
ABSTRACT
Anti-PD-1 and anti-PD-L1 immunotherapy has provided a new therapeutic opportunity for treatment of advanced-stage non-small cell lung cancer (NSCLC). However, overall objective response rates are approximately 15%–25% in all NSCLC patients who receive anti-PD therapy. Therefore, strategies to overcome primary resistance to anti-PD immunotherapy are urgently needed. We hypothesized that the barrier to the success of anti-PD therapy in most NSCLC patients can be overcome by stimulating the lymphocyte infiltration at cancer sites through locoregional virotherapy. To this end, in this study, we determined combination effects of anti-PD immunotherapy and oncolytic adenoviral vector-mediated tumor necrosis factor-α-related apoptosis-inducing ligand (TRAIL) gene therapy (Ad/E1-TRAIL) or adenoviral-mediated TP53 (Ad/CMV-TP53) gene therapy in syngeneic mice bearing subcutaneous tumors derived from M109 lung cancer cells. Both anti–PD-1 and anti–PD-L1 antibodies failed to elicit obvious therapeutic effects in the M109 tumors. Intratumoral administration of Ad/E1-TRAIL or Ad/CMV-TP53 alone suppressed tumor growth in animals preexposed to an adenovector and bearing subcutaneous tumors derived from M109 cells. However, combining either anti–PD-1 or anti–PD-L1 antibody with these two adenoviral vectors elicited the strongest anticancer activity in mice with existing immunity to adenoviral vectors. Dramatically enhanced intratumoral immune response was detected in this group of combination therapy based on infiltrations of CD4+ and CD8+ lymphocytes and macrophages in tumors. Our results demonstrate that resistance to anti–PD-1 immunotherapy in syngeneic mouse lung cancer can be overcome by locoregional virotherapy.
Introduction
Lung cancer is the leading cause of cancer death in men and women, causing approximately 155,870 annual deaths in the United States and 1.6 million annual deaths worldwide.Citation1,2 The overall survival rate in patients with lung cancer has improved only slightly over the past 4 decades despite the use of many new therapeutic modalities.Citation3–5 Recent success in immunotherapy with immune checkpoint inhibitors has provided new opportunities for the treatment of this deadly disease. The high mutation rates observed in lung cancerCitation6 make this disease particularly vulnerable to anti-PD immunotherapy,Citation7 which dramatically reduced risk of death in some patients with advanced non–small cell lung cancer (NSCLC).Citation8,9 Nevertheless, despite the promising results observed in various clinical trials with anti–PD-1Citation9–12 and anti–PD-L1Citation13–15 antibodies, the overall objective response rates in NSCLC patients who received anti-PD immunotherapy were approximately 15%–25%.Citation8–13,15 Therefore, strategies to overcome primary resistance to immune checkpoint inhibitors are urgently needed so that most lung cancer patients can benefit from anti-PD therapy.
It has been demonstrated that the success of anti-PD therapy requires preexisting CD8+ T cells at cancer sites and PD-L1+ tumor microenvironments that mediate evasion of immune surveillance.Citation13,16,17 Efficacy of anti-PD therapy also correlates with a high mutation burden and high neoantigen burden in tumorsCitation7 that can trigger lymphocyte infiltration at tumor sites. PD-L1 expression is absent in normal tissues but can be induced by some cancer driversCitation18,19 or by interferon-γ (IFNγ),Citation20,21 a cytokine produced mainly by inflammatory cells, especially T cells.Citation22,23 Tumor antigens trigger recruitment of tumor-infiltrating lymphocytes (TILs). IFNγ released from TILs enhances TIL effector functions by stimulating TIL proliferation, differentiation, and antigen processing but paradoxically induces TIL apoptosis by triggering PD-L1 expression in the tumor microenvironment, which leads to a negative feedback mechanism that suppresses antitumor immunity.Citation22–24 Although blocking PD-L1/PD-1 interaction abrogates the negative feedback and dramatically enhances TILs' anticancer activity at cancer sites, absence of TILs at cancer sites, which has been observed in a large fraction of cancer patients,Citation24,25 imposes a major barrier for anti-PD therapy.Citation13,26 Thus, strategies that promote recruitment of lymphocytes to tumor sites are needed to overcome resistance to anti-PD therapy.
Inflammatory signals strongly promote activated T cell homing to sites of infection.Citation27,28 In the presence of microbial infection and foreign gene expression, even very weak T-cell receptor–ligand interactions are sufficient to activate T cells, inducing rapid proliferation and generation of effector and memory cells.Citation29 Indeed, our previous study demonstrated that lymphocyte infiltration at tumor sites can be induced dramatically by expression of foreign antigens through locoregional adenovirus- or vaccinia virus–mediated gene transfer.Citation30 A recent study in a melanoma tumor model also demonstrated that intratumoral injection of Newcastle disease virus induced inflammatory responses and anticancer activity, and dramatically enhanced the activity of anti–CTLA-4 antibody.Citation31 These results led us to hypothesize that resistance to anti-PD therapy caused by low or no immune cell infiltration might be overcome by inflammation-mediated stimulation of lymphocyte infiltration at cancer sites. To test our hypothesis, we evaluated combination effects of anti-PD therapy with adenovirus-mediated gene therapy in a syngeneic lung cancer model. Our results demonstrated that resistance to anti–PD-1 immunotherapy in syngeneic mouse lung cancer can be overcome by locoregional virotherapies.
Results
Infectivity of murine lung cancer cell lines with human adenovirus
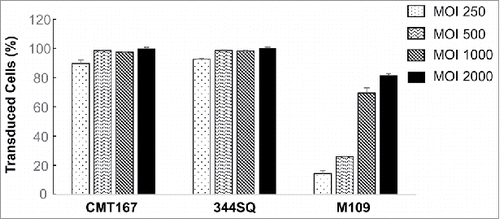
To determine the feasibility of combination therapy of locoregional virotherapy and immunotherapy with an immune checkpoint inhibitor, we first evaluated infectivity of human adenovirus in syngeneic mouse lung cancer cells. To this end, mouse lung cancer cell lines CMT167, 344SQ and M109 were infected with Ad/CMV-GFP at MOIs of 250 to 2000 viral particles (vp). Cells were harvested for quantification of GFP-positive cells by flow cytometric analysis at 48 h after the infection. The results showed that CMT167 and 344SQ cells had similar sensitivity to Ad/CMV-GFP expression (). About 90% of cells were GFP positive even at the lowest MOI tested. In contrast, M109 cells were relatively resistant to adenovirus infection. At the dose range of 250–2000 MOIs, GFP-positive cells increased from 14% to 82% in a dose-dependent manner. Both CMT167 and 344SQ cell lines have Kras mutations,Citation32,33 whereas the genotype of M109 cells is not available. Whether Kras mutations contribute to increased sensitivity to adenovirus infection is not clear.
Figure 1. Sensitivity of mouse lung cancer cell lines to adenoviral vector. Mouse lung cancer cells were treated with Ad/CMV-GFP at the indicated MOIs. Percentages of GFP-positive cells were determined at 48 h after the treatment. The data represent mean + SD of a triplicate assay.
Susceptibility of murine lung cancer cell lines to TRAIL-armed oncolytic adenovirus
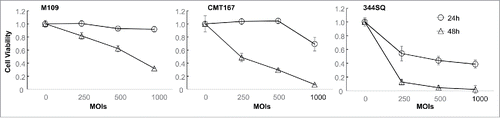
We have previously reported that TRAIL-armed oncolytic adenovirus (Ad/E1-TRAIL) has high oncolytic activity in human lung cancer cell lines in vitro and in vivo.Citation34 To test the cytotoxicity of Ad/E1-TRAIL in murine lung cancer cells, we infected CMT167, 344SQ and M109 cells with Ad/E1-TRAIL at MOIs ranging from 250 to 1000 vp. Mock-infected cells were used as the control. Cell viability was determined at 24 h and 48 h after infection. The results showed that Ad/E1-TRAIL induced cell viability changes in all three cell lines in dose-dependent and time-dependent manners (). Among the three cell lines tested, 344SQ was the most sensitive to Ad/E1-TRAIL–induced cytotoxicity, whereas M109 was the least sensitive.
Figure 2. Cytotoxic effect of Ad/E1-TRAIL in mouse lung cancer cell lines. Mouse lung cancer cells were treated with Ad/E1-TRAIL at the indicated MOIs. Cell viability was determined at 24 h and 48 h after treatment. Mock-infected cells were used as the control (indicated by the 0 in the MOIs), and their value was set as 1. The values represent mean ± SD of a quadruplet assay. The assay was repeated at least thrice with similar results.
Effects of combining Ad/TRAIL-E1 and immune checkpoint inhibitor anti–PD-L1 antibody
To determine combination effects of locoregional virotherapy and immunotherapy with immune checkpoint inhibitors, we performed a pilot experiment to determine in vivo tumor formation of CMT167, 344SQ and M109 cells in mice. For this purpose, we inoculated 1–3 × 106 cells of these three cell lines into their host strains of mouse (C57 BL/6, 129/Sv and BALB/c, respectively). We found that all three cell lines formed tumors within 1 week after tumor cell inoculation. Nevertheless, CMT167 and 344SQ had a tendency to form ulceration when tumors were still small (around the second week of tumor cell inoculation). Therefore, we used M109 cells in the subsequent in vivo studies.
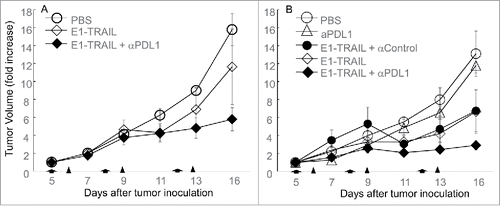
Because the majority of human patients have been exposed to adenovirus infection and have preexisting immunity against adenovirus,Citation35,36 we preexposed BALB/c mice to adenovirus through two sequential subcutaneous administrations of Ad/CMV-LacZ. A group of animals preexposed to PBS were used as the naïve control. Subcutaneous tumors were established by inoculating the mice with M109 cells (1 × 106). Treatment started when tumors reached 5 mm in diameter. Animals were randomized and treated with Ad/E1-TRAIL (1 × 1010 vp/intratumoral injection), anti–PD-L1 antibody (100 μg/mouse/injection, intraperitoneally), or both. Vehicle or Ad/E1-TRAIL plus a control antibody were used as controls. In naïve animals, treatment with Ad/E1-TRAIL resulted in mild but insignificant (P = 0.69) tumor growth inhibition when compared with PBS treated animals, whereas the combination of Ad/E1-TRAIL with anti–PD-L1 resulted in significant inhibition of tumor growth (P <0.001, at day 16) (). In the preexposed animals, treatment with anti–PD-L1 antibody also resulted in mild and insignificant tumor growth inhibition (P = 0.63) compared with vehicle controls (). Treatment with Ad/E1-TRAIL alone or in combination with a control antibody suppressed tumor growth at levels similar to those observed in naïve mice treated with Ad/E1-TRAIL plus anti–PD-L1 antibody (P < 0.05 at day 16 when compared with PBS treated mice). Nevertheless, the strongest anti-tumor activity was observed in the preexposed animals treated with Ad/E1-TRAIL plus anti–PD-L1 antibody (P < 0.05 when compared with all other groups). These results suggested that although anti–PD-L1 alone is ineffective for M109 tumors, it can significantly enhance Ad/E1-TRAIL-induced tumor suppression in M109 tumors and that animals with preexisting immunity against adenovirus benefitted more from this combination therapy than did adenovirus-naïve animals.
Figure 3. Effect of combining Ad/E1-TRAIL and anti–PD-L1 antibody. BALB/c mice were preexposed to PBS (naïve) (A) or Ad/CMV-LacZ (B). Subcutaneous tumors were then established from M109 lung cancer cells and were treated with Ad/E1-TRAIL (E1-TRAIL) and/or anti–PD-L1 antibody (αPDL1), or control antibody (αControl) as indicated. The mice were monitored for tumor growth. Tumor size (volume) for each mouse at the beginning of treatment was set to 1. Tumor suppression was significant in the Ad/E1-TRAIL + anti–PD-L1 treatment in preexposed mice (P < 0.05 at day 16 when compared with all other groups). Arrows and triangles indicate time of adenoviral and antibody treatment, respectively. n = 5 for all groups.
Lymphocyte and macrophage infiltrations in tumor tissues
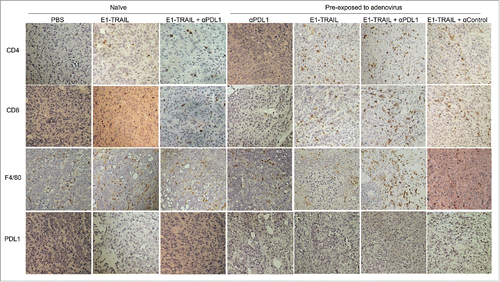
To determine effect of locoregional virotherapy and anti-PD-L1 therapy on lymphocyte and macrophage infiltrations in tumor tissue, we sacrificed all mice in the above-mentioned experiment at day 16, when the first mouse had to be sacrificed because of tumor burden. Tumor tissues from each group were harvested for immunohistochemical analysis on the infiltrations of T lymphocytes and macrophage and the expression of PD-L1. The result showed that PD-L1 expression in tumor tissues was barely detectable in all groups and was not affected noticeably by the different treatments. Macrophage infiltration in tumors was detected by anti-F4/80 antibody in all groups, but was dramatically increased in the animals pre-exposed to adenovirus and treated with Ad/E1-TRAIL + anti-PD-L1 antibody (). CD4+ or CD8+ lymphocytes were not detectable in tumors treated with PBS or anti-PD-L1 antibody, regardless whether animals were naïve or pre-exposed to adenovirus. Treatment with Ad/E1-TRAIL triggered both CD4+ and CD8+ lymphocyte infiltrations, which were more dramatic in animals pre-exposed to adenovirus than in naïve animals. This result is consist with our previous observation that immunized animals responded better to foreign antigen-induced immune response inside tumors.Citation30 Nevertheless, strongest CD4+ and CD8+ lymphocyte infiltrations were observed in animals pre-exposed to adenovirus and treated with Ad/E1-TRAIL plus anti–PD-L1 antibody (), consisting with the strongest anticancer activity observed in this group.
Figure 4. Lymphocyte and macrophage infiltrations in tumors. Formalin fixed paraffin embedded tumor samples were staining for CD4, CD8, F4/80 and PD-L1. Representative pictures of those markers were shown for each group. Strongest CD4, CD8 and F4/80 signals were detected in tumors of animals with pre-existing immunity against adenovirus and treated with Ad/E1-TRAIL plus anti-PD-L1 antibody.
Effects of combining Ad/CMV-TP53 and immune checkpoint inhibitor anti–PD-1 antibody
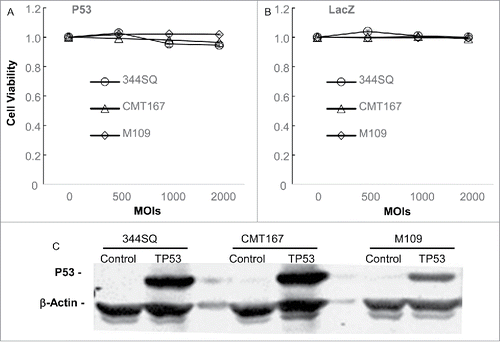
The promising results observed for the combination effects of Ad/E1-TRAIL with anti–PD-L1 antibody led us to test whether the combination therapy works for other adenoviral vectors and immune checkpoint inhibitors. To this end, we determined the combination effect of Ad/CMV-TP53 and anti–PD-1 antibody. We first determined whether the murine lung cancer cell lines were susceptible to Ad/CMV-TP53 that expresses the wild-type human TP53 gene. The cells were infected with Ad/CMV-TP53 or the control vector Ad/CMV-LacZ at MOIs of 500 to 2000 vp. The cell viability was determined at 72 h after the treatment. Cells treated with PBS were used as mock controls. The results showed that neither Ad/CMV-TP53 nor Ad/CMV-LacZ induced any observable cytotoxicity in these cell lines (,B). We then determined human TP53 expression in these cells after treatment. Cells were harvested at 48 h after treatment with adenoviral vectors, and cell lysates were analyzed for human p53 expression by Western blot analysis. The results showed that cells treated with Ad/CMV-TP53 but not the control vector induced substantial expression of human p53 protein (), suggesting the lack of cytotoxicity by Ad/CMV-TP53 was not caused by lack of target gene expression.
Figure 5. Susceptibility of mouse lung cancer cells to Ad/CMV-TP53. Mouse lung cancer cells were treated with Ad/CMV-TP53 (A) or Ad/CMV-LacZ (B) at the indicated MOIs. Cell viability was determined at 72 h after treatment. Mock-infected cells (indicated by the 0 for MOI) were used as controls, and their value was set as 1. The values represent mean ± SD of a triplicate assay. The assay was repeated at least thrice with similar results. (C) Expression of human p53 in mouse lung cancer cells 48 h after treatment with a control vector or Ad/CMV-TP53 at an MOI of 1000 vp. β-Actin was used as a loading control.
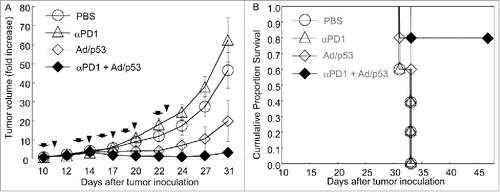
We then determined the combination effects of Ad/CMV-TP53 and anti-mouse PD-1 antibody in the M109 tumor models. BALB/c mice were preexposed to adenovirus as described above. Subcutaneous tumors were then established by inoculating the mice with M109 lung cancer cells (5 × 105). When tumors reached 5 mm in diameter, animals were randomized and treated with Ad/CMV-TP53 (1 × 1010 vp/mouse, intratumoral injection), anti–PD-1 antibody (100 μg/mouse, intraperitoneally), or both, twice a week for 5 treatments. Control animals were treated with vehicle. Tumor growth was monitored every 2–3 days and was normalized to the tumor volume at the beginning of treatment, which was set to 1. Tumors in mice treated with anti–PD-1 antibody grew faster than those in vehicle-treated mice, although the difference was not statistically significant (P = 0.32). Mice treated with Ad/CMV-TP53 had significantly less tumor growth than those treated with vehicle or anti–PD-1 antibody (P < 0.05). The combination group had the least tumor growth. The difference was significantly when compared with the tumor growth in the other three groups at day 24 and after (P <0.05). All mice treated with vehicle, anti–PD-1, or Ad/CMV-TP53 died by day 33, and the mean survival durations for those three groups were 32.2 ± 1.1, 32.2 ± 1.1 and 32.6 ± 0.9 days, respectively. In contrast, 80% of the mice treated with anti–PD-1 + Ad/CMV-TP53 were alive at day 46, the latest time point of observation. As assessed by log-rank test, the combination group had significant improvement in cumulative survival duration when compared with the other three groups (P ≤0.014) (). This result demonstrated that, although M109 tumors were resistant to anti–PD-1 antibody therapy, combination of this agent with adenovirus-mediated gene therapy resulted in profound anticancer activity in vivo.
Figure 6. Combination effect of Ad/CMV-TP53 and anti–PD-1 antibody. BALB/c mice were preexposed to Ad/CMV-LacZ. Subcutaneous tumors were then established from M109 lung cancer cells and were treated with single agents or combinations of Ad/CMV-TP53 (Ad/p53) and anti–PD-1 antibody (αPD1). The mice were monitored for tumor growth and survival. (A) Tumor growth. Tumor size (volume) for each mouse at the beginning of treatment was set to 1. Tumor growth was suppressed significantly in the combination group when compared with the other groups (P < 0.05 at day 22 and after). (B) Cumulative survival. The combination group had significantly improved survival when compared with the other three groups (P≤0.014). Arrows and triangles indicate the times of adenoviral and antibody treatment, respectively. n = 5 for all groups.
Discussion
The anti–PD-1 antibodies nivolumabCitation8,9 and pembrolizumabCitation12,37 and anti–PD-L1 antibody atezolizumabCitation15 have recently been approved for treatment of NSCLC by the United States Food and Drug Administration, providing a new opportunity for lung cancer patients to improve their clinical outcomes and/or quality of life. Nevertheless, the results from clinical trials have shown that only a limited number of lung cancer patients benefited from anti-PD therapy and most patients had primary resistance.Citation8–13,15 The mechanisms underlying this resistance have not been well characterized but likely involve multiple factors. Lack of lymphocyte infiltration at cancer sites, a phenotype that has been observed in many cancer patients,Citation24,25 is one of the major factors of resistance to anti-PD therapy.Citation13,16 In this study, we investigated whether anticancer immunotherapy can be enhanced by adenovirus-mediated gene therapy. Our results showed that the mouse lung adenocarcinoma M109 cell line is resistant to anti–PD-1 and anti–PD-L1 antibody treatment, and this resistance can be overcome by adenovirus-mediated gene therapy.
Immunotherapy with checkpoint inhibitors such as anti–PD-1 and anti–PD-L1 antibodies targets primarily the immune defects at tumor sites by locoregional modulation of immunosuppressive signals induced by PD-1 and PD-L1 interactions;Citation26 presence of immune cells in tumor sites is the premise for the effective anticancer activity of this therapy. This may explain why the efficacy of PD-1 blockage therapy in lung cancer correlates with a high mutation burden and high neoantigen burden in tumors,Citation7 which can trigger lymphocyte infiltration at tumor sites. However, for cancers with a low mutation burden, the neoantigen burden might not be strong enough to induce sufficient lymphocyte infiltration. We previously reported that lymphocyte infiltration at tumor sites can be dramatically induced by locoregional administration of foreign antigen–expressing viral vectors, such as adenovirus and vaccinia virus.Citation30 Lymphocyte infiltration at tumor sites was enhanced in animals preexposed to the viral vectors. Moreover, this approach can also induce tumor-specific immunity, because animals cured by this approach rejected tumor growth after being challenged with parental tumor cells.Citation30 In the present study, we confirmed our previous observation that oncolytic virotherapy induced lymphocyte infiltrations in tumors were enhanced by pre-existing immunity against the viral vector. Although we purposely preexposed mouse to adenovirus to induce pre-existing anti-human adenovirus immunity in this study, this procedure is not required in future clinical study because majority of human patients have preexisting immunity against adenovirus.Citation35,36 We found that mice with preexisting immunity have better treatment response to Ad/E1-TRAIL than naïve mice and that Ad/CMV-TP53 has single-agent activity in M109-bearing mice preexposed to an adenoviral vector, even though Ad/CMV-TP53 did not have a detectable cytotoxic effect in M109 cells in vitro. These results are consistent with our previous observation that redirecting adaptive immunity against foreign antigens to tumors can elicit anticancer activity.Citation30 On the other hand, lymphocyte infiltration at the tumor site is expected to overcome the resistance to PD-1 blockage therapy caused by lack of lymphocytes in tumor tissue. Indeed, our results showed that combination therapy of PD-1 blockage therapy and locoregional virotherapy led to profound in vivo anticancer activity and strongest lymphocyte and macrophage infiltrations in the M109 tumor model. Nevertheless, at an acute phase of locoregional viral infection, majority of immune cells attracted to the infection site are likely specific for viral antigens. However, our previous study revealed that destruction of cancer cells tagged with viral antigens by professional immune cells was able to promote tumor-specific immune response,Citation30 because animals could effectively reject tumor growth of parental tumor cells without viral infections.
Our results revealed that M109 tumors are intrinsically resistant to the treatment of antibodies against PD-1 or PD-L1. PD-L1 expression was barely detectable in M109 tumors, consisting with observation in clinics that most PD-L1 negative tumors were resistant to anti-PD-L1 or PD-1 therapy.Citation13,16,17 Nevertheless, co-administration of either anti–PD-1 or anti–PD-L1 antibody with adenovirus resulted in significantly stronger activity in M109 tumors than administration of the single agent with adenovirus. Similar results have recently been reported by others for the combination therapy of anti–CTLA-4 antibody and localized oncolytic virotherapy with Newcastle disease virus in melanoma tumor models.Citation31 This combination effect is not unexpected, because locoregional lymphocyte infiltration caused by viral infection and/or expression of foreign antigens will result in increased expression of IFNγ, the major inducer of PD-L1 in vivo.Citation38 Increased expression of PD-L1 in the tumor microenvironment will trigger a negative feedback mechanism that limits inflammatory response and tissue damage, but concurrently suppresses anticancer immunity. Treatment with anti–PD-1 or anti–PD-L1 antibody is expected to block this negative feedback and enhance immune response at tumor sites. It is intriguing that intratumoral PD-L1 expression was not detectable in all samples analyzed in this study, regardless dramatic differences in intratumoral infiltrations of lymphocytes and macrophages in different treatment groups. It is possible that induction of PD-L1 expression is transient, and the samples collected at the end of experiment may not reflect dynamic changes of PD-L1 expressions. Alternatively, it is possible that the assay itself is not sensitive enough to detect low levels of PD-L1 expressions. This study also had some limitations as it only tested two adenoviruses in one mouse model and the immunohistochemical assay is qualitative. The roles of TP53 or TRAIL gene expression in enhancing anti-PD therapy were not clear. It is likely that direct cell killing by the transgenes may not play a major role in the in vivo locoregional virotherapy because Ad/CMV-TP53 itself did not induce cytotoxicity in M109 cells in vitro. We expect that any viral vectors or strategies that induce locoregional inflammatory response at tumor sites may have a similar effect in overcoming resistance to immunotherapy with immune checkpoint inhibitors. Because a few viral gene therapy vectors, such as Ad/CMV-TP53Citation39 and herpes simplex virus type 1-derived oncolytic vector talimogene laherparepvec,Citation40 have been approved for cancer treatment in China and the United States of America, respectively, it will be interesting to test whether combination therapy of those approved therapeutic agents with anti-PD immunotherapy will improve the response rates in human patients with lung cancers.
Materials and methods
Cell lines and cell culture
The murine lung carcinoma M109 cell lineCitation41 was obtained from Frederick National Laboratory for Cancer Research, National Cancer Institute (Frederick, MD). The CMT167 cell lineCitation42 was obtained from Sigma-Aldrich Corporation. The 344SQ cell line was established from Kras/Tp53 double-mutant mouse lung cancers as described previously.Citation32 The cells were routinely cultured in RMPI 1640 medium supplemented with 6% heat–inactivated fetal calf serum and 1% Antibiotic-Antimycotic (100X) (ThermoFisher). All cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C.
Recombinant adenovirus and immune checkpoint antibodies
Adenovirus expressing the LacZ protein (Ad/CMV–LacZ), green fluorescent protein (GFP) (Ad/CMV–GFP) and TP53 (Ad/CMV-TP53), and oncolytic adenovirus expressing tumor necrosis factor-α-related apoptosis-inducing ligand (TRAIL) (Ad/E1-TRAIL) were constructed in our laboratory as described previously.Citation34,43 The expansion, purification, titration and quality analyses of both vectors were performed as we previously described.Citation34,43 The titer used in this study was determined by the absorbency of the dissociated virus at A260 nm (1 A260 unit = 1 × 1012 viral particles [vp]/mL), and the infectious units (IU) determined by the median tissue culture infective dose (TCID50) assayCitation44 were used as additive information. The vp:IU ratio was usually between 30:1 and 100:1. Monoclonal antibodies of rat anti-mouse PD-L1 (clone 10 F.9 G2) and master anti-mouse PD-1 (clone J43) and rat control IgG were obtained from Bio X Cell (West Lebanon, NH).
Adenovirus transduction in murine lung cancer cells
Cells were seeded at 5 × 105/well in 6-well plates and incubated for 4 h at 37°C with 5% CO2. Ad/CMV-GFP was added into the supernatant at a multiplicity of infection (MOI) ranging from 250 to 2000 VP. Mock-infected and virus-treated cells were harvested 48 h after the treatment by trypsinization, washed with phosphate-buffered saline (PBS), centrifuged into cell pellets and resuspended in PBS. Flow cytometric analysis for quantification of GFP-positive cells was performed at the Flow Cytometry and Cellular Imaging Facility of MD Anderson Cancer Center.
Cell viability assay
The cytotoxicity of the virus was determined using the sulforhodamine B assay. Cells were seeded at 1 × 103/well in 96-well plates and incubated for 24 h at 37°C with 5% CO2. Recombinant adenoviruses were added into the medium with different MOIs. PBS was used as the mock control. Cell viability was determined 24 h to 72 h after viral infection. After fixation of viable adherent cells with trichloroacetic acid in a 96–well microplate, the cells were stained with sulforhodamine B and quantified with optical density measured at 570 nm. The percentage of viable cells was determined according to the cell viability of the mock control, which was set at 100%. Each experiment was performed in triplicate and repeated at least three times.
Western blot analysis
Western blot analysis was performed to analyze adenovirus-mediated human TP53 protein expression after murine lung cancer cells were treated with Ad/CMV-TP53 or control vector Ad/CMV-LacZ. Briefly, cells were washed in PBS, collected and then lysed in RIPA buffer containing proteinase inhibitor cocktail and phosphatase inhibitor cocktail (Roche, Indianapolis, IN). The lysate was centrifuged at 10,000 g at 4°C for 10 min. The supernatant (30 µg protein) was fractioned by 10% SDS-PAGE gels and electrophoretically transferred to Immobilon-FL PVDF membranes. The membrane was blocked with blocking buffer (Li-Cor) at room temperature for 1 h, then incubated with the goat anti-human P53 IgG (1:1000) for 2 h or with the primary antibody at 4°C overnight. After washing with phosphate-buffered saline with (0.05% Tween 2) (PBST), the membrane was incubated with IRDye infrared secondary antibody (Li-Cor) for 1 h at room temperature. The membrane was washed with PBST again, and protein expression was detected with the Odyssey Infrared Imaging System (Lincoln, NE).
Animal experiments
Animal experiments were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health Publication number 85–23) and the institutional guidelines of MD Anderson Cancer Center. Six- to eight-week-old BALB/c, C57 BL/6 or 129/Sv mice were purchased from Charles River Laboratory or Jackson Laboratory and housed at MD Anderson under conventional conditions. To simulate the situation observed in the clinic, that the majority of patients have previously been exposed to adenovirus infection and have preexisting immunity against adenovirus,Citation35,36 we preexposed mice to adenovirus before tumor implantation through two subcutaneous inoculations of adenovirus Ad/CMV-LacZ (1 × 1010 vp/mouse/injection) at day 1 and day 14. A group of animals preexposed to PBS were used as the naïve control. Subcutaneous tumors were then established by inoculation of 5 × 105 tumor cells. When tumors reached about 5 mm in diameter, animals were randomized and treated with adenovirus (1 × 1010 vp/mouse, intratumoral injection), anti–PD-1 antibody (100 µg/mouse, intraperitoneal injection), or both, twice a week for 2 weeks. Animals treated with vehicle were used as placebo control. Tumor growth was monitored every 2 – 3 days and was normalized with tumor volume at the beginning of treatment, which was set to 1. Subcutaneous tumors were measured with calipers to determine the largest and smallest diameters, while tumor volume was calculated according to the formula V = ab2/2, where a is the largest diameter and b is the smallest. Mice were killed once one of the tumors reached 15 mm in diameter. Tumors harvested at the end of experiment were analyzed for immunohistochemical assay.
Immunohistochemical analysis
Formalin-fixed paraffin-embedded tumor tissue samples were submitted for immunohistochemical analysis at the Research Histology, Pathology and Imaging Core in our institution. The sectioning slides were stained with antibodies specific for mouse CD4 (eBioscience, #14-9766), CD8 (eBioscience, #14-0808), PDL1 (Cell Signaling, #64988) and F4/80 (Abcam, #ab6640) using the standard operation protocols performed in the Core facility. Mouse spleen and lymph node were used as positive controls for CD4, CD8 and F4/80, while mouse thymus was as a positive for PDL1 (see Supplement Fig. 1).
Statistical analysis
Differences among the treatment groups were assessed by analysis of variance using IBM SPSS Statistics V24 software. Survival analysis was done using the Kaplan-Meier test, and differences between curves were assessed using the log-rank test. P < 0.05 was considered statistically significant.
Ethics approval and consent to participate
Not applicable
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgment
We would like to thank Sunita C Patterson and the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center for editorial review of the manuscript.
Funding
This work was supported in part by the National Institutes of Health through grants R01CA190628, Specialized Program of Research Excellence (SPORE) grant CA070907, and The University of Texas MD Anderson Cancer Center support grant P30CA016672 (used the Flow Cytometry and Cellular Imaging Facility, Research Animal Support Facility and Research Histology, Pathology and Imaging Facility); and by endowed funds to The University of Texas MD Anderson Cancer Center, including the Lung Cancer Moon Shot Program and the Goldman Sachs Philanthropy Fund.
References
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67:7-30. doi:10.3322/caac.21387. PMID:28055103
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi:10.3322/caac.21262. PMID:25651787
- Fang B, Mehran RJ, Heymach JV, Swisher SG. Predictive biomarkers in precision medicine and drug development against lung cancer. Chin J Cancer. 2015;34:26. doi:10.1186/s40880-015-0028-4.
- Edwards BK, Noone AM, Mariotto AB, Simard EP, Boscoe FP, Henley SJ, Jemal A, Cho H, Anderson RN, Kohler BA, et al. Annual Report to the Nation on the status of cancer, 1975–2010, featuring prevalence of comorbidity and impact on survival among persons with lung, colorectal, breast, or prostate cancer. Cancer. 2014;120:1290–314. doi:10.1002/cncr.28509. PMID:24343171
- DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–71. doi:10.3322/caac.21235. PMID:24890451
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi:10.1126/science.1235122. PMID:23539594
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi:10.1126/science.aaa1348. PMID:25765070
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39. doi:10.1056/NEJMoa1507643. PMID:26412456
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35. doi:10.1056/NEJMoa1504627. PMID:26028407
- Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2015;33:2004–12. doi:10.1200/JCO.2014.58.3708. PMID:25897158
- Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015;16:257–65. doi:10.1016/S1470-2045(15)70054-9. PMID:25704439
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi:10.1056/NEJMoa1501824. PMID:25891174
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA6 McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280 A in cancer patients. Nature. 2014;515:563–7. doi:10.1038/nature14011. PMID:25428504
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi:10.1056/NEJMoa1200694. PMID:22658128
- Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–65. doi:10.1016/S0140-6736(16)32517-X. PMID:27979383
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. doi:10.1038/nature13954. PMID:25428505
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi:10.1056/NEJMoa1200690. PMID:22658127
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63. doi:10.1158/2159-8290.CD-13-0310. PMID:24078774
- Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, Herter-Sprie GS, Akbay EA, Tchaicha JH, Altabef A, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590–604. doi:10.1016/j.ccr.2014.03.033. PMID:24794706
- Muhlbauer M, Fleck M, Schutz C, Weiss T, Froh M, Blank C, Schölmerich J, Hellerbrand C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol. 2006;45:520–8. doi:10.1016/j.jhep.2006.05.007. PMID:16876901
- Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, Oh S, Shin JG, Yao S, Chen L, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006;580:755–62. doi:10.1016/j.febslet.2005.12.093. PMID:16413538
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion.[Erratum appears in Nat Med 2002 Sep.8(9):1039]. Nat Med. 2002;8:793–800. PMID:12091876
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi:10.1126/scitranslmed.3006504. PMID:23986400
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi:10.1126/scitranslmed.3003689. PMID:22461641
- Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139–45. doi:10.1158/0008-5472.CAN-15-0255. PMID:25977340
- Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J Clin Invest. 2015;125:3384–91. doi:10.1172/JCI80011. PMID:26325035
- Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–6. doi:10.1126/science.272.5258.60. PMID:8600538
- Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–20. doi:10.1038/nri3442. PMID:23598650
- Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–4. doi:10.1038/nature07657. PMID:19182777
- Hu W, Davis JJ, Zhu H, Dong F, Guo W, Ang J, Peng H, Guo ZS, Bartlett DL, Swisher SG, et al. Redirecting adaptive immunity against foreign antigens to tumors for cancer therapy. Cancer Biol Ther. 2007;6:1773–9. doi:10.4161/cbt.6.11.4855. PMID:17986853
- Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra32. doi:10.1126/scitranslmed.3008095. PMID:24598590
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. doi:10.1038/ncomms6241. PMID:25348003
- Justilien V, Regala RP, Tseng IC, Walsh MP, Batra J, Radisky ES, Murray NR, Fields AP. Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PloS One. 2012;7:e35040. doi:10.1371/journal.pone.0035040. PMID:22545096
- Dong F, Wang L, Davis JJ, Hu W, Zhang L, Guo W, Teraishi F, Ji L, Fang B. Eliminating established tumor in nu/nu nude mice by a tumor necrosis factor-alpha-related apoptosis-inducing ligand-armed oncolytic adenovirus. Clin Cancer Res. 2006;12:5224–30. doi:10.1158/1078-0432.CCR-06-0244. PMID:16951242
- Garnett CT, Erdman D, Xu W, Gooding LR. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J Virol. 2002;76:10608–16. doi:10.1128/JVI.76.21.10608-10616.2002. PMID:12368303
- Bauer U, Flunker G, Bruss K, Kallwellis K, Liebermann H, Luettich T, Motz M, Seidel W. Detection of antibodies against adenovirus protein IX, fiber, and hexon in human sera by immunoblot assay. J Clin Microbiol. 2005;43:4426–33. doi:10.1128/JCM.43.9.4426-4433.2005. PMID:16145087
- Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, Tsiouris AJ, Cohen J, Vortmeyer A, Jilaveanu L, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: Early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–83. doi:10.1016/S1470-2045(16)30053-5. PMID:27267608
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi:10.1126/scitranslmed.3006504. PMID:23986400
- Ma G, Shimada H, Hiroshima K, Tada Y, Suzuki N, Tagawa M. Gene medicine for cancer treatment: Commercially available medicine and accumulated clinical data in China. Drug Des Devel Ther. 2009;2:115–22. PMID:19920899
- Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33:2780–8. doi:10.1200/JCO.2014.58.3377. PMID:26014293
- Schultz R, Ruiz P, Chirigos M, Heine U, Nelson-Rees W. Establishment and characterization of a cell line derived from a spontaneous murine lung carcinoma (M109). In Vitro. 1977;13:223–31. doi:10.1007/BF02615079. PMID:194826
- Franks LM, Layton MG. Ultrastructural tumour differentiation and organ specificity in high and low metastatic lines from a mouse lung carcinoma. Br J Cancer. 1984;49:423–9. doi:10.1038/bjc.1984.68. PMID:6324837
- Bouvet M, Ellis LM, Nishizaki M, Fujiwara T, Liu W, Bucana CD, Fang B, Lee JJ, Roth JA. Adenovirus-mediated wild-type p53 gene transfer down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in human colon cancer. Cancer Res. 1998;58:2288–92. PMID:9622060
- Fang B, Ji L, Bouvet M, Roth JA. Evaluation of GAL4/TATA in vivo. Induction of transgene expression by adenovirally mediated gene codelivery. J Biol Chem. 1998;273:4972–5. doi:10.1074/jbc.273.9.4972. PMID:9478943