
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The redirection of T cell activity using bispecific antibodies is one of the most promising cancer immunotherapy approaches currently in development, but it is limited by cytokine storm-related toxicities, as well as the pharmacokinetics and tumor-penetrating capabilities of current bispecific antibody formats. Here, we have engineered the ATTACK (Asymmetric Tandem Trimerbody for T cell Activation and Cancer Killing), a novel T cell-recruiting bispecific antibody which combines three EGFR-binding single-domain antibodies (VHH; clone EgA1) with a single CD3-binding single-chain variable fragment (scFv; clone OKT3) in an intermediate molecular weight package. The two specificities are oriented in opposite directions in order to simultaneously engage cancer cells and T cell effectors, and thereby promote immunological synapse formation. EgA1 ATTACK was expressed as a homogenous, non-aggregating, soluble protein by mammalian cells and demonstrated an enhanced binding to EGFR, but not CD3, when compared to the previously characterized tandem bispecific antibody which has one EgA1 VHH and one OKT3 scFv per molecule. EgA1 ATTACK induced synapse formation and early signaling pathways downstream of TCR engagement at lower concentrations than the tandem VHH-scFv bispecific antibody. Furthermore, it demonstrated extremely potent, dose-dependent cytotoxicity when retargeting human T cells towards EGFR-expressing cells, with an efficacy over 15-fold higher than that of the tandem VHH-scFv bispecific antibody. These results suggest that the ATTACK is an ideal format for the development of the next-generation of T cell-redirecting bispecific antibodies.
Introduction
Redirecting the activity of T cells using bispecific antibodies (bsAbs), which cross-link tumor cells and T cells, independently of their T cell receptor (TCR) specificity, is a potent strategy to cancer therapyCitation1. The core concept of this approach is an antibody's binding to a cell surface tumor-associated antigen (TAA) and simultaneous binding to the CD3ϵ chain of the TCR/CD3 complex. This crosslinking of the TCR activates the T cell, resulting in secretion of cytokines and cytotoxic effector proteins, which induces apoptosis of the target cell.Citation2 Major obstacles to the broad application of T cell-recruiting bsAbs (T-bsAbs) are related to the extraordinary potency of T cell-mediated responses, which can negatively impact non-tumor cells expressing low levels of the TAA, as well as systemic cytokine-associated toxicities. T-bsAbs carrying active IgG Fc domains may crosslink Fc gamma receptor (FcγRs)-bearing cells and T cells, leading to off-target Fc-induced T cell activation and subsequent cytokine storm.Citation3,4 This risk can be avoided by using engineered Fc-free T-bsAbs such as the bispecific T cell engager (BiTE)Citation5 and dual-affinity-re-targeting (DART) antibodies.Citation6 Both the BiTE and DART formats have a single TAA-binding single-chain variable fragment (scFv) and a single CD3-binding scFv. In December 2014, blinatumomab, an anti-CD19 x anti-CD3 BiTE, was approved by the US Food and Drug Administration (FDA) for the treatment of relapsed/refractory B-cell acute lymphocytic leukemia, and several other BiTEs and DARTs have entered clinical development.Citation7 However, although the small size of these molecules (≈55 kDa), which lies below the glomerular filtration threshold (50-60 kDa), might be advantageous with regards to tissue penetration in solid tumors, they require continuous intravenous infusion due to their short circulatory half-life.Citation8 Tandem diabodies (tandAbs; 115 kDa), with two TAA-binding domains and two CD3-binding domains, are sufficiently large to prevent glomerular filtration and therefore have an extended circulatory half-life compared to the smaller BiTE and DART.Citation9 However, their bivalent CD3 binding may crosslink the TCR even without simultaneous binding to TAA-expressing tumor cells, leading to an OKT3-like cytokine release syndrome.Citation10,11
Recently, a number of Fc-free or Fc-attenuated T-bsAbs have been designed with bivalent TAA binding and monovalent CD3 binding, i.e. a 2 + 1 binding stoichiometry.Citation12 For example, a T-bsAb containing one anti-CD3 scFv and two anti-TAA Fabs has been constructed using the dock-and-lock (DNL) method.Citation13 The potential advantages of this molecule include bivalent binding to tumor cells, which strengthens the binding through the avidity effect, and a larger size (≈130 kDa) with the associated pharmacokinetic changes (e.g. preclusion from glomerular filtration and crossing of the blood-brain barrier). Another example of a 2 + 1T-bsAb is CEA TCB, a heterodimeric IgG with two CEA-binding Fabs, one CD3-binding Fab, and a molecular weight of ≈200 kDa. Importantly, it bears an Fc region engineered for abrogated binding to C1q and FcγR, but not to neonatal FcR (FcRn), in order to mitigate complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity while maintaining IgG-like pharmacokinetics.Citation14 The use of IgG-based T-bsAbs can be a double-edged sword. While their large molecular weight and FcRn interactions convey an unequalled circulatory half-life, IgGs have a slow rate of diffusion exactly because of this large molecular weight. To treat non-hematological malignancies, a T-bsAb must efficiently cross the vascular endothelium and traverse the extracellular matrix in order to permeate the tumor. IgGs are restricted to perivascular tumor regions due to their slow diffusivity and the general impermissiveness of tumor physiology.Citation15 Consequently, large tumor masses may be difficult to treat using IgG-based therapies.
By adapting the tandem trimerbody formatCitation16 to the TAA/CD3 crosslinking approach, we have generated a novel class of Fc-free T-bsAb called the asymmetric tandem trimerbody for T cell activation and cancer killing (ATTACK). This tetravalent bispecific antibody combines trivalent binding to the cell surface TAA epithelial growth factor receptor (EGFR), and monovalent binding to CD3 with an intermediate molecular weight of ≈100 kDa, which strikes a compromise between the needs for circulatory half-life extension and tumor penetration. Furthermore, the ATTACK structure positions the anti-TAA and anti-CD3 binding domains on opposite sides of the molecule, oriented in reverse directions, which should allow it to effectively form synapses between T cells and tumor cells. We found that the anti-EGFR x anti-CD3 ATTACK (3 + 1) was more effective than a conventional anti-EGFR x anti-CD3 tandem VHH-scFv bispecific antibody (1 + 1) using the same binding domains at inducing T cell activation and redirecting T cells to lyse EGFR-expressing cancer cells.
Results
Generation of the anti-EGFR x anti-CD3 ATTACK
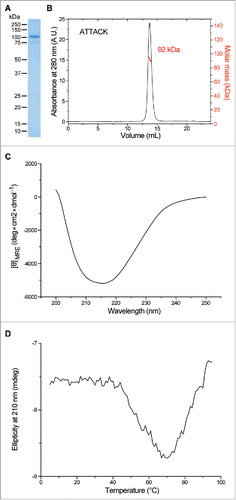
In this study, we generated a bispecific anti-EGFR x anti-CD3 ATTACK consisting of three anti-EGFR VHH-TIEXVIII modules and one anti-CD3 scFv fused to the C-terminus of the C-terminal TIEXVIII domain on a single protein chain ( and ). We used two well-characterized antibodies as building blocks: the inhibitory anti-EGFR VHH domain EgA1Citation17 and the mitogenic anti-CD3ϵ scFv OKT3.Citation18,19 The anti-EGFR x anti-CD3 ATTACK antibody (hereafter referred to as EgA1 ATTACK) was purified from conditioned medium from transfected HEK-293 cells by immobilized metal affinity chromatography (IMAC) followed by protein A chromatography, which yielded proteins that were >95% pure, as determined by coomassie-stained SDS-PAGE (). EgA1 ATTACK was further analyzed by SEC-MALS. The protein eluted from the size exclusion column as a major peak, with no significant absorbance at the exclusion volume of the column, which indicates the absence of large aggregates. MALS from the central part of this peak gave a molecular size corresponding to ≈92 kDa (). This result is consistent with the value of 99.4 kDa predicted from the sequence of amino acids without the signal sequence. Altogether these data show that the EgA1 ATTACK forms intramolecular homotrimers. The circular dichroism spectrum has a single minimum at 216 nm (), indicating that it contains predominantly β-sheet and irregular secondary structure. The EgA1 ATTACK antibody is folded into a stable three-dimensional structure, as seen by the cooperative thermal denaturation (). A major denaturation event occurs with a mid-point temperatures of approximately 54°C, followed by a decrease of signal which is possibly due to precipitation of the denatured protein at high temperatures.
Figure 1. Schematic representation and three-dimensional model of the anti-EGFR x anti-CD3 ATTACK. (A) Schematic diagrams showing the genetic (left) and domain structure (right) of the ATTACK molecule, bearing a signal peptide from the oncostatin M (white box), three anti-EGFR EgA1 VHH genes (red boxes) and three collagen-derived trimerization (TIE) domains flanked peptide linkers (pale green boxes), the anti-CD3 OKT3 scFv gene (blue boxes), and epitope tags (yellow box). Arrows indicate the direction of transcription. (B) Three-dimensional model of the ATTACK molecule.
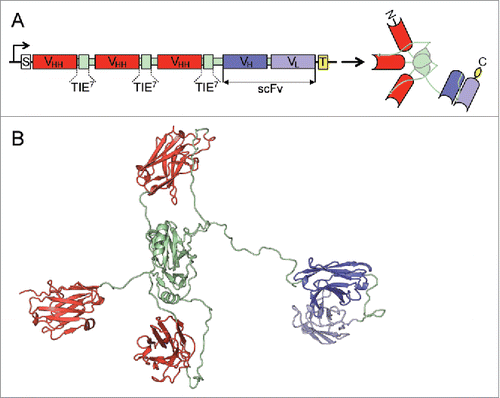
Figure 2. Structural characterization of purified EgA1 ATTACK. Reducing SDS-PAGE (A) and SEC-MALS (B) with the indicated molecular masses measured at the center of the chromatography peaks. The black line corresponds to the UV absorbance (left axis) and the red line to the measured molar mass (right axis). Circular dichroism spectrum (C) and tertiary structure analysis by thermal denaturation measured by the change in ellipticity at 210 nm (D).
EgA1 ATTACK binding specificity and affinity
Two monospecific molecules were used as controls: the monomeric anti-EGFR EgA1 VHH20 and the trimeric anti-EGFR EgA1 VHH-based multichain N-trimerbody (EgA1N) (Fig. S1A).Citation16 The EgA1 VHH was purified from the periplasm of E. coli cells by IMAC and the EgA1N was purified from conditioned medium from transfected HEK293 cells by a two-step chromatography. Both proteins were > 95% pure, as estimated by SDS-PAGE (). The binding kinetics of the EgA1 ATTACK, as well as the two monospecific molecules, were investigated by biolayer interferometry (BLI) using biosensors coated with a human EGFR-Fc fusion ( and ). The anti-EGFR mAb cetuximab and its derived Fab fragment (Fig. S2) were additionally included to verify that the method can demonstrate the avidity effect and that it gave comparable kinetic values to what has previously been reported for cetuximab-Fab21. The monovalent EGFR binders, cetuximab-Fab and EgA1 VHH, showed similar kinetics (Table S1), with nanomolar-order KD values and dissociation half-lives of ≈7 minutes ( and ). In contrast, the intact cetuximab mAb and the EgA1-derived trimerbodies (EgA1N and EgA1 ATTACK) showed little dissociation during the two hours over which dissociation was measured ( and ). Signal fluctuations on the EGFR-coated biosensors, combined with the limited extent of dissociation measured over the two hour-long dissociation step, rules out a precise quantification of the kinetic rate constants of the multivalent binders, although the data implies them to have low picomolar KD values and dissociation half-lives much higher than 2 hours. The severe difference in the rate of dissociation of the monovalent and multivalent EGFR binders is as expected due to the avidity effect.
Figure 3. Functional characterization of purified EgA1 ATTACK. (A) Reducing SDS-PAGE of the purified EgA1 VHH, EgA1 multichain N-trimerbody (EgA1N), EgA1 ATTACK and EGFR-Fc. (B and C) Biolayer interferometry (BLI)-derived sensorgrams (in black) for the interaction between immobilized EGFR-Fc and cetuximab- (B) or EgA1-based (C) antibodies. Fitting curves for cetuximab-Fab and EgA1-VHH are included in red. (D) The binding to EGFR on the cell surface of HeLa cells by cetuximab- and EgA1-based antibodies at 0.1, 0.32, 1, and 10 nM, measured by FACS and normalized to the binding at 10 nM. (E) The binding to CD3 on the cell surface of Jurkat cells by OKT3- and EgA1-based antibodies at 0.1, 1, 3.2, and 10 nM, measured by FACS and normalized to the binding at 10 nM.
EgA1 ATTACK binding specificity and affinity to cell-surface expressed antigens
We have previously generated a tandem bispecific anti-EGFR x anti-CD3 antibody by fusing the single EgA1 VHH and the OKT3 scFv with a five-residue peptide linker, creating an antibody format similar to the BiTE, just with a scFv replaced by a VHH (Fig. S1B). This 1 + 1 antibody, which we called the EgA1 light T-cell engager (EgA1 LiTE), was purified from conditioned medium of transfected HEK-293 cells to >95% purity (Fig. S3A). SEC-MALS showed that the EgA1 LiTE elutes with one major symmetrical peak containing molecules with a mass of 43 kDa (Fig. S3B). This result is consistent with the value of 44.1 kDa predicted from its amino acid sequence.
The ability of both antibodies to detect their antigens as cell surface proteins was studied by flow cytometry. EgA1 LiTE and EgA1 ATTACK were both found to bind the EGFR-expressing human cervix adenocarcinoma cell line HeLa and the CD3-expressing human T cell line Jurkat, without binding the mouse cell line 3T3 (Fig. S4). Flow cytometry was also used to determine the equilibrium saturation of cell surface EGFR by EgA1 LiTE and EgA1 ATTACK at concentrations of 0.1, 0.32, 1, or 10 nM ( and S5A). According to the kinetics values determined by BLI, the saturation of EGFR by a monovalent EgA1 VHH antibody should be highly concentration-dependent in this range, whereas saturation by an EgA1-based trimerbody should be nearly concentration-independent. As in BLI, cetuximab and cetuximab-Fab were included to provide a point of comparison and a control for the avidity effect. EgA1 LiTE and cetuximab-Fab did indeed bind EGFR in a concentration-dependent manner, with EC50 values (i.e. KD) of ≈1 nM and ≈5 nM, respectively ( and S5A, Table S2). On the other hand, the saturation of EGFR by cetuximab was not attenuated even at the low concentrations of 0.32 and 0.1 nM, demonstrating an EC50 value well below the chosen concentration range ( and S5A, Table S2). EgA1 ATTACK showed a consistent drop in signal from 10 nM to 1 nM; as the signal only drops slightly when the concentration is further decreased to 0.32 and 0.1 nM, this is indicative of complex binding with a major low-picomolar functional affinity interaction. The flow cytometry results corroborate those obtained from BLI, but in a cell surface context, supporting a significant and physiologically relevant enhancement of the functional affinity of EgA1 by the trivalence of the ATTACK format.
In a similar manner, the binding of EgA1 LiTE and EgA1 ATTACK to cell surface CD3 was assayed by flow cytometry with Jurkat cells. OKT3 and OKT3-Fab (Fig. S2) were included to define the bivalent and monovalent OKT3-clone response profiles, albeit for the OKT3-Fab binding domain and not for OKT3 scFv. CD3 binding by OKT3-Fab, EgA1 LiTE, and EgA1 ATTACK dropped significantly at 3.2 and 1 nM compared to 10 nM, with little to no apparent binding at 0.1 nM; fitting gave EC50 values of ≈23, ≈3, and ≈8 nM, respectively ( and S5B, Table S2). OKT3 mAb, on the other hand, showed no decrease in binding at 3.2 and 1 nM, and maintained roughly 50% binding at 0.1 nM, suggesting an EC50 of 0.1 nM. The experiment was able to distinguish between monovalent and bivalent OKT3 antibodies, and the parity in dose-response of OKT3-Fab to EgA1 LiTE and EgA1 ATTACK indicates that both engineered antibodies behave as monovalent binders of cell surface CD3.
Effect of EgA1 ATTACK on cell proliferation and EGFR phosphorylation
EgA1 is a well-characterized inhibitory anti-human EGFR VHH domain that binds an epitope near the EGFR domain II/III junction, preventing the conformational changes in the receptor which are required for high-affinity ligand binding and receptor dimerization.Citation17 To further assess the functionality of the EgA1 ATTACK, we studied its capacity to inhibit proliferation and EGFR phosphorylation in the EGFR gene-amplified, inhibitor-responsive cell line A431 (Fig. S6A).Citation22 In proliferation assays, the anti-human EGFR mAb cetuximab, a ligand-competitive inhibitor,Citation21 was used as a positive control, and OKT3 mAb was used as a negative control. As shown in Fig. S7A, cetuximab, but not OKT3, inhibited A431 proliferation in a dose-dependent manner. Compared to OKT3, inhibition of cell growth was statistically significant for every single dose of EgA1 ATTACK tested. In fact, the EgA1 ATTACK proved to be even more effective than cetuximab in reducing the growth of A431 cells. At the highest dose (50 nM), EgA1 ATTACK was more potent than cetuximab (p< 0.01), whereas the EgA1 LiTE had no significant effect on proliferation (Fig. S7 A). Then, the ability of EgA1 ATTACK to inhibit EGF-induced signaling was tested. The phosphorylation status of tyrosine 1068 (Tyr1068) of the EGFR was determined, as this tyrosine is the docking site for Grb2 and its phosphorylation is the initiation of signaling towards Ras.Citation23 In EGF-treated A431 cells, cetuximab inhibited EGFR phosphorylation by 36%, whereas an equimolar quantity of EgA1 ATTACK decreased p-EGFR by 75% (Fig. S7B). The EgA1 LiTE had no effect on the phosphorylation status of EGFR (Fig. S7B). Taken together, these results showed that EgA1 ATTACK performed best in inhibiting mitogenic signaling from EGFR.
EgA1 ATTACK triggers immunological synapse formation
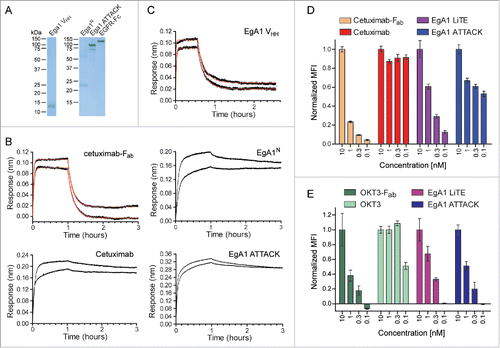
Upon simultaneous binding to EGFR-expressing 3T3 cells (Fig. S6B) and CD3-expressing T cells, both EgA1 LiTE and EgA1 ATTACK crosslink T cells to target cells, leading to the formation of the immunological synapse (IS), as observed by measuring CD3ζ and actin accumulation at the T cell-target cell contact surface (). The IS showed organized CD3ζ and actin accumulation at the center and the periphery, respectively (). IS were not formed between wild-type EGFR-negative 3T3 cells (Fig. S6B) and T cells, showing that the IS formation was antigen-dependent (). IS were likewise not formed when 3T3-EGFR cells were used as target cells in the absence of EgA1 LiTE and EgA1 ATTACK, verifying that the expression of EGFR itself did not promote IS formation ( and ).
Figure 4. Immunological synapse formation is triggered by EgA1 ATTACK. (A) IS formation assessed by confocal microscopy. Green (CD3ζ) and red (actin) channels of a confocal section are shown with a pseudo-color displaying the range of intensity values. Calibration bar for pseudo-color applied in all images is shown in the first panel. Images of merged channels are shown, where CD3ζ is presented in green, actin in red and 3T3-EGFR cells in blue. As a control, Jurkat T cells were incubated together with 3T3-EGFR cells in the absence of antibodies. Scale bar 5 μm. (B) Quantification of CD3ζ (left graph) and actin (right graph) polarization towards the IS in Jurkat T cells. Dots in graphs represent individual cells obtained from several experiments. Results are expressed as mean ± S.D. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns, not significant). The experiments were performed three times and results of one representative experiment are shown.
Induction of early signaling downstream of TCR by EgA1 ATTACK
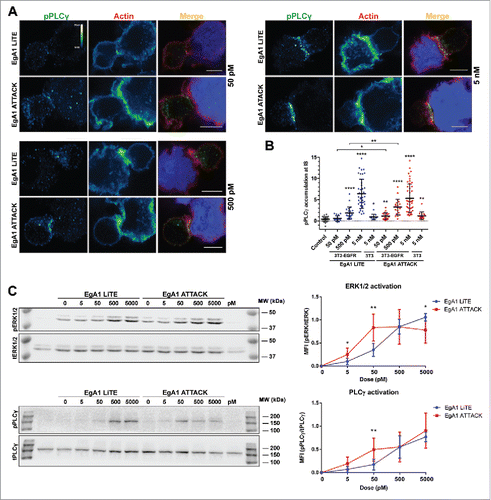
Next, we investigated whether EgA1 ATTACK was able to trigger effective signaling downstream of TCR activation. Confocal microscopy was used to examine cell conjugates formed between Jurkat J77T cells and 3T3-EGFR cells for a range of increasing concentrations of either EgA1 LiTE or EgA1 ATTACK. We then assessed polarized signaling at the IS by measuring the phosphorylation of phospholipase Cγ (pPLCγ), a central signaling component downstream antigenic stimulation. While higher concentrations of both EgA1 LiTE and EgA1 ATTACK induced phosphorylation of PLCγ at the IS, EgA1 ATTACK induced stronger PLCγ signaling at lower concentrations ( and ). PLCγ was not phosphorylated when untransfected 3T3 cells were used, demonstrating that phosphorylation was dependent on EGFR engagement (). Activation efficiency of PLCγ and downstream MAPK signaling was also evaluated by western blot (). EgA1 ATTACK induced higher PLCγ and ERK1/2 activation at concentrations ranging between 5 pM and 50 pM. However, saturating concentrations of EgA1 LiTE reached the same or higher signaling activation compared to EgA1 ATTACK. These data suggest that induction of early signaling downstream of the TCR required lower EgA1 ATTACK concentrations compared to EgA1 LiTE.
Figure 5. Activation of downstream TCR signaling by EgA1 ATTACK. (A) Polarised activation of PLCγ assessed by confocal microscopy. Green (pPLCγ) and red (actin) channels of a confocal section are shown with a pseudo-color displaying the range of intensity values. Calibration bar for pseudo-color applied in all images is shown in the first panel. Images of merged channels are shown, where pPLCγ is presented in green, actin in red and 3T3-EGFR cells in blue. Scale bar 5 μm. (B) Quantification of pPLCγ activation at the immunological synapse. As a control, Jurkat T cells were incubated together with 3T3-EGFR cells in absence of antibodies. Dots represent individual cells of one experiment. (C) Western blot analysis for ERK1/2 and PLCγ activation. Densitometric analyses were performed, and the ratio of the mean fluorescence intensity (MFI) of phosphoproteins and total proteins is shown for different concentrations of EgA1 LiTE or EgA1 ATTACK. Results are expressed as mean ± S.D (n = 3). (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Not significant differences are not displayed. The experiments were performed three times and results of one representative experiment are shown.
T cell activation by EgA1 ATTACK
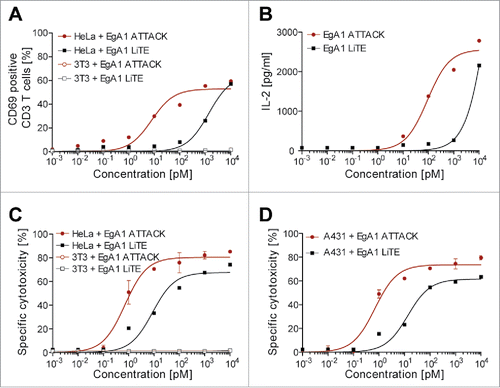
T cell activation is further reflected by expression of the activation marker CD69 and secretion of IL-2. Human T cells derived from unstimulated peripheral blood mononuclear cells (PBMCs) from healthy donors were co-cultured with EGFR-positive HeLa cancer cells in the presence of different amounts of EgA1 LiTE or EgA1 ATTACK. Both antibodies gave a dose-dependent increase in CD69 () and IL-2 (); for CD69, the EC50 values were 1 nM for the EgA1 LiTE and 15 pM for the EgA1 ATTACK, while for IL-2 the EC50 value was 147 pM for the EgA1 ATTACK. The 70-fold lower EC50 for CD69 and the lower EC50 for IL-2 indicate that EgA1 ATTACK is a more potent activator of T cells in vitro than the EgA1 LiTE.
Figure 6. Induction of T cell activation and cytotoxicity by EgA1 ATTACK. EGFR-positive HeLa cells or EGFR-negative 3T3 cells were co-cultured in 96-well plates with PBMCs in the effector:target (E:T) ratio of 5:1, and purified EgA1 LiTE or EgA1 ATTACK. After 24 hours, the surface expression of T-cell activation surface marker CD69 was determined by FACS analysis (A) and IL-2 production was determined by ELISA (B). Specific lysis of HeLaLuc cells and 3T3Luc cells (C) or A431Luc cells (B) incubated with PBMC (5:1) in the presence of purified antibodies and after 48 h. Percent viability was calculated relative to the luminescence from an equal number of input control cells and used to calculate percent specific lysis. Results are expressed as a mean ± SD (n = 3) from 1 of at least 3 separate experiments. The experiments were performed three times and results of one representative experiment are shown.
Redirected lysis of EGFR-positive cancer cells by the EgA1 ATTACK
The EgA1 ATTACK could redirect unstimulated peripheral T cells to lyse EGFR-expressing HeLa and A431 cancer cells in a concentration-dependent fashion with EC50 values of 0.9 and 0.7 pM, respectively ( and ). These values were lower than the EC50 values for T cell activation determined in the same assay ( and ), indicating that only a fraction of T cells needed to be activated for potent redirected lysis. In these assays, the EgA1 ATTACK was able to induce 70–80% lysis of the target cells. The EgA1 LiTE was also able to induce lysis of target cells but with higher EC50 values for both cell types (14.9 pM for HeLa and 15.8 pM for A431 cells) and a lower percentage lysis (≈60% for both cell lines). The EgA1 ATTACK was therefore 15- to 20-fold more potent in the in vitro redirection of T cell towards EGFR-expressing target cells ( and ). Importantly, neither antibody induced the lysis of EGFR-negative 3T3 cells ().
Discussion
Here, we report, for the first time, the generation and characterization of an Fc-free T cell-recruiting antibody with a 3 + 1 stoichiometry intended for cancer immunotherapy. This antibody is the result of further development to the previously reported tandem trimerbody format,Citation16 which has been structurally iterated upon through the addition of a C-terminal scFv, and functionally repurposed as a bispecific T-bsAb. The ATTACK is uniquely able to combine trivalent TAA binding with monovalent CD3 binding in an intermediate MW package that orients the two specificities in opposite directions, in order to optimally engage two different cells.
Recombinant EgA1 ATTACK molecules were efficiently expressed by transfected HEK-293 cells as homogenous, non-aggregating, soluble protein consistent with what we have previously reported for multi-chain and tandem trimerbodies.Citation16,24-26 The EgA1 ATTACK could be purified as a homogeneous intramolecular homotrimer using standard affinity chromatographic methods. The EgA1 ATTACK specifically recognized its TAA target either immobilized on plastic, or expressed in a cell surface context. Biolayer interferometry and flow cytometry studies demonstrated a significant enhancement of the functional affinity of anti-human EGFR EgA1 VHH when incorporated in the ATTACK format. Prior to the binding experiments, the area of influence of the EgA1 VHH domain/s in the LiTE and ATTACK formats were simulated (Fig. S8), and EgA1 in the ATTACK was found to have access to 2.25 times the area and 3.4 times the volume of EgA1 in the LiTE. The results of the binding experiments empirically demonstrate multivalence of the ATTACK and indirectly support the expanded influence of EgA1 in the ATTACK. This was not a foregone conclusion, as the functionality of one or more EgA1 VHH could have been compromised when included in the ATTACK, e.g. due to steric hindrance or linker placement. scFvs and Fabs can lose some or all functionality when N- or C-terminally fused together in certain arrangements, and this can dictate the final layout and included binding domains in a bsAb.Citation12 In assays with A431 cells, EgA1 ATTACK potently inhibited intracellular signaling downstream of EGFR, as well as EGFR-dependent cell proliferation. It has previously been demonstrated that in A431 cells, inhibition of EGF-stimulated proliferation by anti-EGFR mAbs requires antibody bivalence, as the mechanism of action involves EGFR dimerization and subsequent receptor down-regulation,Citation27 and in fact the EgA1 LiTE showed no inhibition of proliferation in our assay. Another flow cytometry experiment defined the monovalent and bivalent binding behavior of OKT3, and showed that EgA1 ATTACK binds cell surface CD3 monovalently. As mentioned earlier, monovalent CD3 binding is critical in order to avoid the dangerous side effects associated with systemic crosslinking of CD3 independent of antibody clustering on a TAA-expressing cell. CD3 monovalence may also play a role in enabling the quick disengagement of a T cell from a target cell, which is necessary for serial tumor cell killing.
The structural layout and binding domain orientation of a bispecific T cell-redirecting antibody are critical to the formation of the initial contact between a tumor cell and a T cell. Our data show that the IS was formed antigen-dependently following the addition of EgA1 ATTACK to co-cultures of EGFR-expressing target cells and T cells, as was the case with the EgA1 LiTE. A normal accumulation of CD3ζ at the center and actin at the periphery of the IS was induced by either EgA1 LiTE or EgA1 ATTACK antibodies. In the BiTE format, the scFvs are highly unimpeded, as the flexible linker can wind, kink, and be compressed as the scFvs bind their antigens.Citation10 The observation that the ATTACK induces the formation of the IS similarly to the BiTE-like LiTE may be evidence of comparable flexibility and compressibility. More rigid or bulkier T-bsAbs might then show a drop-off in performance, especially as molecules with bulky extracellular domains, such as the tyrosine phosphatase CD45, have been shown to be excluded from the IS.Citation28,29 EgA1 ATTACK is able to promote the IS formation and induce early signaling pathways downstream of TCR engagement at lower concentrations than EgA1 LiTE. This may be due to its lower KD allowing a greater saturation of target cell surface EGFR even at low picomolar concentrations. These findings are supported by measuring the upregulation of activation markers, cytokine secretion, and redirected lysis of EGFR-expressing tumor cells. Under identical assay conditions, the ATTACK had a 15- to 20-fold higher potency than the LiTE when redirecting T cells to lyse of EGFR-expressing tumor cells and reached close to 80% target cell lysis, compared to the 60% lysis reached by the LiTE. We found that T cell activation and redirection towards tumor cells mediated by EgA1 LiTE and EgA1 ATTACK was entirely dependent on the expression of EGFR on target cells.
Previous results from our group using scFv-based N-terminal trimerbodies demonstrated the tumor targeting potential of TAA-specific trivalent molecules with a similar molecular weight (110 kDa).Citation24,30 These studies showed that trivalent binding of tumor-related antigens conveyed a massive improvement in tumor accumulation and retention, as compared to monovalent binders of the same antigens, and are likely to be pertinent when comparing the ATTACK with monovalent T-bsAbs. Importantly, the ATTACK format achieves TAA trivalence without acquiring an excessive MW, with a size of ≈100 kDa as compared to the ≈55 kDa of a BiTE or the >150 kDa of an Ig-based T-bsAb. This intermediate size is expected to convey an intermediate rate of diffusion, and correspondingly, tumor penetration. Additionally, ATTACK molecules are larger than the cut-off size for glomerular filtration, which is a clear pharmacokinetic advantage over the smaller BiTE and DARTs, as they may then be suitable for bolus therapy regimes instead of continuous infusion therapy.Citation8 The tumor targeting and the greater efficiency at low doses of the ATTACK molecules for TAA-specific T cell activation and redirection may allow lower doses to be used in future treatment regimens, thereby reducing the general toxicity of the therapy.
In this project, we used EGFR as the targeted TAA, which has been useful due to the availability of recombinant EGFR proteins and EGFR-expressing cell lines. However, EGFR may be a non-ideal target for clinical T cell redirection, as the widespread expression of EGFR in normal tissuesCitation31 can lead to considerable off-tumor toxicity, as documented for both cetuximabCitation32 and EGFR-targeting BiTEs.Citation33 However, in lieu of more tumor-specific antigens, strategies must be found to minimize these toxicities. One approach is inspired by physiological systems where multiple low-affinity interactions are used to distinguish one cell type from another on the basis of receptor density.Citation34 In a similar way, multivalent antibodies with low affinity of individual binding domains may be able to distinguish between normal cells and tumor cells over-expressing a TAA. In fact, this has been demonstrated using an EGFR-binding IgG with a low Fab affinity.Citation35 Based on these results, an ATTACK displaying a low affinity anti-EGFR VHH (KD > 10−8 M) should be able to discriminate between cell surfaces expressing different levels of EGFR. Alternatively, ATTACKs could be generated to target antigens which are more tumor-specific, or expressed only by non-essential cells, such as markers of hematopoietic lineage or receptors like CEA or PSMA,Citation36 although these antibodies would be applicable to a narrower range of cancers.
In summary, the current data show that the 3 + 1 ATTACK is an effective bsAb for retargeting human T cells towards EGFR-expressing cells in vitro, with a potency over 15-fold higher than that of the 1 + 1 LiTE. Despite its complexity, the ATTACK is not prone to lowered expression, stability, or solubility, and its modularity enables the design of combination immunotherapies by a single agent in the future. Synergies between immune checkpoint blockade and T cell redirection strategies are especially intriguing.
Materials and methods
Cells and culture conditions
HEK-293 (CRL-1573), HeLa cells (CCL-2), A431 (CRL-1555) and NIH/3T3 (CRL-1568) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Lonza, Walkersville, MD, USA) supplemented with 2 mM L-glutamine, 10% (vol/vol) heat inactivated Fetal Calf Serum (FCS), and antibiotics (100 units/mL penicillin, 100 μg/mL streptomycin) (all from Life Technologies, Carlsbad, CA, USA) referred as to DMEM complete medium (DCM), unless otherwise stated. Jurkat clone E6–1 cells (TIB-152) were maintained in RPMI-1640 (Lonza) supplemented with 2 mM L-glutamine, heat-inactivated 10% FCS, and antibiotics, referred as to RPMI complete medium (RCM). All of these cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA). The human Jurkat J77 cell lineCitation37 was maintained in RCM. 3T3, HeLa and A431 cells were infected with lentivirus encoding the firefly luciferase (Luc) geneCitation38,39 has been described previously.Citation40 The 3T3 cells stably expressing human EGFR (3T3-EGFR) were kindly provided by Antonio Villalobo (Instituto de Investigaciones Biomédicas Albert Sols, CSIC-UAM, Madrid, Spain) and cultured in DCM. All cell lines were routinely screened for the absence of mycoplasma contamination by PCR using the Mycoplasma Plus TM Primer Set (Stratagene, Cedar Creek, TX, USA).
Construction of expression vectors
The plasmid encoding the multichain anti-EGFR EgA1 VHH-based N-trimerbody (EgA1N) has been previously described.Citation16 To generate the anti-EGFR EgA1 VHH-based tandem trimerbody expression vector a synthetic gene containing two EgA1 VHH antibodies fused at its 5´-end by a 17-mer linker to a human TIEXVIII domain, and at its 3´-end to a 7-mer linker to a human TIEXVIII domain (17EgA17TIE17EgA17TIE) was synthesized by Geneart AG (Regensburg, Germany). The NotI /BamHI cleaved fragment was ligated to into pCR3.1-EgA1-TIECitation7 to obtain the plasmid pCR3.1-scEgA1-TIE. To generate the anti-EGFR x anti-CD3 ATTACK expression vector the BamHI/XbaI fragment from plasmid pCR3.1-hNC1XVIII-OKT326 was ligated into the BamHI/XbaI digested backbone of plasmid pCR3.1-scEgA1-TIE, to obtain the plasmid pCR3.1-scEgA1-TIE-OKT3. To create the anti-EGFR x anti-CD3 VHH-scFv tandem expression plasmid pCR3.1-EgA1-(G4S)-OKT3 the DNA fragment encoding the anti-EGFR VHH EgA1 was ClaI/NotI digested from pCR3.1-EgA1-TIECitation7,16 and ligated into the ClaI/NotI digested backbone of previously described plasmid pCR3.1-MFE23-(G4S)-OKT340. The sequences were verified using primers FwCMV (SEQ ID NO: 31: 5′-CGCAAATGGGCGGTAGGCGTG-3′) and RvBGH (SEQ ID NO: 32: 5′-TAGAAGGCACAGTCGAGG-3′).
Expression and purification of recombinant antibodies
EgA1VHH was produced as described beforeCitation41 but with minor modifications. Briefly, E.coli BL-21 Codon Plus (DE3)-RIL (Agilent Technologies Inc., Santa Clara, CA, USA) cells were transformed with EgA1-myc-his-pAX51 and a single colony was grown overnight at 37°C in 2xTY medium containing 2% (w/v) glucose and 100 µg/ml ampicillin. Protein production was started by inoculating bacteria from the overnight culture in 2xTY medium supplemented with 100 µg/ml ampicillin and 0.2% (w/v) glucose till an OD600 of 0.1, then IPTG was added to obtain a final concentration of 1 mM and growth continued overnight at 25 °C. Periplasmic fractions were prepared by resuspending the pellet in ice cold TES buffer (200 mM Tris-HCl pH8.0, 0.5 mM EDTA, 500 mM sucrose), and the bacteria centrifuged and the supernatant collected. The pellet was resuspended in precooled TES 5 mM MgSO4, incubated on ice for 30 min and centrifuged. The two fractions were pooled, and the EgA1 VHH purified by means of immobilized metal ion affinity chromatography (IMAC) on TALON resin (Clontech, Palo Alto, CA, USA) according to the manufacturer's protocol.
HEK-293 cells were transfected with the appropriate expression vectors using calcium phosphateCitation42 and selected in DCM with 500 μg/ml G-418 (Sigma-Aldrich, St. Louis, MO, USA) to generate stable cell lines. Supernatants from transiently and stably transfected cell populations were analyzed by western blotting and FACS. The EgA1N, EgA1 ATTACK and the EgA1 LiTE were purified in a two-step process from DCM conditioned medium collected from stably transfected HEK293 cells. Collected medium was centrifuged and filtered. Supernatant were first purified 500 mL at a time using a 1 mL HiTrap Excel column (GE Healthcare, Uppsala, Sweden), washing with 30 column volume (CV) of 20 mM imidazole in PBS (pH 7.4) and eluting with 500 mM of imidazole in PBS. Elution fractions containing the relevant proteins were then pooled, diluted 10x in PBS, and further purified on a 1 mL HiTrap Protein A Hp column (GE Healthcare), washing with 45 CV of PBS and eluting with 0.1 M citric acid (pH 2.7). The resulting fractions were pooled, dialyzed against PBS, and concentrated using spintrap columns (Merck Millipore, Billerica, MA, USA).
Western blotting
Samples were separated under reducing conditions on 12% Tris-glycine gels and transferred to nitrocellulose membranes (Life Technologies) and probed with anti-c-myc mAb (clone 9E10 (Abcam, Cambridge, UK), followed by incubation with an IRDye800-conjugated donkey anti-mouse IgG (H&L) (Rockland Immunochemicals, Gilbertsville, PA, USA). Visualization and quantitative analysis of protein bands were carried out with the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Preparation of cetuximab and OKT3 Fab fragments
The mouse anti-human CD3ϵ OKT3 mAb was from Ortho Biotech (Bridgewater, NJ, USA), and the chimeric mouse/human anti-human EGFR cetuximab was from Merck KGaA (Darmstadt, Germany). The Fab fragments were prepared using a Pierce Fab Micro Preparation Kit (Thermo Scientific, Rockford, IL, USA) following the manufacturer's protocol. Briefly, the mAbs were cleaved by papain, and the resulting Fc regions, as well as uncleaved mAbs, were removed using a protein A column. The resulting cetuximab-Fab and OKT3-Fab were then verified by SDS-PAGE, dialyzed against PBS, and concentrated on spintrap columns.
Size exclusion chromatography-multiangle light scattering (SEC-MALS)
Static light scattering experiments were performed at 25°C using a Superdex 200 10/300 GL column (GE HealthCare) attached in-line to a DAWN-HELEOS light scattering detector and an Optilab rEX differential refractive index detector (Wyatt Technology, Santa Barbara, CA, USA). The column was equilibrated with running buffer (PBS 0.1 μm filtered) and the SEC-MALS system was calibrated with a sample of BSA at 1 g/L in the same buffer. Then 100 μL samples of the EgA1 ATTACK at 0.6 g/L in PBS were injected into the column at a flow rate of 0.5 mL/min. Data acquisition and analysis were performed using ASTRA software (Wyatt Technology). Based on numerous measurements on BSA samples at 1 g/L under the same or similar conditions we estimate that the experimental error in the molar mass is around 5%.
Circular Dichroism (CD)
Circular dichroism measurements were performed with a Jasco J-810 spectropolarimeter (JASCO, Tokyo, Japan). The spectra were recorded on protein samples at 0.08 g/L in PBS using 0.2 cm path length quartz cuvettes at 25°C. Thermal denaturation curves from 5 to 95°C were recorded on the same protein samples and cuvette by increasing temperature at a rate of 1°C/minute and measuring the change in ellipticity at 210 nm.
Molecular modeling
The structural modeling of LiTE and ATTACK antibodies and comparative analysis between their space sampling were performed thought comparative homology modeling with MODELLER.Citation43 In both molecules, the structure of a homo-specific diabody (pdb:5GS1)Citation44 was used a template for the anti-EFGR EgA1 VHH domainCitation45 [blast e-value of 3e-81 and a 54% of sequence identity], while the anti-CD3ϵ OKT3 scFv domain was modeled after the MFL-23 Recombinant Ab Fragment (pdb:1QOK)Citation46 (blast e-value of 1e-107 and 75% sequence identity). For ATTACK, the structure of human collagen XVIII trimerization domain (pdb:3HSH)Citation47 was used as template for the trimerization domain as described in previous works.Citation16 To mimic the putative repositioning of the different Ig domains according to their flexible linkers, constraints in the linking regions were removed. Thus, for each construct, a total of 150 structures where generated displaying the internal flexibility of their respective system. The surface area of LITE and ATTACK where approximated through Knud Thomsen´s formula for general ellipsoids with a relative error of 1.061%. The surface area calculated through this method do not represent the per-atom exposed surface area but the simplification of the global shape of that area.
Biolayer interferometry
The kinetic rate constants for the interactions between the human EGFR-Fc chimera (R&D Systems, Minneapolis, USA) immobilized on the surface of biosensors and a panel of antibodies (cetuximab, cetuximab-Fab, EgA1 VHH, EgA1N and EgA1 ATTACK) were determined using biolayer interferometry. Biosensors used for experiments with cetuximab, cetuximab-Fab, EgA1N and EgA1 ATTACK were prepared using amine reactive coupling. Briefly, AR2G biosensors (Fortebio, Menlo Park, CA, USA) were activated with EDC and s-NHS, loaded for 30 minutes with 3 µg/mL EGFR-Fc in a 10 mM acetate buffer at pH 5, and quenched with ethanolamine. Anti-human Fc (hFc) capture biosensors (Fortebio), loaded with 10 nM of EGFR-Fc in kinetics buffer (PBS with 0.1% BSA and 0.05% Tween20) for 45 minutes, were used for experiments with the EgA1 VHH (which was not detectable using AR2G biosensors) and cetuximab-Fab (included as a means of comparison to verify that the anti-hFc biosensors performed similarly to the AR2G biosensors, which they did). Each kinetics experiment included the analyte antibody in kinetics buffer at two different concentrations, as well as a reference biosensor, loaded with EGFR-Fc, which was kept in kinetics buffer without antibody. All antibodies except EgA1 VHH were included at 4 and 2 nM, EgA1 VHH was included at 10 and 4 nM. Kinetic rate constants were determined via fitting to a 1:1 model after subtraction of the reference using the Octet Data Analysis software.
Flow cytometry
The ability of EgA1 LiTE and EgA1 ATTACK to bind to cell surface EGFR and CD3 was studied by flow cytometry as described previously.Citation16 Briefly, 3T3, HeLa or Jurkat cells were incubated with purified antibodies (5 μg/ml) and anti-c-myc mAb for 30 min. After washing, the cells were treated with appropriate dilutions of phycoerytrin (PE)-conjugated goat F(ab′)2 fragment anti-mouse IgG (Fc specific; Jackson Immuno Research, Newmarket, UK). OKT3 and cetuximab mAbs were used as controls on FACS studies, using appropriate dilutions of PE-conjugated goat F(ab′)2 fragment anti-mouse IgG and PE-conjugated goat F(ab′)2 anti-human IgG (Abcam), respectively. The samples were analyzed with a MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec). Flow cytometry was used to determine the saturation of EGFR on the surface of HeLa cells by antibodies at 0.1, 0.32, 1, and 10 nM, allowing the distinction of antibodies binding with a low picomolar or a nanomolar KD. HeLa cells (2 × 105) were incubated for 2 hours in a volume of 5 mL containing FACS buffer (PBS with 0.1% sodium azide and 1% FCS) with 0.1, 0.32, 1, or 10 nM of cetuximab-IgG, cetuximab-Fab, EgA1 LiTE, or EgA1 ATTACK, or FACS buffer only. The cells were then immediately fixed with 1% formaline in PBS for 15 minutes, in order to minimize dissociation of antibodies during the following detection steps. Cell-bound antibodies were then detected with anti-human kappa chain mAb (clone SB81a; Abcam) or anti-(H)5 mAb (penta-His; Qiagen, Hilden, Germany), as appropriate, followed by PE-conjugated goat F(ab’)2 fragment anti-mouse IgG. To measure CD3 saturation on the surface of Jurkat cells by OKT3, OKT3 Fab, EgA1 LiTE, and EgA1 ATTACK at 0.1, 1, 3.2, and 10 nM, the cells (2 × 105) were kept for 2 hours in 1 mL of analyte antibody solution. The cells were then fixed with 1% formaline in PBS for 15 minutes. Cell-bound antibodies were then detected with rabbit anti-mouse IgG H+L (Jackson Immuno Research) followed by PE-conjugated donkey F(ab’)2 fragment anti-rabbit IgG (H+L) (Abcam), or penta-His mAb followed by PE-conjugated goat F(ab’)2 fragment anti-mouse IgG.
The samples were analyzed using a Cell Sorter SH800 (Sony, Tokyo, Japan). The mean fluorescence intensities (MFIs) from each sample were normalized by subtracting the MFI of the appropriate negative control (that is, cells incubated only with FACS buffer, fixed, then detected with the same antibodies) and dividing by the MFI of the antibody at 10 nM minus the MFI of the negative control:
Inhibition of EGFR-mediated cell proliferation
A431 cells were seeded at a density of 2,000 cells/well in 96-well plates in DCM. After 24 hours, medium was replaced by DMEM supplemented with 1% FCS containing equimolar concentrations (0.19–50 nM) of purified antibodies: cetuximab, OKT3, EgA1 LiTE or EgA1 ATTACK. Cell were then incubated for 72 hours in humidified 5% CO2 atmosphere at 37°C, and cell viability was assessed using the CellTiter-Glo luminescent assay (Promega, Madison, USA). Bioluminescence was measured using a Tecan Infinite F200 plate-reading luminometer (Tecan Trading AG, Switzerland). Experiments were performed in triplicates.
Inhibition of EGFR signaling
A431 cells were maintained in DCM for 24 hours and prior to antibody treatment, cells were starved for 18 hours DMEM supplemented with 1% FCS. Before growth factor stimulation, cells were incubated for 4 hours in serum-free DMEM in presence of equimolar concentrations (100 nM) of cetuximab, OKT3, EgA1 LiTE or EgA1 ATTACK, followed by incubation with 25 ng/mL of human EGF (Miltenyi Biotec, Bergisch Gladbach, Germany) for 5 minutes. After stimulation, cells were lysed in Laemmli-lysis buffer (Bio-Rad, Hercules, CA, USA) for 10 minutes on ice and collected by scraping. Samples were separated under reducing conditions on 4–12% Tris-glycine gels and transferred to nitrocellulose membrane using iBlot Dry Blotting System (Life Technologies). Membranes were incubated overnight with a rabbit anti-human phospho EGFR (Tyr1068) mAb (clone D7A5, Cell Signaling Technology Inc., Danvers, MA, USA) followed by incubation with an IRDye800 conjugated donkey anti-rabbit antibody (Rockland Immunochemicals). Simultaneously, anti β-actin mouse mAb (clone mAbcam 8226; Abcam) was added as a loading control, followed by IRDye700-conjugated donkey anti-mouse antibody (Rockland Immunochemicals). Visualization and quantitative analysis of protein bands were carried out with the Odyssey infrared imaging system.
Immunological synapse formation and polarized PLCγ signalling
For imaging, 3T3 and 3T3-EGFR cells were fluorescently labeled with 10 µM cell tracker dye 7-amino-4-chloromethylcoumarin (CMAC; Life Technologies) for 30 minutes and incubated with purified EgA1 LiTE or EgA1 ATTACK at concentrations ranging from 50 pM to 5 nM for one hour. After thoroughly washing, an equal number of Jurkat J77 cells and 3T3 or 3T3-EGFR cells were co-incubated for 20 minutes on poly-L-lysine-coated coverslips at 37°C in a humidified atmosphere with 5% CO2. Cell mixtures were then fixed with 4% paraformaldehyde (PFA) for 5 minutes at room temperature. Specimens were permeabilized with TBS-Triton 0.1% for 5 minuntes at room temperature and blocked with 10 µg/ml human gamma globulin (Sigma-Aldrich) in TBS with 0,5% blocking reagent (Roche Diagnostics, Mannheim, Germany) (TNB) for 30 minuntes at room temperature. Samples were stained with rabbit anti-human CD247 (CD3ζ) antiserum (kindly provided by Balbino Alarcón, Centro de Biología Molecular Severo Ochoa, UAM-CSIC, Madrid, SpainCitation48 or rabbit anti-human phospho-PLCγ1 (Tyr783) antibody (Cell Signaling Technology Inc.) in TNB for 1 hour at room temperature. Cells were then washed with TBS and incubated with Alexa Fluor 488 conjugated donkey anti-rabbit antibody (Life Techonologies) and Phalloidin-Alexa-568 (Thermo Scientific) for 30 minuntes at room temperature. Samples were analyzed by using a FV1200 confocal microscope (Olympus, Tokyo, Japan). Polarization towards the IS was quantified by using Image J software (National Institutes of Health) as previously described.Citation49
TCR early signalling
3T3-EGFR cells were incubated with purified EgA1 LiTE or EgA1 ATTACK at concentrations ranging from 5 pM to 5 nM) for one hour. After thoroughly washing, Jurkat J77 cells were mixed with 3T3-EGFR cells at 10:1 ratio for 20 min at 37°C in a humidified atmosphere with 5% CO2. Cell mixtures were lysated for 40 minutes on ice with lysis buffer (Tris HCl pH 7.4 20 mM, Triton X-100 0.2%, NP40 1%, NaCl 150 mM, MgCl2 1.5 mM, EDTA 2 mM) containing 1X protease inhibitors cocktail (Roche Diagnostics), 1 mM PMSF and phosphatase inhibitors (5 mM b-glycerolphosphate, 1 mM sodium pyrophosphate, 10 mM sodium fluoride, 2 mM sodium ortovanadate). Extracts were preclared, runned in 13% SDS-PAGE and transferred to PVDF membranes (Merck Millipore). The membranes were blocked with 5% BSA and 0.2% Tween-20 in TBS for 1 hour and incubated with unconjugated rabbit anti-human phospho-PLCγ1 (Tyr783) and rabbit anti-human phospho-p44/42 MAPK (ERK1/2, Thr202/Tyr204) antibodies (both from Cell Signaling Technology Inc.) overnight at 4°C. Secondary reagents donkey anti-rabbit IgG IRDye800 (LI-COR Biosciences) or HRP-conjugated goat anti-rabbit IgG (Merck Millipore) were used at room temperature for 1 hour. For total PLCγ membranes were stripped in NewBlot PVDF Stripping Buffer (LI-COR Biosciences) for 20 minutes at room tempearture. Membranes were incubated with primary antibodies rabbit anti-human PLCγ1 (Cell Signaling Technology Inc.), and mouse anti-human p44/42 MAPK (ERK1/2) (Cell Signaling Technology Inc.) followed by secondary reagents goat anti-mouse IgG 680 (LI-COR Biosciences) or HRP-conjugated goat anti-rabbit IgG as before. WB were developed by directly reading fluorescence or by reading quimioluminiscence after using ECL reagents (Thermo Scientific) in a Odyssey infrared imaging system. Data of phospho-proteins were normalized to the total and then, the value of unstimulated samples was subtracted. Normalized data was compared using the Student's t-test.
T cell activation assays
HeLa or 3T3 cells were plated in triplicated in 96-well microtiter plates at 4 × 104/well one day before the assay. Human peripheral blood mononuclear cells (PBMCs), isolated from the buffy coat fraction of healthy volunteers' peripheral blood by density-gradient centrifugation, or Jurkat E6–1 cells were added in 5:1 effector:target (E:T) ratio on HeLaor 3T3 cells in the presence of purified EgA1 LiTE or EgA1 ATTACK antibodies. After 24 hours aliquots of the culture supernatants were harvested and the levels of IL-2 were measured by using a commercially available ELISA test kit (Diaclone, Besançon, France). Determination of CD69 expression was performed after 24 hours incubation using PE-conjugated anti-CD69 mAb (clone FN50, BD Biosciences, San José, CA, USA) and FITC-conjugated anti-CD3 mAb (clone UCHT1, Abcam) by flow cytometry as described.Citation4 The samples were analyzed with a Beckman-Coulter FC-500 Analyzer.
Cytotoxicity assay
Gene-modified luciferase expressing HeLa (HeLaLuc), A431 (A431Luc) and 3T3 cells (3T3Luc) were plated in triplicate in 96-well microtiter plates at 4 × 104/well one day before the assay. Human PBMCs cells were added in 5:1 E:T ratio on luciferase-expressing target cells in the presence of purified EgA1 LiTE or EgA1 ATTACK antibodies. After 48 hours, viability was measured adding D-luciferin (Promega) to a final concentration of 20 μg/ml. A 100% lysis control was included by treating the target cells with 1% Triton-X100 (Sigma-Aldrich), and the value for spontaneous lysis was obtained by incubating the target cells with effector cells only (“spont lysis”). Percent tumor cell viability was calculated as the mean bioluminescence of each sample divided by the mean bioluminescence of the input number of control target cells times 100. Percent cell viability was then plotted against the effector molecule concentration and data were evaluated using Prism 5 (GraphPad Software, San Diego, CA, USA) by fitting a sigmoidal dose-response (three parameter equation).
Statistics
Results were expressed as mean ± S.D. Data were evaluated using the Student's t-test. Statistical analysis was performed using Prism (GraphPad Software) and differences were considered statistically significant when p<0.05.
Conflict of interest
A.A-C. and M.C. are current or former employees of Leadartis SL. No other potential conflict of interest relevant to this article was reported.
Suppl_information_15092017.pdf
Download PDF (6.2 MB)Acknowledgments
J.B. was supported from the ‘EPFL Fellows’ fellowship program co-funded by Marie Skłodowska-Curie, Horizon 2020 (665667). L.S. was supported by grants from the Fondo de Investigación Sanitaria/Instituto de Salud Carlos III (PI13/00090), co-funded by FEDER funds, and the Comunidad de Madrid (S2010/BMD-2312). F.J.B. was supported by the Spanish Ministry of Economy and Competitiveness (CTQ2014-56966-R). PR-N was funded by the Spanish Ministry of Economy and Competitiveness (SAF2016-75656-P), co-funded by FEDER funds. L.A-V. was supported by grants from the Danish Council for Independent Research, Medical Sciences (DFF-6110-00533), and the Novo Nordisk Foundation (NNF14OC0011019).
References
- Alvarez-Vallina L. Genetic approaches for antigen-selective cell therapy. Curr Gene Ther. 2001;1(4):385-397. https://doi.org/10.2174/1566523013348418. PMID:12109064
- Sanz L, Blanco B, Álvarez-Vallina L. Antibodies and gene therapy: teaching old ‘magic bullets’ new tricks. Trends Immunol. 2004;25:85-91. https://doi.org/10.1016/j.it.2003.12.001. PMID:15102367
- Sebastian M, Passlick B, Friccius-Quecke H, Jäger M, Lindhofer H, Kanniess F, Wiewrodt R, Thiel E, Buhl R, Schmittel A. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM x anti-CD3): A phase I study. Cancer Immunol Immunother. 2007;56:1637-1644. https://doi.org/10.1007/s00262-007-0310-7. PMID:17410361
- Linke R, Klein A, Seimetz D. Catumaxomab: Clinical development and future directions. MAbs. 2010;2:129-136. https://doi.org/10.4161/mabs.2.2.11221. PMID:20190561
- Mack M, Riethmuller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:7021-7025. https://doi.org/10.1073/pnas.92.15.7021. PMID:7624362
- Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W, Zhang W, Tuaillon N, Rainey J, Barat B et al. Effector cell recruitment with novel fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. 2010;399:436-449. https://doi.org/10.1016/j.jmb.2010.04.001. PMID:20382161
- Nunez-Prado N, Compte M, Harwood S, Álvarez-Méndez A, Lykkemark S, Sanz L, Álvarez-Vallina L. The coming of age of engineered multivalent antibodies. Drug Discov Today. 2015;20:588-94. https://doi.org/10.1016/j.drudis.2015.02.013. PMID:25757598
- Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, Gökbuget N, Neumann S, Goebeler M, Viardot A et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226-6233. https://doi.org/10.1182/blood-2012-01-400515. PMID:22592608
- Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I, Eser M, McAleese F, Molkenthin V, Gall FL et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs. 2015;7:584-604. https://doi.org/10.1080/19420862.2015.1029216. PMID:25875246
- Molhoj M, Crommer S, Brischwein K, Rau D, Sriskandarajah M, Hoffmann P, Kufer P, Hofmeister R, Baeuerle PA et al. CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis. Mol Immunol. 2007;44:1935-1943. https://doi.org/10.1016/j.molimm.2006.09.032. PMID:17083975
- Chatenoud L, Ferran C, Reuter A, Legendre C, Gevaert Y, Kreis H, Franchimont P, Bach JF. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected. N Engl J Med. 1989;320:1420-1421. https://doi.org/10.1056/NEJM198905253202117. PMID:2785642
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017 9:182-212. https://doi.org/10.1080/19420862.2016.1268307. PMID:28071970
- Rossi DL, Rossi EA, Cardillo TM, Goldenberg DM, Chang CH. A new class of bispecific antibodies to redirect T cells for cancer immunotherapy. MAbs. 2014;6:381-391.. https://doi.org/10.4161/mabs.27385. PMID:24492297
- Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22:3286-3297. https://doi.org/10.1158/1078-0432.CCR-15-1696. PMID:26861458
- Baker JH, Lindquist KE, Huxham LA, Kyle AH, Sy JT, Minchinton AI. Direct visualization of heterogeneous extravascular distribution of trastuzumab in human epidermal growth factor receptor type 2 overexpressing xenografts. Clin Cancer Res. 2008;14:2171-2179. https://doi.org/10.1158/1078-0432.CCR-07-4465. PMID:18381959
- Alvarez-Cienfuegos A, Nuñez-Prado N, Compte M, Cuesta AM, Blanco-Toribio A, Harwood SL, Villate M, Merino N, Bonet J, Navarro R et al. Intramolecular trimerization, a novel strategy for making multispecific antibodies with controlled orientation of the antigen binding domains. Sci Rep. 2016;6:28643. https://doi.org/10.1038/srep28643. PMID:27345490
- Schmitz KR, Bagchi A, Roovers RC, van Bergen en Henegouwen PM, Ferguson KM. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure. 2013;21:1214-1224. https://doi.org/10.1016/j.str.2013.05.008. PMID:23791944
- Kung P, Goldstein G, Reinherz EL, Schlossman SF. Monoclonal antibodies defining distinctive human T cell surface antigens. Science. 1979;206:347-349. https://doi.org/10.1126/science.314668. PMID:314668
- Kjer-Nielsen L, Dunstone MA, Kostenko L, Ely LK, Beddoe T, Mifsud NA, Purcell AW, Brooks AG, McCluskey J, Rossjohn J. Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc Natl Acad Sci U S A. 2004;101:7675-7680. https://doi.org/10.1073/pnas.0402295101. PMID:15136729
- Roovers RC, Laeremans T, Huang L, De Taeye S, Verkleij AJ, Revets H, de Haard HJ, van Bergen en Henegouwen PM. Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR nanobodies. Cancer Immunol Immunother. 2007;56(3):303-317. https://doi.org/10.1007/s00262-006-0180-4. PMID:16738850
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301-311. https://doi.org/10.1016/j.ccr.2005.03.003. PMID:15837620
- Merlino GT, Xu YH, Ishii S, Clark AJ, Semba K, Toyoshima K, Yamamoto T, Pastan I. Amplification and enhanced expression of the epidermal growth factor receptor gene in A431 human carcinoma cells. Science. 1984;224:417-419. https://doi.org/10.1126/science.6200934. PMID:6200934
- Batzer AG, Rotin D, Urena JM, Skolnik EY, Schlessinger J. Hierarchy of binding sites for Grb2 and shc on the epidermal growth factor receptor. Mol Cell Biol. 1994;14(8):5192-5201. https://doi.org/10.1128/MCB.14.8.5192. PMID:7518560
- Cuesta AM, Sánchez-Martín D, Sanz L, Bonet J, Compte M, Kremer L, Blanco FJ, Oliva B, Alvarez-Vallina L. In vivo tumor targeting and imaging with engineered trivalent antibody fragments containing collagen-derived sequences. PLoS One. 2009;4(4):e5381. https://doi.org/10.1371/journal.pone.0005381. PMID:19401768
- Cuesta AM, Sánchez-Martín D, Blanco-Toribio A, Villate M, Enciso-Álvarez K, Álvarez-Cienfuegos A, Sainz-Pastor N, Sanz L, Blanco FJ, Alvarez-Vallina L. Improved stability of multivalent antibodies containing the human collagen XV trimerization domain. MAbs. 2012;4:226-232. https://doi.org/10.4161/mabs.4.2.19140. PMID:22453098
- Blanco-Toribio A, Sainz-Pastor N, Álvarez-Cienfuegos A, Merino N, Cuesta ÁM, Sánchez-Martín D, Bonet J, Santos-Valle P, Sanz L, Oliva B et al. Generation and characterization of monospecific and bispecific hexavalent trimerbodies. MAbs. 2013;5(1):70-79. https://doi.org/10.4161/mabs.22698. PMID:23221741
- Fan Z, Lu Y, Wu X, Mendelsohn J. Antibody-induced epidermal growth factor receptor dimerization mediates inhibition of autocrine proliferation of A431 squamous carcinoma cells. J Biol Chem. 1994;269:27595-27602. PMID:7961676
- Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15:751-761. https://doi.org/10.1016/S1074-7613(01)00234-5. PMID:11728337
- Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43:763-771. https://doi.org/10.1016/j.molimm.2005.03.007. PMID:16360021
- Sanchez-Martin D, Cuesta AM, Fogal V, Ruoslahti E, Alvarez-Vallina L. The multicompartmental p32/gClqR as a new target for antibody-based tumor targeting strategies. J Biol Chem. 2011;286:5197-5203. https://doi.org/10.1074/jbc.M110.161927. PMID:21156793
- Yano S, et al. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res. 2003;23:3639-3650. PMID:14666659
- Ballestrero A, Garuti A, Cirmena G, Rocco I, Palermo C, Nencioni A, Scabini S, Zoppoli G, Parodi S, Patrone F. Patient-tailored treatments with anti-EGFR monoclonal antibodies in advanced colorectal cancer: KRAS and beyond. Curr Cancer Drug Targets. 2012;12:316-328. https://doi.org/10.2174/156800912800190956. PMID:22385512
- Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B, Friedrich M, Thomas O, Lorenczewski G, Rau D et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A. 2019;107:12605-12610. https://doi.org/10.1073/pnas.1000976107
- Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4:696-703. https://doi.org/10.1016/S1367-5931(00)00153-8. PMID:11102876
- Garrido G, Tikhomirov IA, Rabasa A, Yang E, Gracia E, Iznaga N, Fernández LE, Crombet T, Kerbel RS, Pérez R. Bivalent binding by intermediate affinity of nimotuzumab: A contribution to explain antibody clinical profile. Cancer Biol Ther. 2011;11:373-382. https://doi.org/10.4161/cbt.11.4.14097. PMID:21150278
- Sanchez-Martin D, Sørensen MD, Lykkemark S, Sanz L, Kristensen P, Ruoslahti E, Álvarez-Vallina L. Selection strategies for anticancer antibody discovery: Searching off the beaten path. Trends Biotechnol. 2015;3:292-301. https://doi.org/10.1016/j.tibtech.2015.02.008
- Niedergang F, Hémar A, Hewitt CR, Owen MJ, Dautry-Varsat A, Alcover A. The staphylococcus aureus enterotoxin B superantigen induces specific T cell receptor down-regulation by increasing its internalization. J Biol Chem. 1995;270:12839-12845. https://doi.org/10.1074/jbc.270.21.12839. PMID:7759540
- Sanz L, et al. Long-term in vivo imaging of human angiogenesis: Critical role of bone marrow-derived mesenchymal stem cells for the generation of durable blood vessels. Microvasc Res. 2008;75(3):308-314. https://doi.org/10.1016/j.mvr.2007.11.007. PMID:18252255
- Compte M, Cuesta AM, Sánchez-Martín D, Alonso- Camino V, Vicario JL, Sanz L, Alvarez-Vallina L. Tumor immunotherapy using gene-modified human mesenchymal stem cells loaded into synthetic extracellular matrix scaffolds. Stem Cells. 2009;27:753-760. https://doi.org/10.1634/stemcells.2008-0831. PMID:19096041
- Compte M, Alvarez-Cienfuegos A, Nuñez-Prado N, Sainz-Pastor N, Blanco-Toribio A, Pescador N, Sanz L, Alvarez-Vallina L. Functional comparison of single-chain and two-chain anti-CD3-based bispecific antibodies in gene immunotherapy applications. Oncoimmunology. 2014;3:e28810. https://doi.org/10.4161/onci.28810. PMID:25057445
- Heukers R, Altintas I, Raghoenath S, De Zan E, Pepermans R, Roovers RC, Haselberg R, Hennink WE, Schiffelers RM, Kok RJ et al. Targeting hepatocyte growth factor receptor (met) positive tumor cells using internalizing nanobody-decorated albumin nanoparticles. Biomaterials. 2014;35:601-610. https://doi.org/10.1016/j.biomaterials.2013.10.001. PMID:24139763
- Compte M, Blanco B, Serrano F, Cuesta AM, Sanz L, Bernad A, Holliger P, Alvarez-Vallina L. Inhibition of tumor growth in vivo by in situ secretion of bispecific anti-CEA x anti-CD3 diabodies from lentivirally transduced human lymphocytes. Cancer Gene Ther. 2007;14:380-388. https://doi.org/10.1038/sj.cgt.7701021. PMID:17218946
- Webb B, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Protein Sci. 2016;86:2.9.1-2.9.37. https://doi.org/10.1002/cpps.20
- Kim JH, Song DH, Youn SJ, Kim JW, Cho G, Kim SC, Lee H, Jin MS, Lee JO. Crystal structures of mono- and bi-specific diabodies and reduction of their structural flexibility by introduction of disulfide bridges at the fv interface. Sci Rep. 2016;6:34515. https://doi.org/10.1038/srep34515. PMID:27682821
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:421-2105-10-421. https://doi.org/10.1186/1471-2105-10-421
- Boehm MK, Corper AL, Wan T, Sohi MK, Sutton BJ, Thornton JD, Keep PA, Chester KA, Begent RH, Perkins SJ. Crystal structure of the anti-(carcinoembryonic antigen) single-chain fv antibody MFE-23 and a model for antigen binding based on intermolecular contacts. Biochem J. 2000;346Pt(2):519-528. https://doi.org/10.1042/bj3460519. PMID:10677374
- Boudko SP, Sasaki T, Engel J, Lerch TF, Nix J, Chapman MS, Bächinger HP. Crystal structure of human collagen XVIII trimerization domain: A novel collagen trimerization fold. J Mol Biol. 2009;392:787-802. https://doi.org/10.1016/j.jmb.2009.07.057. PMID:19631658
- San Jose E, Sahuquillo AG, Bragado R, Alarcon B. Assembly of the TCR/CD3 complex: CD3 epsilon/delta and CD3 epsilon/gamma dimers associate indistinctly with both TCR alpha and TCR beta chains. evidence for a double TCR heterodimer model. Eur J Immunol. 1998;28:12-21. https://doi.org/10.1002/(SICI)1521-4141(199801)28:01%3c12::AID-IMMU12%3e3.0.CO;2-9. PMID:9485181
- Calabia-Linares C, Robles-Valero J, de la Fuente H, Perez-Martinez M, Martín-Cofreces N, Alfonso-Pérez M, Gutierrez-Vázquez C, Mittelbrunn M, Ibiza S et al. Endosomal clathrin drives actin accumulation at the immunological synapse. J Cell Sci. 2011;124:820-830. https://doi.org/10.1242/jcs.078832