p < 0.0001. See also Supplementary Figure S1.](/cms/asset/b6b3cc72-94de-4e0a-8511-6635cd4b5f5b/koni_a_1365997_f0001_oc.gif)
CTLs are functionally impaired in lymphoma-bearing mice in vivo
To investigate whether CTLs in Eµ-myc+L mice can be activated by a physiologic stimulus in vivo, we immunized Eµ-myc+L mice with mature dendritic cells (DCs) derived from H8 miceCitation23 ubiquitously expressing the MHC-class I restricted peptide gp33 from LCMV (H8 DCs). Eight days after primary immunisation, the frequencies and the absolute numbers of interferon gamma (IFN-γ) and tumor necrosis factor α (TNF-α)-producing CD8+ T cells were substantially lower in lymphoma-bearing mice than in controls (, S2A). Activated CD8+ T cells produced lower levels of IFN-γ and TNF-α per cell when compared with controls as indicated by a lower MFI Index (, ). The lytic function of gp33-specific CD8+ T cells in vivo was analyzed by transferring gp33-loaded CFSElo and unpulsed CFSEhi labeled B cells into Eµ-myc+L mice and BL/6 controls 8 d after immunisation with H8 DCs. Antigen-specific killing was completely absent in Eµ-myc+L mice whereas BL/6 mice eliminated 35% and 50% of antigen-pulsed B cells 1 and 4 h after transfer, respectively ().
Figure 2. Impaired CD8+ T cell function in lymphoma-bearing mice after immunization with dendritic cells (DCs) or LCMV. (A-H) BL/6, Eµ-myc-L and Eµ-myc+L mice were immunized day 0 and day 2 i.v. with 2 × 105 H8 DCs and CD8+ T cells were analyzed in spleen 8 d after primary immunization. (A, B, E) Frequencies, (C, F) absolute numbers and (D, G) MFI index of cytokine expressing CD8+ T cells from BL/6, Eµ-myc-L and Eµ-myc+L mice after 5 h of in vitro re-stimulation with LCMV gp-33 are shown (n = 5 mice/group). (H) 3 × 107 gp33-pulsed CFSElo labeled B cells and unpulsed CFSEhi labeled B cells were injected into H8 DC-immunized BL/6 and Eµ-myc+L mice (n = 3–6 mice/group). Naïve BL/6 and Eµ-myc+L mice served as controls (n = 2 mice/group). In vivo cytolytic activity of CD8+ T cells was determined by measuring antigen-specific elimination of gp33-pulsed B cells 1 and 4 h after transfer by flow-cytometry. (I-P) BL/6 and Eµ-myc+L mice were infected with 200 pfu LCMV-WE i.v. CD8+ and CD4+ T cells were analyzed in the spleen 8 d after infection. (I, J) Frequencies, (K) absolute numbers and (L) MFI indexes of IFN-γ and TNF-α positive CD8+ T cells after re-stimulation with LCMV gp33 in vitro (n = 4 mice/group). (M) LCMV titres in livers and spleens (n = 4 mice/group). Dotted line indicated detection limit of the assay. (N) Frequencies, (O) absolute numbers and (P) MFI indexes of IFN-γ and TNF-α positive CD4+ T cells after re-stimulation with LCMV gp13 in vitro (n = 4 mice/group). For (A)and I, one representative dot plot out of 5 is depicted and frequencies are displayed as Mean ± SEM (n = 5 mice/group). Expression values for D, G, L, and (P) are displayed as MFI Index ( = intensity of expression in cytokine positive / cytokine negative cells). Data are displayed as mean ± SEM. Statistics: (B-G) One-way ANOVA, (H, J-P) Student's t test. *p < 0.05, **p < 0.01, ***p < 0.0001.
LCMV infection in mice leads to a very strong inflammatory stimulus and to a massive expansion of antigen-specific CTLs.Citation24 We therefore asked whether LCMV infection could overcome the T cell dysfunction observed in Eµ-myc+L mice. Upon LCMV infection the number of IFN-γ and TNF-α single positive and IFN-γ/TNF-α double positive antigen specific CD8+ T cells was significantly lower in Eµ-myc+L mice than in controls (, S2B). In addition, activated CD8+ T cells from Eµ-myc+L mice produced less IFN-γ and TNF-α per cell (). This resulted in approximately 1000–10000 fold higher virus titers in livers and spleens of Eμ-myc+L mice 8 d post infection (). Similarly, the LCMV gp13-specific CD4+ T cell function was impaired in Eµ-myc+L mice with reduced numbers of IFN-γ and TNF-α single and double positive CD4+ T cells (, , S2C). Again, each activated CD4+ T cell produced less cytokine in Eµ-myc+L mice than in controls (). These experiments document a severe T cell dysfunction in lymphoma-bearing mice.
CTL dysfunction in B cell lymphoma-bearing mice is reversible after transfer into naive BL/6 mice
Lymphoma cells also infiltrated primary lymphoid organs such as bone marrow and thymus (data not shown) and thereby may affect T cell development. To analyze the effect of the B cell lymphoma on naïve CD8+ T cells that developed in the absence of lymphoma, we transferred naïve p14 CTLs to Eµ-myc+L mice and BL/6 controls. p14 CTLs were activated with either H8 DCs (; S3A) or with 200 pfu LCMV-WE i.v. (; S3B). Significantly fewer p14 CTLs produced IFN-γ and TNF-α in lymphoma-bearing mice (; , S3A, B). Similarly, the IFN-γ and TNF-α production per cell was reduced in Eµ-myc+L mice as indicated by MFI index (, ).
Figure 3. Dysfunctional p14 CTLs from lymphoma-bearing mice regain effector function after adoptive transfer into healthy recipient mice. (A-D) 1 × 106 congenic CD45.1+ p14 T cells were adoptively transferred into lymphoma-bearing Eµ-myc or BL/6 mice (n = 5–8 mice/group). Mice were immunized twice with 2 × 105 H8-DCs (day 0 and day 2). (A, B) Frequency, (C) total numbers and (D) MFI index of IFN-γ− and TNF-α-producing p14 CTLs in the spleen 6 d after immunisation. (E-H) 1 × 106 p14 CTLs were adoptively transferred into Eµ-myc+L or BL/6 mice (n = 9 mice/group). 18 h later p14 CTLs were activated with 1 × 104 pfu LCMV. (E, F) Frequency, (G) total number and (H) MFI index of IFN-γ and TNF-α-producing p14 CTLs in the spleen after re-stimulation in vitro with gp33. One representative dot plot out of 4–5 is depicted. (I) MACS-purified p14 CTLs from Eµ-myc x p14-L and +L mice were isolated and re-stimulated in vitro with gp33-pulsed irradiated splenocytes for 72 h and 3[H]-TdR incorporation of p14 CTLs was measured. (J-M) 1 × 106 MACS-purified p14 CTLs from Eµ-myc x p14-L and +L mice were adoptively transferred i.v. in naive BL/6 recipients and activated 18 h later with 1 × 104 pfu LCMV i.v.. (J) Total number, (K) frequencies (L) absolute numbers and (M) MFI index of IFN-γ and TNF-α producing p14 CTLs after re-stimulation with gp33 in vitro were assessed in the spleen 6 d after LCMV infection (n = 9 mice/group). For A, E, and K one representative dot plot out of 5 or 9 is depicted and frequencies are displayed as mean ± SEM (n = 5 (A); n = 9 (E, K) mice/group). Expression values for D, (H) and, M are displayed as MFI index ( = intensity of expression in cytokine positive / cytokine negative cells). Data are displayed as mean ± SEM. Statistics: Student's t test. *p < 0.05, **p < 0.01, ***p < 0.0001.
![Figure 3. Dysfunctional p14 CTLs from lymphoma-bearing mice regain effector function after adoptive transfer into healthy recipient mice. (A-D) 1 × 106 congenic CD45.1+ p14 T cells were adoptively transferred into lymphoma-bearing Eµ-myc or BL/6 mice (n = 5–8 mice/group). Mice were immunized twice with 2 × 105 H8-DCs (day 0 and day 2). (A, B) Frequency, (C) total numbers and (D) MFI index of IFN-γ− and TNF-α-producing p14 CTLs in the spleen 6 d after immunisation. (E-H) 1 × 106 p14 CTLs were adoptively transferred into Eµ-myc+L or BL/6 mice (n = 9 mice/group). 18 h later p14 CTLs were activated with 1 × 104 pfu LCMV. (E, F) Frequency, (G) total number and (H) MFI index of IFN-γ and TNF-α-producing p14 CTLs in the spleen after re-stimulation in vitro with gp33. One representative dot plot out of 4–5 is depicted. (I) MACS-purified p14 CTLs from Eµ-myc x p14-L and +L mice were isolated and re-stimulated in vitro with gp33-pulsed irradiated splenocytes for 72 h and 3[H]-TdR incorporation of p14 CTLs was measured. (J-M) 1 × 106 MACS-purified p14 CTLs from Eµ-myc x p14-L and +L mice were adoptively transferred i.v. in naive BL/6 recipients and activated 18 h later with 1 × 104 pfu LCMV i.v.. (J) Total number, (K) frequencies (L) absolute numbers and (M) MFI index of IFN-γ and TNF-α producing p14 CTLs after re-stimulation with gp33 in vitro were assessed in the spleen 6 d after LCMV infection (n = 9 mice/group). For A, E, and K one representative dot plot out of 5 or 9 is depicted and frequencies are displayed as mean ± SEM (n = 5 (A); n = 9 (E, K) mice/group). Expression values for D, (H) and, M are displayed as MFI index ( = intensity of expression in cytokine positive / cytokine negative cells). Data are displayed as mean ± SEM. Statistics: Student's t test. *p < 0.05, **p < 0.01, ***p < 0.0001.](/cms/asset/52badbe6-fa6b-4844-9f67-305219438381/koni_a_1365997_f0003_oc.gif)
Next we investigated whether the dysfunction of T cells from Eµ-myc+L was restored in a healthy microenvironment. CTLs from Eµ-myc x p14+L mice incorporated significantly less 3[H]-TdR than CTLs isolated from Eµ-myc x p14-L mice (). Isolated p14 T cells were injected into naïve BL/6 mice and activated with LCMV-WE. p14 CTLs originating from Eµ-myc x p14+L and -L mice expanded similarly (). In addition, the frequency and absolute numbers of IFN-γ and TNF-α producing p14 CTLs in the spleen were comparable ().
These results indicate that BL-like lymphoma induces a severe CD8+ T cell dysfunction that is reversible in a healthy host environment.
Blocking PD-1 signaling partially restores T cell function
Interfering with the PD-L1/PD-1 interaction restores T cell function in different cancer entities and prolongs survival of patients with melanoma, non-small-cell lung cancer and others.Citation25 PD-L1 was expressed on CD19+ B cells and on CD11b+ and CD11c+ cells in the spleen of BL/6 and Eµ-myc+L mice (). Co-culture of naïve CD8+ T cells with B cells from Eµ-myc+L mice inhibited 3[H]-TdR uptake in a dose-dependent manner (). A similar trend was observed in co-culture experiments with B cells from naïve BL/6 mice, but to a much lesser extent (). Blocking PD-1 by mAb did not reduce the inhibitory effect of malignant CD19+ B cells on PD-1 expressing CD8+ T cells isolated form lymphoma-bearing mice (). In contrast, blocking PD-1 signaling by mAb increased 3[H]-TdR uptake of CD8+ T cells in co-culture experiments with antigen-presenting CD11b+/CD11c+ APCs from Eµ-myc+L mice (). To analyze the effect of PD-1 signaling on the immune control of BL-like disease in vivo, we transferred MACS-sorted CD19+ B cells from Eμ-myc+L into naïve BL/6 mice followed by treatment with an αPD-1 mAb or control IgG. Blocking PD-1/PD-L1 axis resulted in significantly higher frequencies of CD4+ and CD8+ T cells and a similar trend in absolute T cell numbers in spleen (, ). In addition, the function of T cells was improved after treatment with αPD-1 mAb as documented by an enhanced expansion of CD8+ T cells ex vivo and higher cytokine production especially of CD4+ T cells after re-stimulation in vitro (, S4A).
Figure 4. Blockade of PD-1 signaling partially restores T cell function. (A) PDL-1 expression was analyzed on CD19+, CD11b+ or CD11c+ cells on splenocytes from BL/6 or Eµ-myc+L mice by flow-cytometry. One representative histogram out of 3 is shown. (black: staining, gray: isotype; n = 3 mice/group) (B) MACS-purified CD8+ T cells from naive BL/6 mice (responders) were activated with plate-bound αCD3 mAb (2 µg/ml) in the presence of titrated numbers of MACS-purified CD19+ B cells from naïve BL/6 or Eµ-myc+L mice for 72 h in triplicate cultures. 3[H]–TdR was added to the culture for the last 18 h of incubation. 1 representative out of 12 experiments is shown. (C) CD8+ T cells were cultured with titrated amounts of MACS-purified CD19+ B cells from Eµ-myc+L in the absence or presence of a blocking αPD-1 mAb (10 μg/ml) or control IgG and proliferation was measured as described in (B). Naïve BL/6 CD8+ T cells were cultured in the presence or absence of a αPD-1 or isotype control mAb served as controls. (D) Titrated numbers of MACS purified, irradiated (1000 rad) CD11b+ and CD11c+ APCs were cultured with CD8+ T cell responders (as described in (B) in the presence or absence (hatched bars) of an αPD-1 mAb (10 μg/ml) or control IgG and 3[H]–TdR incorporation was measured. (E-M) 3 × 106 MACS-purified CD19+ B cells from Eµ-myc+L mice were transplanted into BL/6 recipients and mice were treated with 200 μg αPD-1 mAb (blue bars) or control IgG (yellow bars) i.p. every third day. Day 17 after treatment, splenocytes were isolated and (E) frequencies and (F) absolute numbers of CD4+ and CD8+ T cells were analyzed using flow cytometry. (G) MACS purified CD8+ T cells were activated with plate-bound αCD3 mAb (2 µg/ml) and 3[H]–TdR incorporation was measured. (H-K) MACS purified CD4+ and CD8+ T cells were stimulated with PMA/Ionomycin for 5 h in vitro. Then, IFN-γ and TNF-α production was measured by flow-cytometry. (H, I) Total numbers and (J, K) MFI index of IFN-γ− and TNF-α-producing T cells is shown. (L) Absolute CD19+ B cell numbers (n = 3–5 mice/group). (M) Kaplan-Meier curve (n = 5 mice/group). (N) 3 × 106 MACS-purified CD19+ B cells from Eµ-myc+L mice were transplanted into BL/6 or PD-1−/− recipients and survival was monitored. Kaplan-Meier curve (n = 5 mice/group). Expression values for J, K are displayed as MFI index ( = intensity of expression in cytokine positive/cytokine negative cells). Data are displayed as mean ± SEM. Statistics: (B-L) Student's t test; (M, N) log-rank test. *p < 0.05, **p < 0.01, ***p < 0.0001. See also Supplementary Figure S4.
![Figure 4. Blockade of PD-1 signaling partially restores T cell function. (A) PDL-1 expression was analyzed on CD19+, CD11b+ or CD11c+ cells on splenocytes from BL/6 or Eµ-myc+L mice by flow-cytometry. One representative histogram out of 3 is shown. (black: staining, gray: isotype; n = 3 mice/group) (B) MACS-purified CD8+ T cells from naive BL/6 mice (responders) were activated with plate-bound αCD3 mAb (2 µg/ml) in the presence of titrated numbers of MACS-purified CD19+ B cells from naïve BL/6 or Eµ-myc+L mice for 72 h in triplicate cultures. 3[H]–TdR was added to the culture for the last 18 h of incubation. 1 representative out of 12 experiments is shown. (C) CD8+ T cells were cultured with titrated amounts of MACS-purified CD19+ B cells from Eµ-myc+L in the absence or presence of a blocking αPD-1 mAb (10 μg/ml) or control IgG and proliferation was measured as described in (B). Naïve BL/6 CD8+ T cells were cultured in the presence or absence of a αPD-1 or isotype control mAb served as controls. (D) Titrated numbers of MACS purified, irradiated (1000 rad) CD11b+ and CD11c+ APCs were cultured with CD8+ T cell responders (as described in (B) in the presence or absence (hatched bars) of an αPD-1 mAb (10 μg/ml) or control IgG and 3[H]–TdR incorporation was measured. (E-M) 3 × 106 MACS-purified CD19+ B cells from Eµ-myc+L mice were transplanted into BL/6 recipients and mice were treated with 200 μg αPD-1 mAb (blue bars) or control IgG (yellow bars) i.p. every third day. Day 17 after treatment, splenocytes were isolated and (E) frequencies and (F) absolute numbers of CD4+ and CD8+ T cells were analyzed using flow cytometry. (G) MACS purified CD8+ T cells were activated with plate-bound αCD3 mAb (2 µg/ml) and 3[H]–TdR incorporation was measured. (H-K) MACS purified CD4+ and CD8+ T cells were stimulated with PMA/Ionomycin for 5 h in vitro. Then, IFN-γ and TNF-α production was measured by flow-cytometry. (H, I) Total numbers and (J, K) MFI index of IFN-γ− and TNF-α-producing T cells is shown. (L) Absolute CD19+ B cell numbers (n = 3–5 mice/group). (M) Kaplan-Meier curve (n = 5 mice/group). (N) 3 × 106 MACS-purified CD19+ B cells from Eµ-myc+L mice were transplanted into BL/6 or PD-1−/− recipients and survival was monitored. Kaplan-Meier curve (n = 5 mice/group). Expression values for J, K are displayed as MFI index ( = intensity of expression in cytokine positive/cytokine negative cells). Data are displayed as mean ± SEM. Statistics: (B-L) Student's t test; (M, N) log-rank test. *p < 0.05, **p < 0.01, ***p < 0.0001. See also Supplementary Figure S4.](/cms/asset/a1493c74-ecb6-4e01-8b3f-8bbbbe1ea9e7/koni_a_1365997_f0004_oc.gif)
Interestingly, αPD-1 treatment increased the expression of all analyzed inhibitory receptors on CD8+ T cells and of 2B4 on CD4+ T cells (Fig. S4B-D), indicating partial redundancy and compensation of T cell inhibitory mechanisms. Despite the fact that αPD-1 treatment partially restored T cell function and numbers, this did not result in a reduction of the lymphoma load as indicated by similar B cell numbers in spleen (). In addition, PD-1 blockade by mAb treatment () had only a minor, non-significant, effect on the survival of lymphoma mice (p = 0,1094). Furthermore, we transferred MACS-sorted CD19+ B cells from Eµ-myc+L into naïve PD-1−/− and BL/6 mice and monitored lymphoma development and survival. Genetic ablation of PD-1 signaling on immune cells did not improve survival of lymphoma mice ().
Therefore, although T cells in lymphoma bearing animals express high levels of PD-1, blocking PD-1 signaling did only partially restore T cell function, mainly of CD4+ T cells, but did not significantly improve the immune control of BL in our model.
CD8+ T cell dysfunction in B cell lymphoma is induced by soluble factors < 1kD
To characterize additional immunosuppressive mechanisms we first analyzed whether cell-cell interactions or soluble factors are responsible. Therefore, we performed a trans-well assay using CD8+ T cell responders and B cells from BL/6 or Eµ-myc+L mice. The results revealed a comparable dose-dependent inhibition of T cell proliferation in a trans-well assay () as observed during direct cell-cell interaction (). In line with this, the addition of sera from Eµ-myc+L mice to CD8+ T cells reduced their proliferation in a dose-dependent manner (). Similarly, B cell-conditioned medium (BCM) from Eµ-myc+L mice inhibited the expansion of naïve CD8+ T cells (, ). BCM from Eµ-myc+L mice even supressed the proliferation of human CD8+ T cells from healthy donors, indicating that a conserved molecule mediates T cell inhibition (). Proteins as possible mediators of T cell dysfunction could be excluded as neither heat inactivation () nor Proteinase K treatment (Fig. S5A, B) of Eµ-myc+L BCM prevented inhibition of CD8+ T cell expansion. Filter size exclusion according to molecular size revealed that only the fraction containing molecules <3kDa impaired CD8+ T cell proliferation (). Similarly, the fraction < 3kDa from BL/6 BCM inhibited CD8+ T cell expansion, however, to a significantly lesser extent (). A further filtration step revealed that the inhibitory molecule was of a molecular size <1kDa ().
Figure 5. Lymphoma B cells induce mouse and human CD8+ T cell dysfunction by secreting a soluble compound smaller than 1kDa. (A) MACS-purified CD8+ T cells from naive BL/6 mice (responders) were activated with plate-bound αCD3 mAb (2 µg/ml) in the presence of titrated numbers of MACS-purified CD19+ B cells from naïve BL/6 or Eµ-myc+L mice for 72 h in transwell tissue culture plates in triplicate cultures and 3[H]–TdR incorporation was determined after 72 h of culture. 1 representative out of 3 experiments is shown. (B) BL/6 CD8+ T cells were stimulated with αCD3 mAb as described in (A) in the presence and absence of titrated amounts of serum from Eµ-myc+L mice (n = 2 different sera) and 3[H]–TdR incorporation was determined after 72 h of culture. (C) Mouse and human naïve CD8+ T cells were stimulated with αCD3 mAb as described in (A) or OKT-3 (2 µg/ml) respectively, in the presence and absence of titrated amounts of B cell conditioned medium (BCM), derived from BL/6 or Eµ-myc+L B cell cultures. 3[H]–TdR incorporation was determined after 72 h of culture. One representative out of 3 experiments is shown. (D) CD8+ T cells were labeled with CFSE and CFSE dilution was measured after stimulation of T cells with αCD3 mAb in the presence and absence of indicated BCMs at a dilution of 1:3 for 72 h. 1 representative out of 2 experiments is shown. (E-G) CD8+ T cells were cultured as described in (A) in the presence and absence of titrated amounts of (E) untouched or heat-inactivated BCM and (F, G) size fractionated BCM from BL/6 or Eµ-myc+L B cell cultures and 3[H]–TdR incorporation was measured. 1 representative out of 6 experiments is shown. (H) Naïve BL/6 cells were cultured in the presence or absence of size fractionated BCM together with αCD3 mAb and viability was determined by Annexin-V staining after 24 h of culture. Apoptosis rate of CD8+ T cells in medium was 41% (data not shown). 1 representative out of 2 experiments is shown. (I) Naïve BL/6 cells were cultured in the presence or absence of size fractionated BCM and αCD3 mAb for 72 h. Then, IFN-γ and TNF-α production was measured by flow-cytometry after additional 5 h of in vitro stimulation with PMA/Ionomycin. In medium, TNF-α production of CD8+ T cells was 29%, IFN-γ production was 18% (data not shown). Data are displayed as mean ± SEM. Statistics: Student's t test.*p < 0.05, **p < 0.01, ***p < 0.0001. See also Supplementary Figure S5.
![Figure 5. Lymphoma B cells induce mouse and human CD8+ T cell dysfunction by secreting a soluble compound smaller than 1kDa. (A) MACS-purified CD8+ T cells from naive BL/6 mice (responders) were activated with plate-bound αCD3 mAb (2 µg/ml) in the presence of titrated numbers of MACS-purified CD19+ B cells from naïve BL/6 or Eµ-myc+L mice for 72 h in transwell tissue culture plates in triplicate cultures and 3[H]–TdR incorporation was determined after 72 h of culture. 1 representative out of 3 experiments is shown. (B) BL/6 CD8+ T cells were stimulated with αCD3 mAb as described in (A) in the presence and absence of titrated amounts of serum from Eµ-myc+L mice (n = 2 different sera) and 3[H]–TdR incorporation was determined after 72 h of culture. (C) Mouse and human naïve CD8+ T cells were stimulated with αCD3 mAb as described in (A) or OKT-3 (2 µg/ml) respectively, in the presence and absence of titrated amounts of B cell conditioned medium (BCM), derived from BL/6 or Eµ-myc+L B cell cultures. 3[H]–TdR incorporation was determined after 72 h of culture. One representative out of 3 experiments is shown. (D) CD8+ T cells were labeled with CFSE and CFSE dilution was measured after stimulation of T cells with αCD3 mAb in the presence and absence of indicated BCMs at a dilution of 1:3 for 72 h. 1 representative out of 2 experiments is shown. (E-G) CD8+ T cells were cultured as described in (A) in the presence and absence of titrated amounts of (E) untouched or heat-inactivated BCM and (F, G) size fractionated BCM from BL/6 or Eµ-myc+L B cell cultures and 3[H]–TdR incorporation was measured. 1 representative out of 6 experiments is shown. (H) Naïve BL/6 cells were cultured in the presence or absence of size fractionated BCM together with αCD3 mAb and viability was determined by Annexin-V staining after 24 h of culture. Apoptosis rate of CD8+ T cells in medium was 41% (data not shown). 1 representative out of 2 experiments is shown. (I) Naïve BL/6 cells were cultured in the presence or absence of size fractionated BCM and αCD3 mAb for 72 h. Then, IFN-γ and TNF-α production was measured by flow-cytometry after additional 5 h of in vitro stimulation with PMA/Ionomycin. In medium, TNF-α production of CD8+ T cells was 29%, IFN-γ production was 18% (data not shown). Data are displayed as mean ± SEM. Statistics: Student's t test.*p < 0.05, **p < 0.01, ***p < 0.0001. See also Supplementary Figure S5.](/cms/asset/a1498b62-336e-40d0-a6e5-d0e0d793a115/koni_a_1365997_f0005_oc.gif)
The Eµ-myc+L BCM fraction <3kDa increased apoptosis of naïve CD8+ T cells in vitro as analyzed by Annexin-V staining (). In contrast, BCM fractions >3kDa did not induce apoptosis. In addition, the Eµ-myc+L fractions <3kDa, <1kDa inhibited TNF-α and IFN-γ production of αCD3 mAb activated naïve CD8+ T cells ( and data not shown). However, co-incubation with BCM did not affect PD-1 expression on CD8+ T cells (data not shown).
These data indicate that secreted molecules form malignant B cells smaller than 1kDa inhibit CD8+ T cell proliferation, effector function and induce apoptosis of naïve CD8+ T cells.
Purine degradation products induce CD8+ T cell dysfunction in B cell lymphoma
To characterize the molecule(s) inducing CD8+ T cell dysfunction in lymphoma-bearing mice, the BCM fraction <1kDa from Eµ-myc+L and BL/6 mice was analyzed by Mass Spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy (). The MS analysis identified 32 different masses that were present at significantly higher concentration in Eµ-myc+L BCM compared with BL/6 BCM including 9 metabolites of documented inhibitory effects on T cells (, ). NMR analysis identified higher concentrations of hypoxanthine (, ), a member of the purine metabolism, and lactic acid in Eµ-myc+L BCM (Fig. S6A). Lipids including sphingosine-1-phosphate (S1P), lactic acid and metabolites of the purine pathway have been shown to inhibit T cell function.Citation26-30 We therefore analyzed if one or several of these molecules are responsible for the T cell dysfunction induced by malignant B cells.
Figure 6. Lymphoma B cells produce and secrete purines that inhibit T cell function. (A) <1kDa fractions of BCM from BL/6 or Eµ-myc+L mice (n = 4 mice/group) were analyzed for metabolites using mass spectrometry. Results are indicated as S-Plot correlation of equal masses, detected in BL/6 ( = −1) or Eµ-myc+L ( = +1) mice. Masses with a very high (Eµ-myc+L = +1) or very low (BL/6 = −1) correlation reflect the highest significance. Masses selected for further screening are displayed in red and depicted in . (B) NMR analysis. (C) NMR signal after spike in of Hypoxanthine standard or Eµ-myc+L BCM (n = 4 mice/group). (D, E) Serum concentration of (D) ATP and (E) uric acid (n = 3 mice/group). (F) ATP concentrations in MACS-purified B cell lysates (n = 3 lysates/group). Concentrations of indicated purines in BCM (n = 3–5 BCM/group). Proliferation of purified, naïve CD8+ T cells from BL/6 mice was determined after addition of titrated amounts of different purines. (G) Titrated amounts of ATP, 5′ADP or AMP were added separately or combined (ATP + 5′ADP and ATP+5′ADP+AMP) to purified, naïve CD8+ T cells from BL/6 mice and 3[H]-TdR incorporation was determined. Data are presented as mean value of triplicate measurements and one representative experiment out of 2 independent experiments is shown. Data are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.0001 (Student's t test); ((G) One-way ANOVA). See also Supplementary Figure S5-S7.
![Figure 6. Lymphoma B cells produce and secrete purines that inhibit T cell function. (A) <1kDa fractions of BCM from BL/6 or Eµ-myc+L mice (n = 4 mice/group) were analyzed for metabolites using mass spectrometry. Results are indicated as S-Plot correlation of equal masses, detected in BL/6 ( = −1) or Eµ-myc+L ( = +1) mice. Masses with a very high (Eµ-myc+L = +1) or very low (BL/6 = −1) correlation reflect the highest significance. Masses selected for further screening are displayed in red and depicted in Table 1. (B) NMR analysis. (C) NMR signal after spike in of Hypoxanthine standard or Eµ-myc+L BCM (n = 4 mice/group). (D, E) Serum concentration of (D) ATP and (E) uric acid (n = 3 mice/group). (F) ATP concentrations in MACS-purified B cell lysates (n = 3 lysates/group). Concentrations of indicated purines in BCM (n = 3–5 BCM/group). Proliferation of purified, naïve CD8+ T cells from BL/6 mice was determined after addition of titrated amounts of different purines. (G) Titrated amounts of ATP, 5′ADP or AMP were added separately or combined (ATP + 5′ADP and ATP+5′ADP+AMP) to purified, naïve CD8+ T cells from BL/6 mice and 3[H]-TdR incorporation was determined. Data are presented as mean value of triplicate measurements and one representative experiment out of 2 independent experiments is shown. Data are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.0001 (Student's t test); ((G) One-way ANOVA). See also Supplementary Figure S5-S7.](/cms/asset/5206d434-2162-41c5-bfaa-7fb282fadf01/koni_a_1365997_f0006_oc.gif)
Table 1. Identification of lipids and members of the purine pathway in BCM from Eµ-myc+L mice. <1kDA fractions of BCM from Eµ-myc+L mice or BL/6 mice were analyzed for metabolites using Mass Spectrometry (as described in material and methods). Masses that appeared with highest intensity and significance in Eµ-myc+L BCM were further verified by fragmentation analysis. Identified masses contained bioactive lipids and members of the purine pathway, listed in , (n = 4 mice/group).
Lactic acid levels were elevated in Eµ-myc+L BCM by NMR (Fig. S6A) as well as by a multiple biochemical analyzer (Fig. S6B). Indeed, lactic acid inhibited T cell proliferation in a concentration-dependent manner (Fig. S6C). Importantly, the concentration of lactic acid necessary to reduce T cell expansion was substantially higher (6.7–20 mM) than the concentrations found in BCM form Eμ-myc+L mice (0.87 mM). Inhibition of lactic acid by the lactic acid dehydrogenase (LDH) inhibitor Sodium Oxamate (S.O.) in Eµ-myc+L BCM did not restore T cell proliferation (Fig. S6D). This suggests that this single pathway does not substantially contribute to the observed T cell dysfunction in our model.
Titrated amounts of S1P did not inhibit CD8+ T cell proliferation in vitro (Fig. S7A). In addition, degradation of S1P in Eµ-myc+L BCM did not reduce the inhibitory effect on CD8+ T cell proliferation (Fig. S7B). Similarly, the polar lipid containing fractions of Eµ-myc+L BCM did not inhibit T cell proliferation (Fig. S7C). These experiments excluded polar lipids such as S1P, psychosine and D-glucosyl-sphingosine as mediators of CD8+ T cell inhibition in our model.
Increased production of purine metabolites and elevated serum levels of uric acid are a hallmark of rapidly proliferating lymphoma cells and frequently detected in patients with aggressive NHL.Citation31,32 We confirmed increased levels of ATP and uric acid in the sera of Eµ-myc+L mice (, ). Moreover, we found substantially higher ATP concentrations in lysates and in supernatants of Eµ-myc+L B cells than of normal BL/6 B cells ( and data not shown). All degradation products of ATP analyzed such as adenosine, adenine, hypoxanthine, xanthine and uric acid were present at higher concentrations in Eµ-myc+L BCM than in BL/6 BCM (). Next, we analyzed which of these molecules potentially inhibit CD8+ T cell proliferation in vitro. We found that ATP, ADP, AMP, adenosine, adenine and uric acid had an inhibitory effect on T cell expansion (). The role of adenosine was also tested with its stable functional analog 2-Chloro-Adenosine (2-CADO) (). No effect on T cell proliferation was found for hypoxanthine, xanthine and uridine (). Moreover, the combination of ATP, 5'ADP, and AMP suppressed T cell function significantly stronger than each of these purine metabolites alone ().
These data indicate that adenine containing purines are elevated in sera and BCM of Eµ-myc+L mice and that ATP degradation products are potent suppressors of CD8+ T cell proliferation in vitro.
Blocking apoptosis or purine receptors partially restores T cell function in B cell lymphoma
It is well documented that purines are released by apoptotic cells.Citation33,34 In addition, an increased cell proliferation accompanied by an increased apoptosis rate is a hallmark of BL.Citation35 We therefore asked if blocking apoptosis of Eµ-myc+L B cells reduced the inhibitory effect on T cells. Treatment of Eµ-myc+L B cells with the pan-caspase inhibitor QVD significantly reduced the concentration of ATP and of its degradation products (hypoxanthin, xanthine and thymidine) in the BCM (). Indeed, incubation of Eµ-myc+L B cells with QVD restored 3[H]-TdR uptake of naïve CD8+ T cells (). Similarly, degradation of ATP by the enzyme apyrase partially reduced the T cell suppressive effect of Eµ-myc+L BCM (). The ectonucleotidases CD39 and CD73 act as extracellular degradation enzymes of purines and have increased activity in cancer.Citation26,36,37 In line with this, we found an approximately 100-fold higher expression of CD39 on CD4+ and CD8+ T cells of Eµ-myc+L mice when compared with controls, but only a slight difference in CD73 expression (). To further confirm the role of ATP degradation products in T cell dysfunction, we used naive CD8+ T cells from CD39−/− () or A2A−/− mice () in a 3[H]-TdR incorporation assay in the presence of BL/6 or Eµ-myc+L BCM. BL/6 BCM and Eµ-myc+L BCM inhibited the expansion of CD39−/− or A2A−/− T cells to a lesser extent than the expansion of BL/6 T cells (, ).
Figure 7. Metabolites of the purine degradation pathway are released by apoptotic lymphoma B cells and induce CD8+ T cell dysfunction. (A, B) MACS-purified B cells from Eµ-myc+L mice were cultured in the presence or absence of the caspase inhibitor QVD (50 µM) and BCM was harvested after indicated time points. (A) The concentrations of ATP, hypoxanthine, xanthine and thymidine in BCM from Eµ-myc+L mice were determined 1 h (ATP) and 24 h (hypoxanthine, xanthine and thymidine) after QVD or control (DMSO) treatment. (B) BCM was harvested 6 h after incubation and added to CD8+ T cell responders at a 1:1 ratio and 3[H]–TdR incorporation was measured. 1 representative out of 2 experiments is shown. (C) MACS-purified B cells from Eµ-myc+L mice were cultured in the presence or absence of the ATP degrading enzyme apyrase at different concentrations. BCMs were harvested 24 h after in vitro culture. BCMs were added to BL/6 CD8+ T cell responders at a 1:3 ratio and 3[H]–TdR incorporation was measured. 1 representative out of 3 experiments is shown. (D) CD4+ and CD8+ T cells from spleens of BL/6 or Eµ-myc+L mice were analyzed for the expression of ATP ectonucleotidases CD39 and CD73 using flow cytometry ex vivo. (n = 2; black: staining, gray: isotype, MFI: MFI staining – MFI isotype). One representative FACS plot out of 4 independent experiments is shown. (E, F) CD8+ T cells responders from naïve BL/6 or (E) CD39−/− (red bars) or (F) A2A−/− (green bars) mice were cultured in the presence or absence of Eµ-myc+L BCM at a 1:3 ratio and 3[H]–TdR incorporation was measured. Data are presented as mean value of triplicate measurements and one representative experiment out of 2 independent experiments is shown. (G) Spleen weights, (H) total splenocyte numbers and (I) T cell/B cell ratios of Eµ-myc x CD39−/−+L or Eµ-myc x CD39+/−+L. (J, K) MACS-purified (J) CD4+ and (K) CD8+ T cells were isolated from Eµ-myc x CD39−/−+L or Eµ-myc x CD39+/−+L and 3[H]–TdR incorporation was measured (n = 2 mice/group). (L) Kaplan-Meier survival curve of Eµ-myc x CD39+/−+L (n = 30) and Eµ-myc x CD39−/−+L (n = 4). Survival is depicted as Kaplan-Meier curve (n = 5 mice/group). Data are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.0001 (Student's t test). See also Supplementary Figure S8 and S9.
![Figure 7. Metabolites of the purine degradation pathway are released by apoptotic lymphoma B cells and induce CD8+ T cell dysfunction. (A, B) MACS-purified B cells from Eµ-myc+L mice were cultured in the presence or absence of the caspase inhibitor QVD (50 µM) and BCM was harvested after indicated time points. (A) The concentrations of ATP, hypoxanthine, xanthine and thymidine in BCM from Eµ-myc+L mice were determined 1 h (ATP) and 24 h (hypoxanthine, xanthine and thymidine) after QVD or control (DMSO) treatment. (B) BCM was harvested 6 h after incubation and added to CD8+ T cell responders at a 1:1 ratio and 3[H]–TdR incorporation was measured. 1 representative out of 2 experiments is shown. (C) MACS-purified B cells from Eµ-myc+L mice were cultured in the presence or absence of the ATP degrading enzyme apyrase at different concentrations. BCMs were harvested 24 h after in vitro culture. BCMs were added to BL/6 CD8+ T cell responders at a 1:3 ratio and 3[H]–TdR incorporation was measured. 1 representative out of 3 experiments is shown. (D) CD4+ and CD8+ T cells from spleens of BL/6 or Eµ-myc+L mice were analyzed for the expression of ATP ectonucleotidases CD39 and CD73 using flow cytometry ex vivo. (n = 2; black: staining, gray: isotype, MFI: MFI staining – MFI isotype). One representative FACS plot out of 4 independent experiments is shown. (E, F) CD8+ T cells responders from naïve BL/6 or (E) CD39−/− (red bars) or (F) A2A−/− (green bars) mice were cultured in the presence or absence of Eµ-myc+L BCM at a 1:3 ratio and 3[H]–TdR incorporation was measured. Data are presented as mean value of triplicate measurements and one representative experiment out of 2 independent experiments is shown. (G) Spleen weights, (H) total splenocyte numbers and (I) T cell/B cell ratios of Eµ-myc x CD39−/−+L or Eµ-myc x CD39+/−+L. (J, K) MACS-purified (J) CD4+ and (K) CD8+ T cells were isolated from Eµ-myc x CD39−/−+L or Eµ-myc x CD39+/−+L and 3[H]–TdR incorporation was measured (n = 2 mice/group). (L) Kaplan-Meier survival curve of Eµ-myc x CD39+/−+L (n = 30) and Eµ-myc x CD39−/−+L (n = 4). Survival is depicted as Kaplan-Meier curve (n = 5 mice/group). Data are displayed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.0001 (Student's t test). See also Supplementary Figure S8 and S9.](/cms/asset/fabfda10-6bb0-4e17-b50b-69ec2cca17b6/koni_a_1365997_f0007_oc.gif)
In CD39−/− mice, extracellular ATP cannot be converted into ADP or AMP leading to approximately 1000-fold higher concentrations of ATP and reduced levels of hypoxanthine, xanthine and inosine in serum when compared with BL/6 mice (Fig. S8A, B). To investigate T cell function and lymphoma development in the absence of ATP degradation products, we crossed Eµ-myc mice with CD39−/− mice. Spleen weights and absolute numbers of splenocytes of Eµ-myc x CD39−/−+L mice were significantly lower when compared with Eµ-myc x CD39+/−+L controls (, ). CD4+ and CD8+ T cell to B cell ratios were significantly increased in Eµ-myc x CD39−/−+L compared to Eµ-myc x CD39+/−+L mice (). Moreover, 3[H]-TdR incorporation of ex vivo isolated T cells from Eµ-myc x CD39−/−+L mice was significantly higher than from Eµ-myc x CD39+/−+L mice (, ). However, absence of ATP degradation products in Eµ-myc x CD39−/−+L mice did not improve survival of lymphoma mice when compared Eµ-myc x CD39+/−+L controls (). Of note, T cells from Eµ-myc x CD39−/−+L mice expressed higher levels of PD-1 (Fig. S9A, B), most likely due to pro-inflammatory environment induced by high ATP concentrations.Citation38,39
These results suggest that the T cell dysfunction in NHL is induced by several redundant pathways and that blocking one major immune inhibiting pathway is not sufficient to treat aggressive NHL.
Discussion
As lymphomas grow in lymphoid organs, it would be expected that they should be efficiently recognized and eliminated by the immune system. Indeed, experimental studies and clinical data clearly document an important role of T, NK and probably NKT cells in the immune control of lymphoma.Citation2,5-7,9Clinically manifest lymphoma in immune-competent hosts must have evaded immune surveillance. One well-documented mechanism of immune escape of lymphoma cells is the loss of antigen-presentation. Approximately 60% of DLBCL have an aberrant expression of β2-microglobulin preventing the presentation of antigenic peptides on MHC class I (HLA class I) to specific CD8+ T cells.Citation10 Similarly, loss of MHC II protein expression and thereby escape of recognition by CD4+ T cells has been documented in B-cell neoplasms and is associated with an aggressive clinical course.Citation40-42
In addition, experiments with lymphoma cells expressing a model antigen indicate that in advanced lymphoma specific T cells are dysfunctional.Citation43,44 How malignant B cells induce T cell dysfunction in vivo has not been documented so far. A siRNA library screen revealed that inhibitory molecules of the B7 family such as CD200, CD274 (PD-L1) and CD276 (B7-H3) and the TNF-receptor superfamily member CD270 (HVEM) impair the formation of the immunological synapse between T cells and chronic lymphatic leukemia cells in vitro.Citation45 Similarly, PD-L1 is expressed in DLBCL.Citation45 In the present study, we document a high expression of PD-L1 on normal and on malignant B cells and on non-malignant CD11c+ and CD11b+ myeloid cells. This is comparable to data from solid tumors where the expression of PD-L1 has been documented on cancer cells and on tumor-infiltrating immune cells.Citation46 In addition, a large fraction of CD8+ and CD4+ T cells expressed PD-1. Blocking PD-1 signaling increased the proliferation of CD8+ T cells in vitro when incubated with CD11c+CD11b+ cells isolated from lymphoma-bearing mice. This is in line with and extends a study by Rehm et al., indicating that a C/EBPb driven upregulation of immunomodulatory cytokines and growth factors in dendritic cells contributes to lymphoma progression in Eµ-myc mice.Citation47
Blocking PD-1 signaling by treatment with mAbs is efficacious in solid tumors and in refractory Hodgkin's lymphoma with alterations in the PD-L1- and PD-L2-encoding chromosome 9p24.1.Citation48-50 Furthermore, there is early evidence of efficacy in follicular lymphoma and in high-grade lymphoma after autologous haematopoietic stem cell transplantation.Citation51,52 Surprisingly, although blocking PD-1 did increase the frequencies and partially also the function of T cells, it did not significantly prolong survival in our model. This is in agreement with a recent study documenting that PD-1 blockade does not improve the control of Myc-driven lymphoma whereas a α-GalCer vaccine together with a stimulating 4–1BB mAb prolonged survival.Citation53 Importantly, 20–25% of total T cells in Eμ-myc+L mice expressed 2 or 3 immune inhibitory receptors including PD-1, 2B4 and LAG-3 and PD-1 blockade increased the expression of other immune inhibitory receptors.
An unbiased screen of BCM from lymphoma cells using MS and NMR revealed some candidate molecules that we functionally tested in vitro. Interestingly, molecules that are associated with an increased cell turn-over and cell death such as lactate and the purine hypoxanthine were detected in high concentrations. Similarly, Johnston H.E. et al. recently documented in a proteomic analysis of plasma from lymphoma-bearing mice a tumor lysis signature.Citation54
In addition, MS and NMR analysis indicated the presence of purines in BCM from lymphoma cells. ATP and its degradation products ADP, AMP, adenosine, adenine, hypoxanthine, xanthine, uric acid and uridine were present at elevated concentrations in lymphoma BCM or serum when compared with BL/6 controls. ATP, ADP, AMP, adenosine, adenine inhibited T cell proliferation when added to the culture in a concentration-dependent manner, whereas hypoxanthine, xanthine, uric acid and uridine had no effect, suggesting that adenosine/adenine-containing purines were the main molecules that inhibit T cell proliferation in our model. In accordance, P2X7 stimulation by high ATP concentrations has been shown to trigger cell death and inhibit T cell functions in different models.Citation38,55,56 However, depending on the concentrations, ATP has opposite effects on immune cells.Citation38,39,56,57 It has been shown that low ATP concentrations have immune-stimulating properties due to activation of the NALP3-inflammasome and caspase-1, resulting in the release of pro-inflammatory cytokines such as IL-1β, IL-18 and IL-33, important inducers of innate and adaptive immune responses.Citation58
Experiments with apyrase that leads to the degradation of ATP to ADP and AMP, with CD39−/− T cells that prevent the degradation of ATP and with A2A−/− T cells indicated that ATP itself and its degradation products reduce T cell proliferation. Moreover, T cells isolated from Eµ-myc x CD39−/−+L mice proliferated more efficiently ex vivo than T cells isolated from control lymphoma-bearing mice. These data indicate that although CD39 deficiency results in substantially higher concentrations of ATP, the lack of its degradation products leads to an improved T cell response in lymphoma bearing CD39−/− x Eµ-myc mice. However, although T cell function was partially improved, there was no significant survival difference between CD39-competent or -deficient lymphoma mice. Of note, absence of CD39 resulted in substantially higher concentrations of ATP that by itself mediates T cell suppression.
Recent studies suggest that only combinatorial blockade of different T cell inhibitory mechanisms enhance anti-tumoral T cell immune responses.Citation59,60 For example, blocking PD-1 leads to increased A2A expression.Citation61 In line with this, Kobayashi et al. identified a successful vaccine approach using the lipid antigen α-GalCer in combination with stimulating α4–1BB against a transplantable B cell clone derived from the Eµ-myc model.Citation62 These successful vaccination approaches,Citation53,62 and the reduced survival of RAG-deficient mice challenged with malignant B cells,Citation63 indicate that T cell immunosurveillance at least partially controls Eµ-myc-driven lymphoma. Nevertheless, the antigenicity of Myc-driven lymphoma might be substantially lower than that of EBV associated lymphoma in humans.Citation64
In summary, our study revealed that in advanced aggressive lymphoma different and partially redundant mechanisms contribute to a severe dysfunction of CD4+ and CD8+ T cells. This includes the expression of inhibitory cell-surface receptors such as PD-1, LAG-3 and 2B4. However, purine degradation products released by cells undergoing apoptosis crucially contribute to the suppression of the T cell response. It is well documented that c-myc activates pathways that induce cell proliferation but also sensitize cells to apoptotic stimuli.Citation65 Blocking apoptosis of lymphoma cells in vitro reduced the T cell inhibitory potential. Our findings are in accordance with a recent study that indicated that the “immunological visibility” and the immunosurveillance of Eμ-myc mice critically depend on the expression of the anti-apoptotic protein Bcl−2.Citation66 Therefore, successful immunotherapy of advanced aggressive NHL may depend on the combined blockade of several immune inhibitory pathways.
Material and methods
Mice
C57 BL/6 J (BL/6) mice were from RCC Ltd. (Füllinsdorf, Switzerland) and Charles River Laboratories (Sulzfeld, Germany). CD45.1+ mice, RAG-1−/− mice, p14 TCR transgenic miceCitation67 specific for the LCMV glycoprotein epitope 33–41 (gp33) (CD45.1+ x RAG−/− x Vα2+Vβ8.1+) and H8 transgenic miceCitation23 ubiquitously expressing LCMV-gp33 under the control of a MHC class I promotor were from the Institute for Laboratory Animals (Zürich, Switzerland). Eµ-myc mice were obtained from Prof. Dr. med. Frank Heppner and Dr. Roland Kälin Institute for Neuropathology at Charité University of medicine (Berlin, Germany).Citation20 CD39−/− mice were provided by Prof. Simon C. Robson, Division of Gastroenterology and Hepatology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts, USA.Citation68 A2A−/− mice were provided by Karen Dwyer and were bred at St Vincent's hospital, Melbourne, Victoria, Australia.Citation69 All mice were on a BL/6 background and were sex and age matched before entering the experiment. Animal experiments were approved by the experimental animal committee of the Canton of Bern, Switzerland, and performed according to Swiss laws for animal protection.
Virus
LCMV strain WE was provided by R.M. Zinkernagel (University of Zürich), propagated and titrated as described previously.Citation70 Virus was injected intravenously (i.v.) at a dosage of 2 × 102 or 1 × 104 pfu per mouse. T cell function was analyzed 6–8 d after immunisation.
Reagents
Brefeldin-A (Cat.#B5936), Phorbol-12-myristate-13-acetate (PMA; Cat.#P8139), ATP (Cat.#34369–07–8), 5'ADP (Cat.#20398–34–9), AMP (Cat.# 4578–31–8), adenosine (Cat.# 58–61–7), cAMP, 2-chloro-adenosine (2-CADO; Cat. #146–77–0), adenine (Cat. #73–24–5), uridine (Cat.#58–96–8), hypoxanthine (Cat.#H9636), xanthine (Cat.# 1196–43–6), uric acid (Cat.#U2875), apyrase (Cat.# A6237), lactic acid (Cat.# 252476) and sodium oxamate (S.O.) (Cat.# O2751) were purchased from SIGMA-Aldrich. Ionomycine (Cat.#sc-300835; Santa Cruz Biotech); LCMV peptides gp33 (amino acid sequence: KAVYNFATM) and gp13 (amino acid sequence: GLKGPDIYKGVYQFKSVEFD) (Cat.#SC1347; NeoMPS SA.); interleukin-2 (BioXCell); Q-VD-OPH (QVD) (Cat.# 03OPH109; MP Biomedicals);. sphingosine-1-phosphate (S1P), S1P -lyase (wt/mt), S1P1 and S1 P3 were a gift from Prof. A. Huwiler, Bern.
Antibodies and flow cytometry
αIFNγ-PE (Clone: XMG1.2; Cat.#127311), αTNFα-FITC (Clone: MP6-XT22; Cat.#117321), αCD80-PE (Clone: 16–10A1; Cat.#120801) were purchased from eBioscience. αCD8-FITC, -PE, -PerCP-Cy5.5, -PE-Cy7, -APC-Cy7 and -PacificBlue (Clone: 53–6.7; Cat.#1007XX), αCD4-FITC, -PE, -PerCP-Cy5.5, -PE-Cy7,-APC (Clone: GK1.5; Cat.#1004XX), αCD19-APC-Cy7 (Clone: 6D5; Cat.#115530), αCD45.1-PE, -PerCP-Cy5.5, -PE-Cy7, -APC (Clone: A20; Cat.#1107XX), αPD-1-PE-Cy7 (Clone: RMP1–30; Cat.#109110), αCD223 (LAG3)-PE (Clone: C9B7 W; Cat.#125208), αCD244.2-FITC (2B4) (Clone: M2B4; Cat.#133504), αCD39-PE (Clone: Duha59; Cat.#143804), αCD73-AlexaFluor647 (Clone: TY/11.8; Cat.# 127208), αCD11 PE-Cy7 (Clone: N418; Cat.#117318), αMHC-II-PE-Cy7 (Clone: M5/114.15.2; Cat.# 107630), αCD86-APC (Clone: GL1; Cat.#105012), αVα2-PerCP-Cy5.5 (Clone: B20.1; Cat.#127814), αCD62 L-PacificBlue (Clone: MEL-14; Cat.#104423); αCD44-APC-Cy7 (Clone: IM7; Cat.#103028) were from BioLegend. Annexin-V-PE was from Immunotools and αVβ8.1,8.2-PE (Clone: MR5–2; Cat.#140104) was from BD PharMingen. All samples were aquired on a BD LSRII (BD Bioscience). Data were analyzed using FlowJo™ software (TreeStar). Mouse αPD-1 mAb (Clone: 2A3; Cat.#BE0146) was from BioXCell and rat IgG2 a (Cat.#I4131) from Sigma.
Transplantation of lymphoma B cells
Isolation of CD19+ lymphoma B cells from the spleen of lymphoma-bearing Eμ-Myc mice was performed using MACS™ αCD19 MicroBeads and LS columns (Milteny Biotec). 3 × 106 purified lymphoma B cells were injected into BL/6 recipient mice, which developed lymphoma 3 to 4 weeks after transplantation. In some experiments, mice were treated with anti-mouse PD-1 mAb (RMP1–14) or rat IgG2 a (2A3) as isotype control (200 µg; i.p.; every third day).
Intracellular IFN-γ and TNF-α assay
1–2 × 106 splenocytes were re-stimulated in vitro for 5 h with 1 μM gp33, 1 μM gp13 or with 1 µg/ml Ionomycin and 100 ng/ml PMA, in the presence of 5 µg/ml Brefeldin A and 25 U/ml of recombinant mouse interleukin-2 (rmIL-2). Cells were stained for surface molecules and fixed with 4% paraformaldehyde. Cell membranes were permeabilized with Perm-Buffer (PBS, 2% FCS, 5 mM EDTA, 0.1% saponin, 0.2% NaN3) and stained with αIFN-γ and αTNF-α antibodies. The fluorescence intensity index was calculated by dividing the intensity of expression in cytokine positive cells by that of negative cells.
H8 DC immunisation protocol
BM cells from H8 mice (7–10 × 107 cells) were cultured in 20 ml of DC medium (RPMI 10%, GM-CSF 10%) in non-tissue-culture-treated petri dishes (VWR) for 10 days, performing half medium changes every 3 d. At day 10 of culture, the non-adherent cells were washed away and resuspended. 1–3 × 107 cells were re-plated per tissue-culture-treated Petri dish (Falcon) and 9 ml of RPMI 10%, GM-CSF 5% and 10 µg/ml LPS was added for DC maturation. At day 12 of culture, the maturation status of DCs was determined by flow cytometry. Mice were immunized twice i.v. with 2 × 105 H8 DCs at an interval of 2 d.
Generation of effector CTLs
1 × 106 splenocytes from p14 mice were adoptively transferred intravenously (i.v.) into BL/6 or Eµ-myc recipient mice. 12–18 h after injection cells were activated by i.v. injection of 2 × 105 H8 DCs or 1 × 104 pfu LCMV-WE. Six days after immunization, spleens were isolated and T cell function of adoptively transferred cells was analyzed. Alternatively, 1 × 106 MACS-purified Eµ-myc x p14 T cells were adoptively transferred into BL/6 mice. 18 h after transfer, p14 cells were activated with 1 × 104 pfu LCMV-WE. Six days after infection, spleens were isolated and T cell function was analyzed.
In vivo cytotoxicity assay
MACS-purified splenic CD19+ lymphoma B cells from malignant Eµ-myc mice were either labeled with 0.1 µM Carboxyfluorescein succinimidyl ester (CFSElo) or with 5 µM CFSE (CFSEhi) for 10 min at 37°C. Cells were then washed and only the CFSElo B cell population was loaded with gp33 peptide (1 μM) for 90 min at 37°C. 15 to 30 × 106 peptide loaded CFSElo and non-peptide loaded CFSEhi B cells were adoptively transferred at a 1:1 ratio into BL/6 or Eµ-myc mice, that were immunised 8 d earlier with or without H8 DCs. Spleens were isolated 1 h and 4 h after i.v. injection of CFSE+ B cells and specific killing was analyzed by flow cytometry. The percent killing was calculated as: 100 − (((% peptide pulsed in infected / % un-pulsed in infected)/(% peptide pulsed in uninfected / % un-pulsed in uninfected)) × 100).
LCMV immunoplaque assay
Virus titers were determined after homogenization of total organs from Eµ-myc mice or BL/6 mice 8 d after infection with 200 pfu LCMV-WE or 6 d after adoptive transfer of 1 × 106 p14 TCR transgenic T cells followed by infection with 1 × 104 pfu LCMV-WE. Dilutions of supernatants from organs were used to infect a monolayer of MC57 cells after adding an methylcellulose (Sigma) overlay in MEM 10% FCS. 2 d later, cells were fixed and stained with VL-4 rat αLCMV mAb and a color reaction of ortho-phenylenediamine (Sigma) served for determining the focus-forming units. Virus titers were calculated by counting the focus-forming units and were divided by the weight of the respective organs from individual mice.
Proliferation assay
To measure the proliferation of T cells 2 × 105 MACS-purified naïve human PBMC-derived or mouse splenic CD8+ T cells were stimulated in vitro. Briefly, 96-U-bottom-well-plates were coated with plate bound anti-human OKT-3 or anti-mouse CD3ϵ-antibody at a concentration of 2 µg/ml overnight in PBS at 4°C. T cells were plated for in vitro expansion of 48 h. 0.5 µCi / well tritiated thymidine (3[H]-TdR) was added to each well for the last 17 h of culture. Cells were harvested with an Omnifilter-96-Harvester (PerkinElmer) on UniFilterJ GF/C plates (PerkinElmer). Incorporation of the radioactively labeled nucleoside was measured after pre-wetting filters with Microsynth 0 Scintillation liquid (PerkinElmer) on a top count microplate scintillation counter (PerkinElmer). In vitro proliferation of naïve Eμ-myc x p14 TCR transgenic T cells was analyzed by co-culturing 2 × 105 MACS-purified CD8+ T cells with 2 × 105 gp33-pulsed and irradiated splenocytes (1000 rad, γ-irradiator; Gammacell 1000, Nordion). After 3 d of co-culture proliferation was measured as described above.
CFSE-dilution assay
MACS-purified splenic CD8+ T cells were labeled with 0.1 µM CFSE for 10 min at 37°C. Cells were then washed and transferred into αCD3ϵ-coated 96-well plates. BCM from lymphoma-bearing Eµ-myc or BL/6 mice was added and CFSE dilution was assessed 72 h later by flow cytometry.
Co-culture of T cells with B cells or APCs
2 × 105 MACS-purified splenic T cells were co-cultured with MACS-purified and irradiated (1000 rad) splenic CD19+ B cells or with irradiated (1000 rad) CD11b/CD11c double positive APCs at different dilutions (1:1; 1:3; 1:10). APCs were isolated from spleens using biotinylated CD11b and CD11c primary antibodies and subsequent MACs purification with streptavidine Microbeads. In some experiments a blocking mouse αPD-1 mAb or control IgG was added (10 μg/ml). Trans-well plates (3.0 µm pore size and 6.5 mm diameter, Corning Costar) were used to separate B and T cell populations, whereas B cells were cultured in the upper chamber and T cells in the αCD3-coated lower chamber.
Filtration of B cell-conditioned media (BCM)
Splenic B cells were cultured at a concentration of 5 × 106 cells/ml in tissue culture-treated 6-well plates. 24 h later, BCM was removed and filtered through a 0.22 µm filter (VWR). The supernatants were then either directly added at different dilutions (1:1; 1:3; 1:10) to naive T cell responders or additional filter steps were performed: 100.000 NMWL (Nominal molecular weight limit); 10.000 NMWL; 3.000 NMWL; Amicon ultra centrifugal filter devices (Merck, Millipore, Billerica, Massachusetts); high recovery ultracel ultrafiltration membrane (1.000 NMWL, 25 mm diameter, Merck, Millipore, Billerica, Massachusetts) suited in a Amicon filter chamber (model 8003, stirred cell, Merck, Millipore, Billerica, Massachusetts). Different fractions of BCM (>100 kDa, 100–10 kDa, 10–3 kDa, <3 kDa, <1 kDa) were then separately added to T cells and proliferation was assessed.
Determination of ATP concentration
ATP concentration in mouse-sera, in whole B cell lysates and in BCM was determined using the ATP Bioluminescence Assay Kit HSII from Roche. Bioluminescence was detected using a Tecan Infinite 200 Plate Reader.
Determination of purine metabolites
Mouse serum levels of Uric Acid and BCM levels of Lactic Acid were determined at the Center of Laboratory Medicine, University Institute of Clinical Chemistry, Inselspital, Bern University Hospital and University of Bern, Switzerland using a multiple biochemical analyzer (cobas® 8000 modular analyzer, Roche). Hypoxanthine, xanthine, uridine, adenine and inosine concentrations were determined by high performance liquid chromatography (HPLC) at the University Institute of Clinical Chemistry, Inselspital, Bern University Hospital and University of Bern, Switzerland.
Protein digestion and measurement
Proteins in BCM were digested by using immobilized Proteinase K containing reaction columns, on 200 µl of matrix G3 m (MoBiTec). After enzymatic treatment, the amount of residual protein in the supernatants was assessed using NanoDrop™ 2000.
MS analysis using a UHPLC-TOF mass spectrometer
BCM and conditioned cell media were injected into a reverse-phase Acquity UPLC HSS T3 C18 Column, 100 Å, 1.8 µm, 1 mm x 100 mm (Waters Corp.) with a gradient mobile phase comprising 0.1% formic acid and acetonitrile containing 0.1% formic acid. Each sample was resolved for 15 min at a flow rate of 0.28 mL/min with the linear gradient consisted of 2% B for 1 min, 1–9.5 min from 2 to 98% B; 9.5–12 min 98% B, and 2% B for 3 min. The column temperature was 60°C. The eluent was introduced by electrospray ionization into the MS (Waters Synapt G2 QTOF), operating in either positive ion (ESI+) or negative ion (ESI-) electrospray ionization modes. In both modes the capillary voltage was set to 700 V and the sampling cone to 30 V. The dissolving gas flow was set to 950 L/h and the temperature to 550°C. The cone gas flow was 50 L/h, and the source temperature was set to 140°C. To maintain mass accuracy, Leucin-Enkephalin ((M-H)+ = m/z 556.2771 or (M-H)− = m/z 554.2615) at a concentration of 2 µg/ml in 50% acetonitrile was used as a lock mass and injected at a rate of 20 µl/min. Data were acquired in continuum mode from 50 to 1200 m/z at a scan time of 0.3 sec. The MS collision energy was scanned from 15 to 40 V.
The continuum data was converted to centroid with the integrated program in MassLynx. Centroided and integrated chromatographic mass data from 50 to 1200 m/z were processed by MarkerLynx (Waters) to generate a multivariate data matrix. Pareto-scaled MarkerLynx matrices including information on sample identity were analyzed by principal components analysis (PCA) and partial least-squares discriminant analysis (PLS-DA) using Markerlynx XS (Waters).To determine which ions contribute to the difference between healthy (y = −1) and lymphoma-bearing (y = +1) mice orthogonal partial least squares (OPLS) was used. The loadings scatter S-plots and the contribution lists were used to describe the candidate markers that were significantly different between healthy and lymphoma-bearing mice. To identify the structure of high-contribution score metabolites, elemental compositions were generated with MassLynx (Waters) based on the exact masses of the top ions. The exact masses and the generated formula were used to screen for biomarkers in different databases like Metacyc and Metlin. Results were compared with the literature to define bioactive compounds with known immune modulatory effects. Other masses with unknown effects were excluded. The remaining possible biomarkers were analyzed using a MS/MS fragmentation spectrum with collision energy ramping from 15 to 40 V.
Nuclear magnetic resonance (NMR) spectroscopy
550 µl of filtered supernatants of Eµ-myc or BL/6 B cells or the non-conditioned medium control were mixed with 50 µl of 0.2% 3-(4,4-dimethyl-4-silapentane-1-ammonium trifluoroacetate) (DSA) (w/v) in an eppendorf tube. After being thoroughly mixed, eppendorf contents were transferred to a labeled NMR tube (WILMAD Labglass). Prepped samples were stored at −40°C. Prior to NMR runs, samples were thawed and inverted several times to mix. Samples were run on a Bruker DRX600 spectrometer fitted with a 5 mm BBI Probe (Bruker Biospin) with a BACS 60 automated sample changer. 1D Carr-Purcell-Meiboom-Gill (CPMG) 1H spectra were acquired for each media sample with a T2 relaxation time of 32 ms to suppress any broad resonances. Final obtained spectra for each sample were the sum of 128 scans. Sample spectra were imported into Matlab (release 2010, The MathWorks, USA), phased, baseline corrected and calibrated to the DSA resonance (0 ppm). Metabolite assignments were made using Chenomx NMR Suite 7.0 software, online metabolome databases and previously published.Citation71-73 Using Matlab, resonances from metabolites of interest were integrated and normalized to the integral of DSA to give relative metabolite concentrations.
Statistical analysis
Statistical analysis was performed by GraphPad Prism 5.0 (GraphPad Software). Results are represented as mean ± SEM. Data were analyzed using Two-tailed student's t-test, one-way ANOVA or log-rank test. P ≤ 0.05 was considered significant.
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Authorship contribution
T. Hilmenyuk designed and performed experiments, interpreted the data, prepared the figures and wrote the paper; C.A. Ruckstuhl designed and performed experiments, interpreted the data, prepared the figures and wrote the paper; M. Hayoz and C. Berchtold performed MS measurements; J. M. Nuoffer performed HPLC experiments, S. Solanki and H. C. Keun performed NMR measurements; P. A. Beavis performed experiments and provided us with materials, facilities and A2A−/− mice; C. Riether prepared figures, designed experiments and wrote the paper; A.F. Ochsenbein designed and supervised experiments and wrote the paper.
downloadFromZipFile.pdf
Download PDF (240.6 KB)Acknowledgments
We thank Andrea Huwiler for providing us with S1P, S1P Lyases and S1P receptor inhibitors as well for the determination of S1P levels in mouse serum and BCM by MS (Institute of Pharmacology, University of Bern); Thomas Kaufmann for providing us with QVD and other apoptosis inhibitors (Institute of Pharmacology, University of Bern) and Simon C. Robson for providing us CD39−/− mice (Division of Gastroenterology and Hepatology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts, USA). This work was supported by the Swiss National Science Foundation, the Swiss Cancer League, the Cancer League of the Canton of Bern, the Werner und Hedy Berger-Janser-Stiftung, the Bernese Foundation for Clinical Cancer Research and the Marlies Schwegler-Stiftung.
References
- Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Adv Immunol. 2011;110:1-26. doi:10.1016/B978-0-12-387663-8.00005-3. PMID:21762814
- Kallies A. T cell immunosurveillance controls B lymphoma development. Oncoimmunology. 2014;3:e28697. doi:10.4161/onci.28697. PMID:25050223
- Grulich AE, Vajdic CM, Cozen W. Altered immunity as a risk factor for non-Hodgkin lymphoma. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2007;16:405-8. doi:10.1158/1055-9965.EPI-06-1070
- Shapiro RS. Malignancies in the setting of primary immunodeficiency: Implications for hematologists/oncologists. American journal of hematology 2011;86:48-55. doi:10.1002/ajh.21903. PMID:21120868
- Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, Wang Z, Derudder E, Li S, Chakraborty T, et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell. 2012;148:739-51. doi:10.1016/j.cell.2011.12.031. PMID:22341446
- Afshar-Sterle S, Zotos D, Bernard NJ, Scherger AK, Rodling L, Alsop AE, Walker J, Masson F, Belz GT, Corcoran LM, et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat Med. 2014;20:283-90. doi:10.1038/nm.3442. PMID:24487434
- Bolitho P, Street SE, Westwood JA, Edelmann W, Macgregor D, Waring P, Murray WK, Godfrey DI, Trapani JA, Johnstone RW, et al. Perforin-mediated suppression of B-cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:2723-8. doi:10.1073/pnas.0809008106. PMID:19196996
- Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755-60. doi:10.1084/jem.192.5.755. PMID:10974040
- Street SE, Hayakawa Y, Zhan Y, Lew AM, MacGregor D, Jamieson AM, Diefenbach A, Yagita H, Godfrey DI, Smyth MJ. Innate immune surveillance of spontaneous B cell lymphomas by natural killer cells and gammadelta T cells. J Exp Med. 2004;199:879-84. doi:10.1084/jem.20031981. PMID:15007091
- Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, Dalla-Favera R. Combined genetic inactivation of beta2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20:728-40. PMID:22137796
- Rimsza LM, Roberts RA, Miller TP, Unger JM, LeBlanc M, Braziel RM, Weisenberger DD, Chan WC, Muller-Hermelink HK, Jaffe ES, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251-8. PMID:14976040
- Stopeck AT, Gessner A, Miller TP, Hersh EM, Johnson CS, Cui H, Frutiger Y, Grogan TM. Loss of B7.2 (CD86) and intracellular adhesion molecule 1 (CD54) expression is associated with decreased tumor-infiltrating T lymphocytes in diffuse B-cell large-cell lymphoma. Clin Cancer Res. 2000;6:3904-9. PMID:11051236
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64. PMID:22437870
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533-8.
- Zhu D, Qi CF, Morse HC, 3rd, Janz S, Stevenson FK. Deregulated expression of the Myc cellular oncogene drives development of mouse “Burkitt-like” lymphomas from naive B cells. Blood 2005;105:2135-7. PMID:15522957
- Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harbor perspectives in medicine 2014;4:a014282.
- Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15:45-56. PMID:25534622
- Waggoner SN, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front Immunol. 2012;3:377. PMID:23248626
- Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene 2008;27:5932-43. doi:10.1038/onc.2008.267. PMID:18836474
- Sidman CL, Denial TM, Marshall JD, Roths JB. Multiple mechanisms of tumorigenesis in E mu-myc transgenic mice. Cancer research 1993;53:1665-9. PMID:8453639
- Wherry EJ. T cell exhaustion. Nature immunology 2011;12:492-9. doi:10.1038/ni.2035. PMID:21739672
- Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Olive D, Kuchroo V, Zarour HM. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72:887-96. doi:10.1158/0008-5472.CAN-11-2637. PMID:22205715
- Ehl S, Hombach J, Aichele P, Rulicke T, Odermatt B, Hengartner H, Zinkernagel R, Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J Exp Med. 1998;187:763-74. doi:10.1084/jem.187.5.763. PMID:9480986
- Moskophidis D, Battegay M, van den Broek M, Laine E, Hoffmann-Rohrer U, Zinkernagel RM. Role of virus and host variables in virus persistence or immunopathological disease caused by a non-cytolytic virus. J Gen Virol. 1995;76 (Pt 2):381-91. doi:10.1099/0022-1317-76-2-381. PMID:7531218
- Dolan DE, Gupta S. PD-1 pathway inhibitors: changing the landscape of cancer immunotherapy. Cancer Control. 2014;21:231-7. PMID:24955707
- Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201-12. doi:10.1038/nri2938. PMID:21331080
- Jin Y, Knudsen E, Wang L, Bryceson Y, Damaj B, Gessani S, Maghazachi AA. Sphingosine 1-phosphate is a novel inhibitor of T-cell proliferation. Blood. 2003;101:4909-15. doi:10.1182/blood-2002-09-2962. PMID:12586615
- Hoskin DW, Reynolds T, Blay J. Adenosine as a possible inhibitor of killer T-cell activation in the microenvironment of solid tumours. Int J Cancer. 1994;59:854-5. doi:10.1002/ijc.2910590625. PMID:7989130
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812-9. doi:10.1182/blood-2006-07-035972. PMID:17255361
- Burnstock G, Boeynaems JM. Purinergic signalling and immune cells. Purinergic Signal. 2014;10:529-64. doi:10.1007/s11302-014-9427-2. PMID:25352330
- Fini MA, Elias A, Johnson RJ, Wright RM. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin Transl Med. 2012;1:16. doi:10.1186/2001-1326-1-16. PMID:23369448
- Primikirios N, Stutzman L, Sandberg AA. Uric acid excretion in patients with malignant lymphomas. Blood 1961;17:701-18. PMID:13738036
- Chen J, Zhao Y, Liu Y. The role of nucleotides and purinergic signaling in apoptotic cell clearance – implications for chronic inflammatory diseases. Front Immunol. 2014;5:656. doi:10.3389/fimmu.2014.00656. PMID:25566266
- Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516-21. doi:10.1038/nature01991. PMID:14520412
- Milner AE, Grand RJ, Waters CM, Gregory CD. Apoptosis in Burkitt lymphoma cells is driven by c-myc. Oncogene. 1993;8:3385-91. PMID:8247541
- Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355-67. doi:10.1016/j.molmed.2013.03.005. PMID:23601906
- Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MW, et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell. 2016;30:391-403. doi:10.1016/j.ccell.2016.06.025. PMID:27622332
- Trabanelli S, Ocadlikova D, Gulinelli S, Curti A, Salvestrini V, Vieira RP, Idzko M, Di Virgilio F, Ferrari D, Lemoli RM. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J Immunol. 2012;189:1303-10. doi:10.4049/jimmunol.1103800
- Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal. 2009;2:pe6. doi:10.1126/scisignal.256pe6. PMID:19193605
- Amiot L, Onno M, Lamy T, Dauriac C, Le Prise PY, Fauchet R, Drenou B. Loss of HLA molecules in B lymphomas is associated with an aggressive clinical course. Br J Haematol. 1998;100:655-63. doi:10.1046/j.1365-2141.1998.00631.x. PMID:9531330
- Riemersma SA, Jordanova ES, Schop RF, Philippo K, Looijenga LH, Schuuring E, Kluin PM. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood. 2000;96:3569-77. PMID:11071656
- Wilkinson ST, Vanpatten KA, Fernandez DR, Brunhoeber P, Garsha KE, Glinsmann-Gibson BJ, Grogan TM, Teruya-Feldstein J, Rimsza LM. Partial plasma cell differentiation as a mechanism of lost major histocompatibility complex class II expression in diffuse large B-cell lymphoma. Blood. 2012;119:1459-67. doi:10.1182/blood-2011-07-363820. PMID:22167754
- Prato S, Mintern JD, Lahoud MH, Huang DC, Villadangos JA. Induction of antigen-specific effector-phase tolerance following vaccination against a previously ignored B-cell lymphoma. Immunol Cell Biol. 2011;89:595-603. doi:10.1038/icb.2010.131. PMID:21079642
- Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, Hemmi S, Hengartner H, Zinkernagel RM. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 2001;411:1058-64. doi:10.1038/35082583. PMID:11429607
- Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood 2012;120:1412-21. doi:10.1182/blood-2012-02-411678. PMID:22547582
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280 A in cancer patients. Nature. 2014;515:563-7. doi:10.1038/nature14011. PMID:25428504
- Rehm A, Gatjen M, Gerlach K, Scholz F, Mensen A, Gloger M, Heinig K, Lamprecht B, Mathas S, Bégay V, et al. Dendritic cell-mediated survival signals in Emu-Myc B-cell lymphoma depend on the transcription factor C/EBPbeta. Nat Commun. 2014;5:5057. doi:10.1038/ncomms6057. PMID:25266931
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-30. doi:10.1056/NEJMoa1412082. PMID:25399552
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123-35. doi:10.1056/NEJMoa1504627. PMID:26028407
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311-9. doi:10.1056/NEJMoa1411087. PMID:25482239
- Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Holland HK, Winter JN, Mason JR, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31:4199-206. doi:10.1200/JCO.2012.48.3685. PMID:24127452
- Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, Koren-Michowitz M, Shimoni A, Nagler A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:3044-51. doi:10.1158/1078-0432.CCR-07-4079. PMID:18483370
- McKee SJ, Doff BL, Soon MS, Mattarollo SR. Therapeutic Efficacy of 4-1BB Costimulation Is Abrogated by PD-1 Blockade in a Model of Spontaneous B-cell Lymphoma. Cancer Immunol Res. 2017;5:191-7. doi:10.1158/2326-6066.CIR-16-0249. PMID:28115358
- Johnston HE, Carter MJ, Cox KL, Dunscombe M, Manousopoulou A, Townsend PA, Garbis SD, Cragg MS. Integrated Cellular and Plasma Proteomics of Contrasting B-cell Cancers Reveals Common, Unique and Systemic Signatures. Mol Cell Proteomics. 2017;16:386-406. doi:10.1074/mcp.M116.063511
- Zheng LM, Zychlinsky A, Liu CC, Ojcius DM, Young JD. Extracellular ATP as a trigger for apoptosis or programmed cell death. J Cell Biol. 1991;112:279-88. doi:10.1083/jcb.112.2.279. PMID:1988462
- Adinolfi E, Pizzirani C, Idzko M, Panther E, Norgauer J, Di Virgilio F, Ferrari D. P2X(7) receptor: Death or life? Purinergic Signal. 2005;1:219-27. doi:10.1007/s11302-005-6322-x. PMID:18404507
- Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33:231-7. doi:10.1016/j.it.2012.02.009. PMID:22487321
- Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659-62. doi:10.1016/j.cell.2006.08.002. PMID:16923387
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275-80. doi:10.1073/pnas.0915174107. PMID:20160101
- Janakiram M, Pareek V, Cheng H, Narasimhulu DM, Zang X. Immune checkpoint blockade in human cancer therapy: lung cancer and hematologic malignancies. Immunotherapy. 2016;8:809-19. doi:10.2217/imt-2016-0001. PMID:27349980
- Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, Kershaw MH, Stagg J, Darcy PK. Adenosine Receptor 2 A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol Res. 2015;3:506-17. doi:10.1158/2326-6066.CIR-14-0211. PMID:25672397
- Kobayashi T, Doff BL, Rearden RC, Leggatt GR, Mattarollo SR. NKT cell-targeted vaccination plus anti-4-1BB antibody generates persistent CD8 T cell immunity against B cell lymphoma. Oncoimmunology 2015;4:e990793. doi:10.4161/2162402X.2014.990793. PMID:25949907
- Al Sayed MF, Ruckstuhl CA, Hilmenyuk T, Claus C, Bourquin JP, Bornhauser BC, Radpour R, Riether C, Ochsenbein AF. CD70 reverse signaling enhances NK cell function and immunosurveillance in CD27-expressing B-cell malignancies. Blood. 2017. doi:10.1182/blood-2016-12-756585. PMID:28495792
- Schlee M, Schuhmacher M, Holzel M, Laux G, Bornkamm GW. c-MYC impairs immunogenicity of human B cells. Adv Cancer Res. 2007;97:167-88. doi:10.1016/S0065-230X(06)97007-9. PMID:17419945
- Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462-72. doi:10.1038/onc.2008.312. PMID:18955973
- Schuster C, Berger A, Hoelzl MA, Putz EM, Frenzel A, Simma O, Moritz N, Hoelbl A, Kovacic B, Freissmuth M. et al. The cooperating mutation or “second hit” determines the immunologic visibility toward MYC-induced murine lymphomas. Blood. 2011;118:4635-45. doi:10.1182/blood-2010-10-313098. PMID:21878673
- Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559-61. doi:10.1038/342559a0. PMID:2573841
- Enjyoji K, Sevigny J, Lin Y, Frenette PS, Christie PD, Esch JS, 2 nd, Imai M, Edelberg JM, Rayburn H, Lech M, et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med. 1999;5:1010-7. doi:10.1038/12447. PMID:10470077
- Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2 a receptor. Nature. 1997;388:674-8. doi:10.1038/41771. PMID:9262401
- Matter M, Odermatt B, Yagita H, Nuoffer JM, Ochsenbein AF. Elimination of chronic viral infection by blocking CD27 signaling. J Exp Med. 2006;203:2145-55. doi:10.1084/jem.20060651. PMID:16923852
- Ellis JK, Athersuch TJ, Cavill R, Radford R, Slattery C, Jennings P, McMorrow T, Ryan MP, Ebbels TM, Keun HC. Metabolic response to low-level toxicant exposure in a novel renal tubule epithelial cell system. Mol Biosyst. 2011;7:247-57. doi:10.1039/C0MB00146E. PMID:21103459
- Zheng XY, Ling ZY, Tang ZY, Liu YK, Feng XL, Zhuang W. The abundance of NM23-H1 mRNA is related with in situ microenvironment and intrahepatic metastasis in hepato-cellular carcinoma. Journal of experimental & clinical cancer research: CR 1998;17:337-41.
- Yakoub D, Keun HC, Goldin R, Hanna GB. Metabolic profiling detects field effects in nondysplastic tissue from esophageal cancer patients. Cancer Res. 2010;70:9129-36. doi:10.1158/0008-5472.CAN-10-1566. PMID:20884633