
ABSTRACT
The tumor infiltration of immune cells in solid cancers can profoundly influence host antitumor responses. In recent years, immunotherapeutic regimens, that target immune checkpoints, demonstrated significant antitumor response by increasing intra-tumoral immune cell populations, including CD8+ effector T cells. However, administration of such immune checkpoint inhibitors is largely inefficacious in inducing immunogenicity and treating breast cancer. Currently, there is a great need to better understand cell autonomous mechanisms of immune evasion in breast cancer to identify upstream therapeutic targets that increase the efficacy of immunotherapy. Here we show that Crk, an SH2 and SH3 domain-containing adaptor protein implicated in focal adhesion signaling, cell migration, and invasion, and frequently up-regulated in human cancers, has an important role in regulating the tumor immune microenvironment. Using a murine 4T1 breast adenocarcinoma model of spontaneous metastasis in immune-competent BALB/C mice, we show that genetic ablation of Crk by CRISPR-Cas9 leads to enhanced anti-tumor immune cell populations, cytotoxic effector and immune surveillance cytokines in primary tumor. Pathologically, this leads to a significant reduction in tumor growth and lung metastasis. Mechanistically, Crk KO suppresses EMT and PD-L1 expression on tumor cells and acts additively with anti-PD1 therapy to suppress tumor growth and metastasis outcomes. Taken together, these data reveal a previously un-described function of Crk adaptor protein expression in tumor cells for cell autonomous regulation of tumor immune microenvironment.
Introduction
Crk adaptor proteins, comprised of two alternatively spliced isoforms that contain SH2 and SH3 modular domains, have critical functions in signal transduction downstream from cell adhesion molecules and growth factor receptors by assembling protein-protein interactions downstream of tyrosine kinases.Citation1-7 By binding to tyrosine phosphorylated focal adhesion proteins that include paxillin and p130cas via its SH2 domain, and Guanine Nucletoide Exchange Factors (GNEFs) for GTPases such as Rap1, Rac1 and Ras via its SH3 domain, Crk regulates evolutionarily conserved cell biological processes that impinge on cell adhesion and motility in metazoan cells.Citation8-11
Pathophysiologically, Crk is overexpressed in a wide array of human cancers, including but not limited to ovarian cancer, glioblastoma multiformae (GBM), lung cancer, breast cancer, and sarcoma; whereby the level of expression often correlates with tumor grade and inversely segregates with overall patient survival.Citation8 Although the effects of Crk overexpression are complex and multifactorial, a recurring hallmark of Crk overexpression in solid cancers is ability to induce cell transformation, promote epithelial-to-mesenchymal transition (EMT), and increase metastatic activity in mouse models.Citation12 More recent studies showed that Crk synergizes with TGF-β in an autocrine activation loop to promote EMT in lung cancerCitation13 which is consistent with previous studies showing that Crk can facilitate HGF-induced EMT-like phenotypes in canine MDCK cells and T47D human breast cancer cells.Citation14 Although the roles of Crk in driving transformation and cell migration is reasonably well understood at the biochemical and molecular level, the role of Crk in regulation of cell extrinsic functions with respect to tumor immune microenvironment has not been thoroughly investigated. Recent data showing that FAK, which like Crk regulates focal adhesion signaling and tyrosine phosphorylation, is an important regulator of an immunosuppressive tumor microenvironment suggests that key oncogenic networks that regulate cell adhesion and EMT may also impinge on the cell extrinsic tumor microenvironment.Citation15
In the present study, using an orthotopic syngeneic immune-competent murine model, we show that knockout of Crk in the poorly immunogenic breast adenocarcinoma 4T1 cells results in an increase of intra-tumoral immune cell infiltrations associated with favorable host anti-tumor outcomes. Crk depleted tumors exhibited increased intra-tumoral CD8+ and CD3+ T-cells, NK cells, and neutrophils, as well as a decrease in PDL-1 expression and reversal of Zeb-1 induced EMT program. Cytokine profiling of the primary tumors from Crk knockout revealed an elevation of tumor suppressing and immune-surveillance suggesting that infiltrating cells are functional to create an inflamed tumor microenvironment. Additionally, combinatorial in vivo results indicate that Crk KO acts additively with anti-PD1 in suppressing tumorigenesis and lung metastasis. In effect, the reprogramming of immune microenvironment by Crk KO results in reduction of both tumor growth and lung metastasis. These results establish a previously undescribed role of Crk in tumor immunology and immune evasion in an immune-competent mouse model.
Materials and methods
Generation of Crk knockout 4T1 murine breast cancer cells
4T1 murine adenocarcinoma cell lines were grown in RPMI supplemented with 10% FBS. Two individual guide RNAs targeting exon 1 and exon 2 of cellular murine Crk were synthesized and cloned into All-in-one plasmid vector (U6-gRNA/CMV-Cas9-RFP) (Sigma). Both vectors containing the guide RNAs were individually transfected into 4T1 cells and sorted for RFP expression after 48 hrs. Cas9-2A peptide-RFP fusion protein expression enabled monitoring of transfection efficiency and FACS based single cell sorting. Single cell clones were grown and screened by western blotting for Crk expression. A total of four individual clones were tested in in vitro experiments and representative data were shown.
In vivo experiments
For the in vivo studies, 6-week-old, female BALB/c from Jackson laboratory were used. All the procedures involving animal care and use were approved by IACUC of Rutgers University. 100,000 Wild-type or Crk KO cells were injected in the mammary fat pad of each mice. The tumors were palpated every 3 days, and body weight and tumor volumes were measured. At the end of the 6 weeks, the mice were sacrificed and tumors and lungs were harvested for immune phenotyping, IHC, western blotting or RNA extraction. Anti-PD1 or isotype antibodies were administered (i.p.) every 3 days at 200 mg/kg/day dosage in the combination experiments with total 4 administrations per group/study.
NanoString immunoprofiling analysis
Total RNA was isolated from four primary tumors for each group (WT and Crk KO) using RNeasy Plus™ total RNA Isolation kit (QIAGEN). RNA samples were subjected to direct gene expression analysis by measure counts of mRNA/per sample using murine nCounter® PanCancer Immune Profiling Panel. Multiplex assay consisting of 770 murine inflammatory response genes were analyzed using nSOLVER™ Analysis software 3.0 by the methods described previously.Citation16
Immunohistochemistry
Immunohistochemistry was performed on a Bond Rx autostainer (Leica Biosystems). Primary tumor sections were stained for anti-CD3 (Abcam16669, 1:100), FoxP3 (Novus NB100-39002, 1:1000), PD-L1 (Proteintech 17952-1-AP, 1:200), F4/80 (eBioscience 14–4801, 1:200), PD-1 (Abcam ab52587, 1:100), CD31 (Abcam ab28364, 1:100), Ly-6G (Abcam ab2557, 1:300), Granzyme B (Bond TM Ready-To-Use, Leica Biosystems PA 029), Ki67 (Abcam ab15580, 2 μg/ml). All rabbit primaries were detected using anti Rabbit-Polymer- HRP followed by DAB. Biotinylated anti-CD8 (Ebio 130808, 1:200) was detected by Streptavidin-HRP (Leica Biosystems) and followed by DAB. All the sections were then counterstained with hematoxylin, dehydrated and film coverslipped using a TissueTek-Prisma and Coverslipper (Sakura). Whole slide scanning (40x) was performed on an Aperio AT2 (Leica Biosystems). At least 3 mice/group/antibody were used for the analyses and a minimum 5 × 105 cells were analyzed per specimen. Results were represented as percentage positively staining and strongly positively staining live cells. Error bars represent +/-SD. P < 0.001.
Cancer Inflammation & Immunity Crosstalk RT2 Profiler PCR Array
Total RNA was isolated from WT and Crk KO primary tumors (3 tumors/group) using the RNeasy Plus™ total RNA Isolation kit. cDNA was prepared from respective samples using QIAGEN RT2 First Strand Kit that were subsequently used to perform gene expression analysis using the SABiosciences Mouse Cancer Inflammation & Immunity Crosstalk RT2 Profiler PCR Array (Cat. PAMM-181Z) and RT2 SYBR® Green qPCR mastermix on Bio-Rad CFX-96® per manufacturer's instructions. The data were analyzed using the RT2 Profiler PCR array data analysis software (SABiosciences) and normalized to GAPDH, β-actin and β2-microglobulin expression levels.
Cell culture, flow cytometry and MTT assay
4T1 cell line was purchased from Perkin-Elmer and maintained at 70% confluence in RPMI-1640 medium supplemented with 10% FBS. Cell were harvested by Accutase and stained with anti-human PD-L1-PE (Biolegend). BD FACS Calibur was used for data acquisition, and the resulting data was analyzed by FlowJo Software (TreeStar). MTT assay to test the cell proliferation of WT or Crk KO cells was performed using MTT cell growth assay kit purchased from EMD-Millipore using manufacturer's protocol.
Protein isolation and immunoblotting
Whole cell lysates were prepared in HNTG buffer (HEPES, NaCl, Triton X-100, Glycine) supplemented with protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail (Cell Signaling Technology, CST). Cell were scraped, incubated on ice for 10 min and centrifuged at 12,000 rpm for 10 min. Cleared lysates were isolated, mixed with SDS containing Laemmli buffer boiled for 5 min and resolved by SDS-PAGE. Immunoblotting was performed using monoclonal Crk (Abcam), PD-L1 (CST), Vimentin (CST), β-catenin (CST), Zeb-1 (CST), β-actin (CST) antibodies.
Statistical analysis
Differences between groups in all in vivo experiments were examined for statistical significance using a two-tailed Student's t-test. P < 0.05 was considered significant.
Results
Crk adaptor protein promotes tumor growth and metastatic colonization
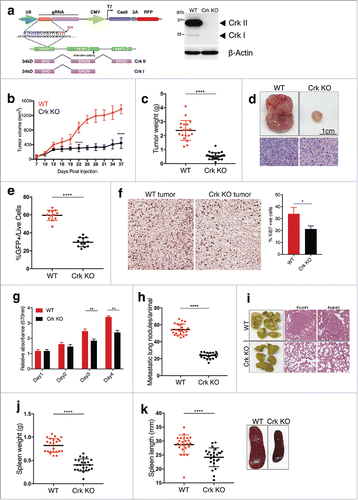
Previously, xenograft experiments transplanting Crk knockdown human cells into non-syngeneic immune-compromised nude mice demonstrated an intrinsic role for Crk in breast cancer growth and metastasis.Citation17 However, to investigate the role of Crk on tumor growth, metastasis, and immune evasion in a syngeneic model, Crk KO 4T1 cells were generated using CRISPR-Cas9 and validated Crk KO at genetic and protein level (). Subsequently, when WT or Crk KO 4T1 cells were injected into the mammary fat pad of immunocompetent virgin BALB/c mice, transplantation of Crk knockout cells led to ∼ 80% decrease in tumor growth () and tumor weights (). Using FACS, GFP+ tumor cells in tumors were also found to be suppressed by more than 20% in Crk KO tumors group, indicating decreased in vivo tumor cell proliferation (). To further test differences in cell proliferation of tumor cells from both groups, we performed Ki67 staining and in vitro MTT assay. We found that Crk KO decreases tumor cell proliferation both in vivo () and in vitro (). Additionally, metastasis of 4T1 KO cells to lung was reduced to about 70% as compared to the WT cells (). While all metastatic nodules in WT tumor-bearing mice were macro-metastatic (>1 mm), most metastatic nodules found in lungs of Crk KO tumor bearing mice were micro-metastatic (<0.2mm). Tumor growth and metastasis in 4T1/BALB/c model was also associated with significant splenomegaly, consistent with previous reports.Citation18 Consistent with overall decreased lung metastatic burden, Crk KO tumor-bearing mice showed concomitant decrease in splenomegaly KO (). Together, these results identify an important role for Crk, noted to be frequently overexpressed in human breast cancers, in controlling tumor growth and metastatic colonization in a triple negative breast model of adenocarcinoma.
Figure 1. Crk promotes tumor growth and metastatic colonization: (a) Western blot analysis of Crk gene knockout in 4T1-luc2-GFP cells by CRISPR-Cas9 vector targeting exon 1 of murine Crk gene. (b) Tumor volume plot indicating in vivo tumor growth of 1.0 × 105 WT or Crk KO 4T1 cells when injected in mammary fat pad of mice; n = 24/group. (c) Tumor weight quantification between both groups; n >18/group. (d) Representative images (top) and H&E staining (bottom) of primary WT or Crk KO tumors shown. (e) Quantification of GFP+ live cells in each tumor type. (f) Representative Ki67 staining images from WT and Crk KO tumors (left) and quantification of % Ki67 positive cells from three individual images from 2 mice tumors (right). (g) MTT cell growth assay to test the differences in cell growth between WT and Crk KO 4T1 cells. (h) Quantification of lung metastatic nodules/mice by 1.0 × 105 WT or Crk KO 4T1 cells; n >18/group. (i) Representative images (left) and H&E staining (right) of lungs from WT or Crk KO tumors bearing mice shown. Assessment of spleen weight (j) and spleen length (k) in WT or Crk KO tumor bearing mice; n >18/group. Representative images of spleen from WT or Crk KO tumor-bearing mice. Error bars, S.D.; all P values are based on one-sided Student's t-tests. * P < 0.01, ** P < 0.001, **** P < 0.0001. Scale bar: 1 cm.
Genetic ablation of Crk increases tumor infiltration in murine orthotopic breast cancer model
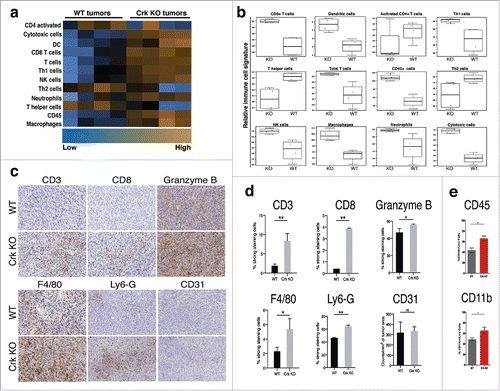
The above-mentioned decrease in splenomegaly in the spontaneous syngeneic tumor model suggested that Crk KO may regulate a tumor-specific host immune response. To investigate this experimentally, we analyzed the molecular landscape of tumor infiltrating immune cells by Nanostring analysis using the whole RNA extracted from the primary tumors. Interestingly, in the Crk KO tumors, we observed that CD8+, and total T cells, DC, NK cells, neutrophils and macrophages were elevated, while CD4+ activated T cells and T helper cells were concomitantly down-regulated (). Additionally, Crk KO tumors showed increased gene expression status of genes frequently associated with dendritic cell functions such as CD86, CD40, CD40lγ, and CCL5 (Suppl. Fig. 2). Cumulatively, in Crk KO bearing tumor associated tumor suppressive Th1 cells populations were increased while tumor-promoting cell populations were decreased by Crk KO in tumor cells. Using IHC analysis of formalin-fixed, paraffin-embedded (FFPE) sections of primary tumors as an unbiased alternative strategy, we validated major findings from Nanostring based RNA analysis, whereby a decrease in tumor suppressive populations of T cells (CD3+, CD8+ T cells) () and a marked increase in GranzB+ immune cell populations were noted in Crk KO tumors. F4/80+ macrophages and Ly6G+ neutrophils populations were also upregulated in tumors from Crk knockout 4T1 cells. While we did not detect any changes in CD31+ endothelial cells (tumor angiogenesis) in Crk KO tumors, there was a significant reduction in vascularity of the tumors by visual inspections (not shown). Finally, by flow cytometric analysis of the primary tumors, we found that CD45+ myeloid cell and CD11b+ macrophage populations were potentiated in KO group (). Together, these results suggest that Crk expression in 4T1-derived primary murine breast adenocarcinomas contributes to a poorly immunogenic tumor microenvironment that, in part, leads to reduced host-anti-tumor immune responsivity and suppressed tumor cell killing of Crk-expressing tumors.
Figure 2. Crk suppresses tumor immune infiltration: (a) Heat map analysis of immune cell population by nCounter® PanCancer Immune Profiling Panel in WT and KO tumors (n = 4/group). (b) Relative RNA expression-based signatures of tumor suppressive immune cell populations in WT and KO tumors (n = 4/group). (c-e) Immuno-phenotyping of the WT and Crk KO tumors: (c) Immunohistochemical analysis of CD3+, CD8+, GranzB+ T cells, F4/80+ macrophages, Ly6-G+ neutrophils and CD31+ endothelial cells. (d) Quantitative analysis of IHC slides of tumors from WT and KO groups by Aperio AT2 Digital Whole Slide Scanning system. At least 500,000 cells were counted for analysis. (n = 3/group). (e) Flow cytometric analysis of CD45+ and CD11b+ immune cell pollutions in WT and Crk KO tumors (n = 8/group). Error bars, S.D.; all P values are based on one-sided Student's t-tests. * P < 0.05, ** P < 0.001.
Crk knockout alters intra-tumoral cytokine and chemokine profiles in favor of anti-tumor immunity
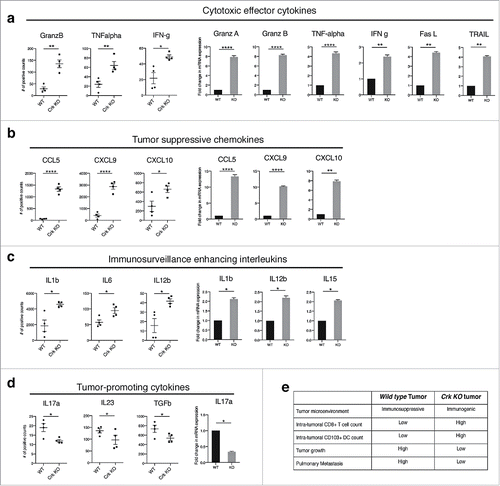
Tumor associated inflammation, associated with specific regulation of cytokine and chemokine profiles in the tumor microenvironment (TME), are predictive features of anti-tumor immune responses.Citation19,Citation20 To better access functional outcomes associated with the immune infiltrated cell types (), we next investigated the cytokine profiles using total RNA from whole primary WT and Crk KO tumors (analyzed by Nanostring and shown as dot plots) and by RT2 PCR profiling using cDNA prepared from replicate total RNA (shown as bar graphs). Consistent with overall immune subsets noted above, we observed that immunoregulatory and cytotoxic cytokines, granzyme A and B, TNF-α, IFN-γ, Fas-L, and TRAIL, that are implicated to enhance tumor antigen presentation, cytotoxicity, and induce tumor cell apoptosis were markedly upregulated in Crk KO tumor microenvironment as compared with native WT tumors (). Moreover, tumors from Crk KO 4T1 cells showed increased expression of CCL5, CXCL9 and CXCL10, chemokines associated with tumor infiltrating lymphocytes (), as well as subsets of interleukins (IL1β, ΙL6, ΙL12β, αnd ΙL15) that enhance in T-cell activation, B-cell function, TH1 immunity and tumor cell cytotoxicity (). In contrast, tumor-promoting cytokines, IL17a, IL23 and TGF-β were downregulated in Crk KO tumors (). Cumulatively, these results suggest that Crk KO, in a poorly immunogenic 4T1 model, reprograms a non-inflamed immunosuppressive cytokine microenvironment into an inflamed immunogenic cytokine profile characterized by infiltration of effector T-cells (summarized in ).
Figure 3. Crk knockout generates anti-tumor cytokine profile in tumor micro-environment: Total RNA extracted from WT and Crk KO tumors were used to analyze cytokine and chemokine gene signatures by Nanostring analysis (showed in dot plots) and were further validated using the RT2 PCR profiler assay (shown in bar graphs). Cytotoxic effector cytokines (a), tumor suppressive chemokines (b), immunosurveillance enhancing interleukins (c), and tumor promoting cytokines (d) were estimated from each tumor type. (n = 4/group). (e) Tabular summary of role of Crk gene knockout by CRISPR-Cas9 indicating a switch from immunosuppressive to immunogenic tumor microenvironment and increase tumor infiltrating CD8+ T cells that DC leading to tumor regression and metastatic suppression. Error bars, S.D.; all P values are based on one-sided Student's t-tests. * P < 0.05, ** P < 0.001, **** P < 0.0001.
Crk promotes Zeb-1 mediated EMT and PD-L1 expression
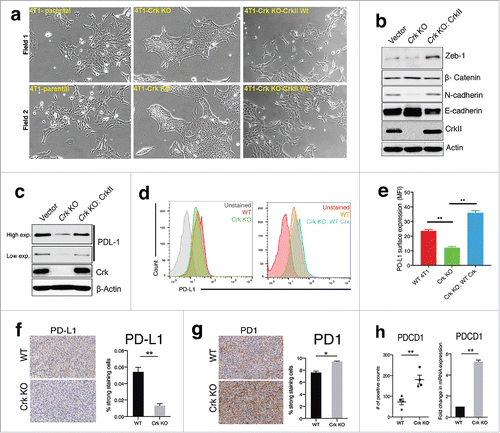
To better understand the biochemical events of Crk in the tumor microenvironment, we examined intrinsic biomarkers for Crk associated with epithelial-mesenchymal transition (EMT). Indeed, in recent years, a relationship between EMT and tumor immune evasion has been identified, including models of bi-directional cross-talk between the transcription factor Zeb-1 and EMT, and the regulation of PD-L1 and tumor immune evasion.Citation21,Citation22 While previous studies have implicated Crk in the regulation of HGF/MET-mediated EMT,Citation14 a role of Crk per se in regulation of EMT and EMT-induced immune evasion mechanisms are not well understood. Consistent with reports of other cancer cell types, Crk KO in the 4T1 cells under investigation in this study showed a contrasting morphological change associated with a loss of mesenchymal-like spindle shaped cells with cell scattering morphology (). Notably, in the Crk KO cells, cell-to cell adhesion increased whereby cells grew in sheet-like structures resembling epithelial morphology, while reconstitution of WT Crk II in Crk KO cells reverted this phenotype (). Interestingly, Western blot analysis of whole-cell detergent lysates from WT/KO cells showed that expression of N-cadherin was suppressed while E-cadherin was enhanced following Crk KO (), while reconstitution of CrkII reversed the N- and E-cadherin switching. Similarly, expression of Zeb-1 (a potent EMT-inducing transcription factor) was also suppressed by Crk KO and rescued by Crk reconstitution in Crk KO cells (). As EMT has been shown to modulate PD-L1 expression on cancer cells,Citation23,Citation24 we investigated the effect of Crk ablation (that suppresses EMT) on PD-L1 expression. We found that PD-L1 (total and surface) expression were ablated by Crk KO and rescued by Crk II reconstitution (). Consistently, in Crk KO primary tumors expression of PD-L1 was downregulated (), while population of PD-1+ immune cells was upregulated (). as observed by IHC (n = 4), Finally, Nanostring and RT2 PCR profiler data showed that PD1 expression was upregulated in Crk KO tumors (n = 4) (). These results support a role for Crk in direct regulation of EMT by controlling Zeb-1 expression and in EMT induced PD-L1 expression on 4T1 breast cancer cells.
Figure 4. Crk promotes EMT and PD-L1 expression on tumor cells: (a) Representative micrographs depicting the morphological changes pertaining to epithelial or mesenchymal morphology in 4T1 cells (left) upon Crk gene knockout (middle) and reconstitution by WT Crk (right). (b) Assessment of changes in expression of EMT markers by Western blotting in WT or Crk KO 4T1 cells. (c) Western blotting assessment of PD-L1 expression by Crk KO and Crk re-expression in Crk KO 4T1 cells. (d-e) Determination of changes in surface expression of PD-L1 by Crk KO and re-expression by flow cytometry. Representative PD-L1 surface expression data shown (d), MFI quantification from three independent experiments (e). f-g. Immunohistochemical analysis of PD-L1+ tumor cells (f) and PD1+ T cells (g), in WT or Crk KO primary tumors. (h) Assessment of PD-1 expression in WT and Crk KO tumors by Nanostring (dot-plot) and RT2 PCR profiler (bar-graph) (n = 4). Error bars, S.D.; all P values are based on one-sided Student's t-tests. * P < 0.05, ** P < 0.001.
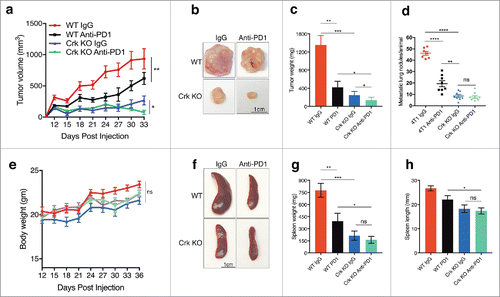
Crk knockout acts additively with anti-PD1 in suppression of primary tumor growth
To more formally implicate a role for EMT-inducible PD-L1 up-regulation, we experimentally tested WT and Crk KO cohorts (n = 8) in synergy with anti-PD1 checkpoint therapeutics. As shown in , mice were administered either 200μg/mice (i.p.) isotype control or anti-PD1 twice a week for 4 times after palpable tumors were formed (7 days post injection). Under these conditions, both anti-PD1 and Crk KO independently and significantly decreased tumor growth and weight, while Crk KO cohort administered together with anti-PD1 showed an additive effect on decrease in tumor growth or tumor weight, due to the robust effects of Crk ablation on this phenotype (). Likewise, both anti-PD1 monotherapy and Crk KO also decreased lung metastasis, spleen weight and size (, ). No significant change in body weight was observed in mice cohorts treated with isotype or anti-PD1 (). Together, the results suggest that Crk KO increases tumor-suppressive immune cell populations, at least in part, by regulation of the PD-L1 and PD-1 axis.
Figure 5. Crk knockout acts additively with anti-PD1 therapeutics in suppression of primary tumor weight: (a) WT or Crk KO 4T1 cells derived tumor growth in mice injected with either anti-PD1 antibody or isotype control. (b) Representative tumors from WT or Crk KO groups administered with IgG (isotype) or anti-PD1. (c-d) Primary tumor weight (c) and metastatic lung tumors quantification (d) analyses among the four groups. (e) Body weight analysis of WT or Crk KO tumor bearing mice upon administration of isotype or anti-PD1. f. Representative spleens from WT or Crk KO groups administered with IgG (isotype) or anti-PD1. Spleen weight (g) and length (h) analysis from the 4 groups. (n = 8/group). Error bars, S.D.; all P values are based on one-sided Student's t-tests. * P < 0.05, ** P < 0.001, **** P < 0.0001.
Discussion
Over the past decade, immune-oncology and immunotherapy have emerged into frontline cancer therapeutics with clear demonstrated clinical benefit in some cancer patients. Unlike classical radiotherapy, chemotherapy, and oncogene-directed inhibitors that target the growth and survival of tumor cells, immune-therapies typically target negative regulatory immune checkpoint molecules that break tolerance, and when successful, can promote immune mediated tumor destruction by T cell effector functions or NK cells and enhance disease-free survival in multiple cancers.Citation25,Citation26 However, by reasons that are not completely understood, immune checkpoint inhibitors (ICI) in some cases can also have poor experimental and clinical efficacy, due in part to low mutational burden of neo-antigens in certain cancer types, as well as low intrinsic immunogenicity in some cancers (so-called cold tumors) via the expression of checkpoint inhibitory ligands that include PD-L1.
Concerning the notion that certain intrinsic oncogenic networks might contribute to suppression of tumor infiltrating immune cell population outcomes in the tumor microenvironment, several oncogenic pathways that can crosstalk and impinge on expression of checkpoint inhibitors, such as PD-L1, have been recently uncovered. For example, Myc, AKT-mTOR Ras-MAPK, HIF-1α, c-MET, and EGFR signaling have been shown to regulate PD-L1 gene expression,Citation27,Citation28 as well as surface expression on tumor cells.Citation21,Citation22,Citation29-33 In breast cancer, including models of basal type and triple negative, PD-L1 expression has a poor prognostic value with respect to therapeutic outcomes.Citation34 Therapeutic interventions that consider oncogenic pathways that regulate PD-L1 are predicted to synergize with ICI-based therapeutics.Citation35
In the present study, by developing a syngeneic mouse model to study Crk tumorigenicity in a poorly immunogenic 4T1 triple-negative breast cancer model, we identified an unrealized role for Crk in regulating an immuno-suppressive TME that inhibits host anti-tumor immunity. Previously, Crk has been intensely studied in the context of cancer cell transformation and shown to be an important regulator of proliferation, cell migration, and invasive phenotypes, and in human cancers expression correlates with metastasis and poor survival outcomes.Citation8 Mechanistically, Crk binds to tyrosine phosphorylated focal adhesion proteins, i.e., paxillin and p130cas, and complexes of Crk and focal adhesion proteins (FAK and p130cas) are elevated in oncogenically transformed cells by Src and Ras.Citation1 The implication that Crk may impinge on the activation of focal adhesion kinase (FAK) and downstream signaling is potentially noteworthy, given the recent evidence that FAK (activation) also appears to promote an immunosuppressive TME in pancreatic ductal adenocarcinoma (PDAC).Citation15 Consequently, FAK inhibitors are shown to enhance the immune-surveillance and increase the efficacy of anti-PD1 immunotherapy in PDAC. The fact that both Crk and Crk-interacting proteins are also part of FAK signaling complexes and participate in cell migration and invasion suggests that Crk may function in parallel pathways to FAK in promoting immunosuppressive signals. However, in addition to FAK, recent reports indicate a role of oncogenes, such as c-Myc, Ras-MAPK, and c-MET in regulation of tumor immunity in different human cancer models,Citation32,Citation33,Citation36 suggesting that common “oncogenic hubs” may control extrinsic immunogenic components of the TME. By extension, these insights might also imply that Crk by acting as an adaptor protein, via its assemblages of multi-protein complexes, could act coordinately to control both cell intrinsic malignant features of tumor cells, such as EMT and invasion, as well as cell extrinsic tumor microenvironment features that impinge on host anti-tumor responses.
Increasing evidence also suggests that aberrant activation of EMT, a hallmark for the development and progression of tumors, can also manifest evasion in anti-tumor immunity.Citation21,23,24,37 While the link between EMT and the immunosuppressed TME is likely to be multi-factorial that may involve down-regulation of MHC class I and other recognition modules utilized by TILs and NK cells, recent evidence also suggests that epithelial versus mesenchymal cancer subtypes have distinct interactions with the TME. Moreover, in global mRNA expression data from TCGA and PROSPECT datasets, mesenchymal type tumors have elevated PD-L1 and PD-L2 that may associate with poorly immunogenic tumors. With respect to Crk and Crk knockout 4T1 cells under investigation in this study, Crk KO cells clearly exhibit more epithelial marker E-cadherin and decreased expression of mesenchymal markers N-cadherin and Zeb1. The fact that Crk KO concomitantly suppressed EMT markers and PD-L1 expression in the poorly immunogenic 4T1 model, is consistent with the idea that Crk-mediated alterations in EMT status may be linked to its capacity to regulate the immunosuppressive TME (summarized in ). This is also substantiated by our finding that that tumors harboring Crk knockout 4T1 cells show increased CD3+ and CD8+ T cells, cytokines associated with infiltrating T cells and their activation status such as IFN-γ and interleukins, as well as cytotoxic genes that include granzyme B and TRAIL. Finally, the observations that Crk KO tumors may act additively with anti-PD1 immunotherapy with respect to tumor growth and metastasis outcomes opens the possibility that the development of Crk inhibitors and siRNA strategies may increase the efficacy of anti-PD-1 immunotherapy in breast cancer. Agents, such as EphB4 receptor inhibitorsCitation38 and Cyclophilin A inhibitors,Citation39 that target the Crk signaling axis, might be considered as promising therapeutic target, possibly akin to the development of FAK inhibitors mentioned above.Citation15
While the aforementioned discussion has exclusively focused on Crk in cancer cells, it is important to note that Crk, and its related gene product CrkL, are also expressed in infiltrating T cells (including TILs) and myeloid-derived NK cells, where they are likely to be affirmatively involved in host tumor immunity. While in T cells, Crk plays a role in T cell extravasation and motility, it is also enhances activation of NK cells.Citation40,Citation41 Such studies suggest Crk may have a dual role, first on tumor cells as signals for EMT and immune escape, and second on immune cells that control anti-tumor immune functions. The fact that we tested Crk knockout activity in an orthotopic tumor model clearly supports a tumor-specific role of Crk in controlling the immunosuppressive TME; however further studies, using conditional KO's of Crk in T- cells and NK cells will be of equal importance to assess the role of lymphocytic and monocyte-specific functions of Crk in syngeneic mouse models. In summary, while previous studies have shown that Crk can drive cell intrinsic cancer phenotypic changes, such as increased invasion and EMT that are associated with malignant transformation and metastasis, our present results indicate that Crk has an extrinsic immunosuppressive role in poorly immunogenic tumors; thus, implicating Crk as the first reported adaptor protein that promotes immune evasion in cancer.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed by the authors.
Authors' contributions
Sushil Kumar designed, performed and analyzed experiments. Viralkumar Davra, Alison Obr and Ke Geng helped in mice experiments and analyzed results. Teresa L. Wood helped analyze the data and reviewed the manuscript. Sushil Kumar and Raymond B. Birge conceived and coordinated the study, designed experiments and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank members of the Birge lab for helpful discussion and Stanley Kimani for providing technical assistance. This work was supported in part by NIH R01 CA165077 to RBB, NIH R01 CA204312 to TLW and NJCCR postdoctoral fellowship (#DFHS15PPC039) to AEO. We acknowledge Rutgers Society of Research Scholars award to SK.
References
- Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. https://doi.org/10.1186/1478-811X-7-13. PMID:19426560
- Akagi T, Shishido T, Murata K, Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci U S A. 2000;97:7290-5. https://doi.org/10.1073/pnas.140210297. PMID:10852971
- Birge RB, Fajardo JE, Mayer BJ, Hanafusa H. Tyrosine-phosphorylated epidermal growth factor receptor and cellular p130 provide high affinity binding substrates to analyze Crk-phosphotyrosine-dependent interactions in vitro. J Biol Chem. 1992;267:10588-95. PMID:1375224
- Birge RB, Fajardo JE, Reichman C, Shoelson SE, Songyang Z, Cantley LC, Hanafusa H. Identification and characterization of a high-affinity interaction between v-Crk and tyrosine-phosphorylated paxillin in CT10-transformed fibroblasts. Mol Cell Biol. 1993;13:4648-56. https://doi.org/10.1128/MCB.13.8.4648. PMID:7687742
- Fajardo JE, Birge RB, Hanafusa H. A 31-amino-acid N-terminal extension regulates c-Crk binding to tyrosine-phosphorylated proteins. Mol Cell Biol. 1993;13:7295-302. https://doi.org/10.1128/MCB.13.12.7295. PMID:7504172
- Feller SM, Knudsen B, Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 1994;13:2341-51. PMID:8194526
- Feller SM, Knudsen B, Hanafusa H. Cellular proteins binding to the first Src homology 3 (SH3) domain of the proto-oncogene product c-Crk indicate Crk-specific signaling pathways. Oncogene. 1995;10:1465-73. PMID:7731701
- Kumar S, Fajardo JE, Birge RB, Sriram G. Crk at the quarter century mark: perspectives in signaling and cancer. J Cell Biochem 2014;115:819-25. https://doi.org/10.1002/jcb.24749. PMID:24356912
- Akakura S, Kar B, Singh S, Cho L, Tibrewal N, Sanokawa-Akakura R, Reichman C, Ravichandran KS, Birge RB. C-terminal SH3 domain of CrkII regulates the assembly and function of the DOCK180/ELMO Rac-GEF. J Cell Physiol. 2005;204:344-51. https://doi.org/10.1002/jcp.20288. PMID:15700267
- Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol. 2000;2:899-905. https://doi.org/10.1038/35046549. PMID:11146654
- Antoku S, Saksela K, Rivera GM, Mayer BJ. A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. J Cell Sci. 2008;121:3071-82. https://doi.org/10.1242/jcs.031575. PMID:18768933
- Tsuda M, Tanaka S. Roles for crk in cancer metastasis and invasion. Genes Cancer. 2012;3:334-40. https://doi.org/10.1177/1947601912458687. PMID:23226571
- Elmansuri AZ, Tanino MA, Mahabir R, Wang L, Kimura T, Nishihara H, Kinoshita I, Dosaka-Akita H, Tsuda M, Tanaka S. Novel signaling collaboration between TGF-beta and adaptor protein Crk facilitates EMT in human lung cancer. Oncotarget 2016;7:27094-107. https://doi.org/10.18632/oncotarget.8314. PMID:27027347
- Lamorte L, Royal I, Naujokas M, Park M. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell. 2002;13:1449-61. https://doi.org/10.1091/mbc.01-10-0477. PMID:12006644
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851-60. https://doi.org/10.1038/nm.4123. PMID:27376576
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317-25. https://doi.org/10.1038/nbt1385. PMID:18278033
- Fathers KE, Bell ES, Rajadurai CV, Cory S, Zhao H, Mourskaia A, Zuo D, Madore J, Monast A, Mes-Masson AM, et al. Crk adaptor proteins act as key signaling integrators for breast tumorigenesis. Breast Cancer Res. 2012;14:R74. https://doi.org/10.1186/bcr3183. PMID:22569336
- DuPre SA, Hunter KW Jr. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp Mol Pathol. 2007;82:12-24. https://doi.org/10.1016/j.yexmp.2006.06.007. PMID:16919266
- Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11-22. https://doi.org/10.1038/nrc1252. PMID:14708024
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99. https://doi.org/10.1016/j.cell.2010.01.025. PMID:20303878
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. https://doi.org/10.1038/ncomms6241. PMID:25348003
- Alsuliman A, Colak D, Al-Harazi O, Fitwi H, Tulbah A, Al-Tweigeri T, Al-Alwan M, Ghebeh H. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol Cancer. 2015;14:149. https://doi.org/10.1186/s12943-015-0421-2. PMID:26245467
- Ock CY, Kim S, Keam B, Kim M, Kim TM, Kim JH, Jeon YK, Lee JS, Kwon SK, Hah JH, et al. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget. 2016;7:15901-14. https://doi.org/10.18632/oncotarget.7431. PMID:26893364
- Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, Mami-Chouaib F, Thiery JP, Chouaib S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology. 2017;6:e1263412. https://doi.org/10.1080/2162402X.2016.1263412. PMID:28197390
- Couzin-Frankel J. Cancer Immunotherapy. Science. 2013;342:1432-3. https://doi.org/10.1126/science.342.6165.1432. PMID:24357284
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64. https://doi.org/10.1038/nrc3239. PMID:22437870
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-34. https://doi.org/10.1084/jem.192.7.1027. PMID:11015443
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8:793-800. https://doi.org/10.1038/nm0902-1039c. PMID:12091876
- Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781-90. https://doi.org/10.1084/jem.20131916. PMID:24778419
- Balan M, Mier y, Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G, Pal F. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015;290:8110-20. https://doi.org/10.1074/jbc.M114.612689. PMID:25645920
- Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, et al. Upregulation of PD-L1 by egfr activation mediates the immune escape in egfr-driven NSCLC: Implication for optional immune targeted therapy for NSCLC patients with egfr mutation. J Thorac Oncol 2015;10:910-23. https://doi.org/10.1097/JTO.0000000000000500. PMID:25658629
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227-31. https://doi.org/10.1126/science.aac9935. PMID:26966191
- Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, Combs S, Rimm DL, Giltnane JM, Estrada MV, et al. RAS/MAPK Activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499-509. https://doi.org/10.1158/1078-0432.CCR-15-1125. PMID:26515496
- Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, Viens P, Caldas C, Birnbaum D, Bertucci F, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget. 2015;6:5449-64. https://doi.org/10.18632/oncotarget.3216. PMID:25669979
- Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237-51. https://doi.org/10.1038/nrc3237. PMID:22437869
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell. 2015;162:1271-85. https://doi.org/10.1016/j.cell.2015.07.061. PMID:26359985
- Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, Byers LA, Wang J, Papadimitrakopoulou VA, Behrens C, et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res. 2016;22:3630-42. https://doi.org/10.1158/1078-0432.CCR-15-1434. PMID:26851185
- Noren NK, Foos G, Hauser CA, Pasquale EB. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat Cell Biol. 2006;8:815-25. https://doi.org/10.1038/ncb1438. PMID:16862147
- Saleh T, Jankowski W, Sriram G, Rossi P, Shah S, Lee KB, Cruz LA, Rodriguez AJ, Birge RB, Kalodimos CG, et al. Cyclophilin A promotes cell migration via the Abl-Crk signaling pathway. Nat Chem Biol. 2016;12:117-23. https://doi.org/10.1038/nchembio.1981. PMID:26656091
- Liu D, Peterson ME, Long EO. The adaptor protein Crk controls activation and inhibition of natural killer cells. Immunity. 2012;36:600-11. https://doi.org/10.1016/j.immuni.2012.03.007. PMID:22464172
- Huang Y, Clarke F, Karimi M, Roy NH, Williamson EK, Okumura M, Mochizuki K, Chen EJ, Park TJ, Debes GF, et al. CRK proteins selectively regulate T cell migration into inflamed tissues. J Clin Invest. 2015;125:1019-32. https://doi.org/10.1172/JCI77278. PMID:25621495