
ABSTRACT
Since tumors are often infiltrated by macrophages, it would be advantageous to turn these types of cells into cytotoxic effector cells. Here, we have designed a novel bispecific antibody (BsAb) that targets both tumor antigen (CD20) and the FcαRI receptor (CD89). This antibody could be used to lyse tumors by connecting tumor cells to CD89-expressing immune effector cells such as macrophages and neutrophils. Previously there were very limited attempts to exploit FcαRI-expressing cells as effector cells for tumor cell-killing, largely due to the lack of an appropriate in vivo model, since mice do not express a human CD89 homolog. In this study, we used a transgenic mouse strain with specific expression of CD89 on macrophages and monocytes. In this transgenic mouse model, the CD89 bispecific antibody showed significant anti-tumor activities, demonstrating that bispecific antibodies can redirect macrophages, including M2 macrophages, to mediate additional effector function in the tumor microenvironment. This approach realized the full potential of the innate immune system and could be applied to other tumor-associated antigens especially the solid tumors, thus has potential to translate into clinical benefits in human cancers.
KEYWORDS:
Introduction
Antibodies have the potential to increase the specificity of cancer therapy by targeting selected tumor-associated antigens. Human IgG1 has been shown to effectively interact with human IgG Fc receptors, especially FcγRIIIa (CD16) involved in natural killer (NK) cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC), and activate human complement.Citation1 However, single-target antibodies do not seem to destroy cancer cells sufficiently in cancer therapy.
Bispecific antibodies (BsAbs) have been used as potential cancer therapeutic methods for decades.Citation2 The anti-tumor effects of BsAbs can be divided into direct effects, such as blocking growth factors, inhibition of proliferation, or induction of apoptosis,Citation3 and indirect effects involving recruitment of immune effector mechanisms such as cell-mediated lysis of target cells with immunoglobulin receptors on effector cells.Citation4 These mechanisms are used in the therapeutic concept of bispecific antibodies; antibodies are directed to cytotoxic trigger molecules such as effector cell receptors. A common strategy used to enhance specific tumor lysis is to design a bispecific antibody that binds a tumor cell surface antigen and T cell marker CD3 in order to recruit T cells as effector cells.Citation5 A well-established example of this type of design is the FDA-approved bispecific antibody blinatumomab. Bispecific antibodies engaging FcγRI or FcγRIII are currently being tested in clinical trials with promising results; these may help to overcome some of the above-mentioned limitations of single-target conventional antibody therapy.Citation6 Cytotoxic Fc receptors on effector cells can be triggered by antibodies reacting with Fc receptors via their variable regions.Citation7 However, many Fc receptor-expressing cells are not cytolytic for tumor cells (e.g., B cells and platelets) and some Fc receptor isoforms (e.g., FcγRIIb and FcγRIIIb) bind antibodies, but do not trigger cytolytic cascades.
FcαRI (CD89) is constitutively expressed on monocytes/macrophages, eosinophils, and neutrophils; however, importantly, it is not expressed on non-effector cell populations.Citation8 The known functions of FcαRI include ADCC, phagocytosis, induction of respiratory bursts, and inflammatory mediator or cytokine release.Citation9 Notably, FcαRI has been proven to be more efficient in triggering tumor cell killing than FcγRI. The capacity of FcαRI and FcγRI to initiate polymorphonuclear (PMN) cell signaling has been investigated. Cross-linking of FcαRI resulted in a more rapid induction of an increase in intracellular free calcium levels than cross-linking of FcγRI.Citation10
In this study, we aimed to design novel bispecific antibody to achieve an improved ADCC effect via CD89. Anti-FcαRI bispecific monoclonal antibodies (mAbs) have been previously explored as tools for antibody-based anti-cancer therapy. Targeting FcαRI has been shown to lead to efficient recruitment of neutrophils as effector cells in vitro in the presence of anti-(CD20 × FcαRI) BsAb.Citation9 Both neutrophils and macrophages efficiently induced ADCC and antibody-dependent phagocytosis of tumor cells in the presence of the antibody. However, in vivo evidence regarding the potential of FcαRI remains limited because mice do not express an FcαRI homolog. Therefore, we used a transgenic mouse strain with specific expression of CD89 on macrophages and monocytes. Macrophages are among the most abundant normal cells in the tumor microenvironment. However, substantial evidence indicates that macrophages showed protumoral characteristics in vivo rather than being tumoricidal.Citation11 These activities include suppression of T cell responses.Citation12,13 In addition, macrophages promote many important features of tumor progression including angiogenesis, tumor cell invasion, motility, and intravasation.Citation14
Our results indicate that the CD89 bispecific antibody shows significant anti-tumor activities in the generated CD89 transgenic mouse model, demonstrating that bispecific antibodies can redirect macrophages, including M2 macrophages, to mediate additional effector functions in the tumor microenvironment. Although bispecific antibodies targeting macrophages and tumors have yet to be demonstrated in clinical trials, this approach holds much promise.
Results
Generation of a heterodimeric one-arm bispecific CD89-CD20 antibody
The bispecific antibody targeting CD89 and CD20 generated in this study is based on a human IgG1 isotype with heavy chains comprised of a variable VH domain and three constant domains: CH1, CH2, and CH3. The antibody is composed of a single chain Fab fragment linked with a scFv that is attached to the C terminus of the heavy chain (). Heterodimerization of the two heavy chains in this novel format was achieved by knobs-into-holes technology. The knob chain (T366W) and hole chain (T366S, L368A, and Y407V) mutations were introduced into the CH3 domain.Citation15 The hole chain was constructed with a disulfide-stabilized scFv fused to the C terminus of the heavy chains by a 15-amino acid (G4S)3 linker.
Figure 1. Structural models of bispecific antibody formats generated in this study and SDS-PAGE analysis. (A) An illustrative representation of the initial antibody and final bispecific antibody format. The format is comprised of IgG-Fc linked to two different Fv domains (CD89/CD20) via 15-amino-acid (G4S)3 linkers. (B) SDS-PAGE analysis of the purification of the bispecific antibody proteins.
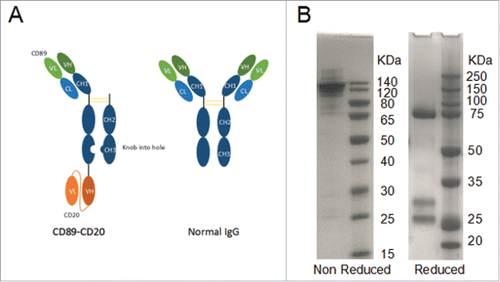
The bispecific antibody was produced by three plasmids co-transient expression in HEK293F cells. Co-transient expression and purification yielded the novel one-arm bispecific CD89-CD20 antibody (5 mg/liter). the antibody was purified to homogeneity by protein A from cell culture supernatants, as demonstrated by SDS-PAGE analysis under non-reducing and reducing conditions (). Reduced SDS-PAGE analysis of CD89-CD20 shows a 77-kDa hole heavy chain band, a 29.3-kDa knob heavy chain, and a 26-kDa light chain ( right).
Binding affinities of the CD89-CD20 bispecific molecule
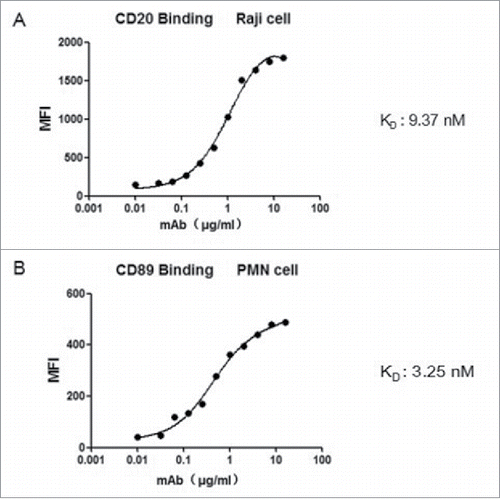
Flow cytometry analysis was used to test whether the bispecific antibody retained its ability to bind to the respective target antigens. The results showed that both components reacted with cells expressing CD89 or CD20, respectively( and ). The apparent affinities for the binding of the bispecific molecule to cell surface CD89 and CD20 were determined using FACS analysis. The bispecific antibody bound to CD20 and CD89 with a KD of 9.37 nM () and 3.25 nM (), respectively. Thus, both components Fab-scFv retained their ability to bind specifically to their target antigens when contained in the fusion antibody protein.
Figure 2. Apparent binding affinities of the Fv components of the bispecific antibody. Increasing concentrations of bispecific antibody were incubated with CD89-positive PMN cell. The binding of both antigens on the CD20 × CD89 molecule was assessed by flow cytometry using (A) A CD20-expressing cell line (Raji cells) and (B) CD89-expressing cell (PMNs). All data are presented as the mean ± SEM (n = 3) from one of three representative experiments.
The CD89-CD20 bispecific antibody mediates ADCC via human and Tg mouse effector cells in vitro
A non-radioactive cytotoxicity assay was performed to investigate tumor lysis mediated by the bispecific antibody in the presence of activated human PMNs and peripheral blood mononuclear cells (PBMCs). For this purpose, a panel of CD20-positive B cell lymphoma Raji cells was used as targets with an E:T ratio of 10:1. The bispecific antibody was added at different concentrations. The CD89-CD20 antibody containing the N297A mutation, which excludes Fc function, was generated as the control antibody. Following a 4 h reaction time, target cell lysis was measured by LDH release, as previously described.Citation16
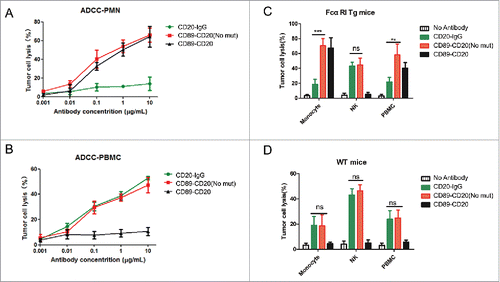
When using PMN cells (consisting of >85% neutrophils) as effector cells, both the CD89-CD20 and CD89-CD20 (No mut) antibodies effectively promoted ADCC, while the control rituximab antibody could not mediate tumor lysis under the same conditions (). These results indicate that the cytotoxic activity was fully dependent on the presence of FcαRI, but not the Fc fragment because CD89-CD20 could also lyse tumor cells effectively. ADCC killing was further examined using PBMCs. This effector cell population contains multiple cell types capable of eliciting cytotoxicity, including NK cells and monocytes; because monocytes can only play a role in overnight assays,Citation17 NK cells were supposed to be the only effector cells. Accordingly, both the CD89-CD20 (No mut) antibody and rituximab mediated potent tumor lysis, while the CD89-CD20 antibody could not lyse target cells (); indicating that ADCC by NK cells was fully dependent on the Fc fragment. Taken together, these results indicate that the CD89-CD20 has efficient tumor cell killing activity via human PMNs.
Figure 3. ADCC of antibody variants mediated by human or mouse effector cells ex vivo. (A) CD20+ Raji cells were incubated with the anti-CD20 × anti-CD89 molecule or ADCC mutated antibody, together with human PMNs for 4 h. (B) Raji cells were incubated with the anti-CD20 × anti-CD89 molecule or ADCC mutated antibody, together with human PBMC cells for 4 h. Data are the average of three different experiments with three separate donors. (C-D) Raji cells were incubated separately with the isolated CD14+ monocytes, mouse monocyte-depleted PBMC fraction (NK), or PBMCs (E:T = 40:1) from FcαRI Tg mice (C) or wild-type C57BL/6 mice (D) using 1 μg/mL anti-CD20 × anti-CD89 molecule or ADCC mutated antibody. For monocytes group and PBMCs groups, a 20-h ADCC assay was used. For monocyte-depleted PBMCs (NK), a 4-h ADCC assay was used. All data are presented as the mean ± SEM (n = 3) from one of three representative experiments. *CD89-CD20(Not mut) group versus CD20-IgG group. **P < 0.01; ***P < 0.001; ns, not statistically significant by two-way ANOVA.
The CD89-CD20 bispecific antibody mediates ADCC by mouse effector cells in vitro
To investigate whether the bispecific antibodies mediate antitumor activity by mouse blood effector cells, we generated a human FcαRI (CD89) Tg mouse strain using an authentic murine CD14 promoter with CD89 expression restricted to blood and tissue monocytes/macrophages.Citation18 Our previous study showed that the FcαRI Tg mouse monocytes/macrophages could mediate ADCC in an overnight assay.Citation16 Therefore, we used isolated CD14 positive monocytes from FcαRI Tg mice as effector cells in a 20-h assay. CD89-CD20 and CD89-CD20 (No mut) mediated potent tumor lysis, which exceeded tumor lysis mediated by CD20-IgG (), indicating that the cytotoxic activity of IgA Fc receptor-FcαRI was stronger than FcγR. Wild-type (WT) monocytes lysed target cells similarly using both CD89-CD20 (No mut) antibody and CD20 IgG (). Monocyte-depleted PBMCs were able to efficiently lyse target cell utilizing both CD20-IgG and CD89-CD20 (No mut), through mechanisms likely mediated by NK cells, indicating that NK mediated ADCC activity could be induced by bispecific antibodies (). As expected, CD89-CD20 (No mut) exhibited greater cytotoxicity than CD20-IgG using PBMCs from Tg mice (which include monocytes and NK cells) as effector cells against Raji cells in an overnight assay. Because mouse PMNs do not express FcγRIa or FcγRIIIa, mouse PMN assays were not carried out.
PK analysis of the CD89-CD20 bispecific antibody
Human IgG-Fc can bind to FcRn and the half-life of human IgG in mice is estimated to be > 100 h.Citation19 Because the bispecific antibody contains IgG1 Fc, the bispecific antibody is expected to be recycled efficiently by FcRn, resulting in IgG1-like serum persistence. To determine the serum half-life of the bispecific antibody in C57BL/6 mice, we intravenously i.v. injected 200 μg of CD20-IgG (rituximab) or the CD89-CD20 bispecific antibody and measured the serum antibody concentration at the indicated time points (). As expected, the bispecific antibody exhibited a half-live of 73.6 hours (), a substantially longer mouse serum half-life than that of common artificial bispecific monoclonal antibodies such as BiTE (half-life of 2.1 h),Citation20 suggesting that FcRn recycling was not influenced in the novel antibody structure.
Figure 4. Pharmacokinetic profiles of antibody variants in C57/BL mice. Antibodies (200 µg) were administered to female mice (four mice per group) by intravenous injection. Serum was harvested from terminal bleeds at the indicated time points and antibody concentrations were determined by ELISA assay. All data are presented as the mean ± SEM (n = 3) from one of three representative experiments. The pharmacokinetic disposition parameters of the proteins were estimated by non-compartmental analysis using PK solve software.
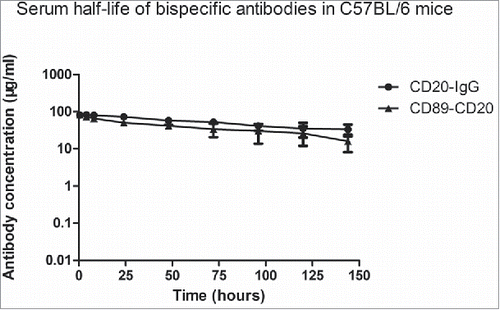
Table 1. Parameter of pharmacokinetics.
In vivo tumor lysis by the CD89-CD20 bispecific antibody
Because both CD89-CD20 bispecific antibodies exhibited strong ADCC in vitro with human and mouse effector cells, we next performed in vivo studies to assess the antitumor activities of the CD89-CD20 bispecific antibody panel. For avoid the negative effect by NK cells, we chose the CD89-CD20 antibody, which have a loss of FcγR induced ADCC activity. Raji cells (2 × 106) were inoculated into the female NOD/SCID mice. Animals were then given an intravenously i.v. injection of the CD89-CD20 bispecific antibody at a 10 mg/kg dose. Treatment of tumor-bearing mice with PMNs and the CD89-CD20 antibody induced significant and persistent tumor regression in this model, while the injection of PMNs alone showed almost no effect (), indicating that in vivo antitumor activity was strictly dependent on the presence of FcαRI-BsAb.
Figure 5. Xenograft models of the bispecific antibody in tumor-bearing mice. (A) Effect of the CD89-CD20 bispecific antibody on the growth of Raji Burkitt's lymphoma in SCID mice. (B) WT and (C) FcαRI Tg mice; 1 × 106 LLC-CD20 cells were mixed in solubilized basement membrane matrix and injected subcutaneously into female mice (n = 6–8/group). Antibodies were administered by i.v. injection on days 0, 4, and 8. Animals were monitored for tumor growth. Tumor volumes are plotted as the mean ± standard error. *CD89-CD20 group versus CD20-IgG group. **P < 0.01; ***P < 0.001 by two-way ANOVA.
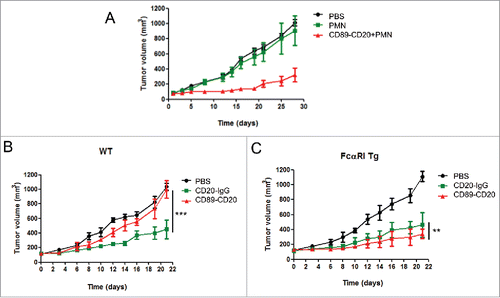
To further assess the anti-tumor effects mediated by mouse effector cells, LLC-CD20 CD89 Tg mice were generated by transducing the mouse LLC cell line with a human CD20 gene to facilitate tumor growth in CD89 transgenic mice. Mice were inoculated with LLC-CD20 cells and treated with either the CD89-CD20 antibody or Rituximab as the control antibody. Rituximab exhibited notable antitumor activity in WT mice, while no tumor killing was observed following the CD89-CD20 treatment (). In contrast, treatment of tumor-bearing mice with CD89-CD20 induced significant and persistent tumor regression in CD89 Tg mice; this antitumor activity was stronger than that of CD20-IgG (), indicating that in vivo antitumor activity was strictly dependent on the presence of FcαRI.
Tumor-associated macrophage-mediated tumor phagocytosis in the presence of BsAb (CD89-CD20)
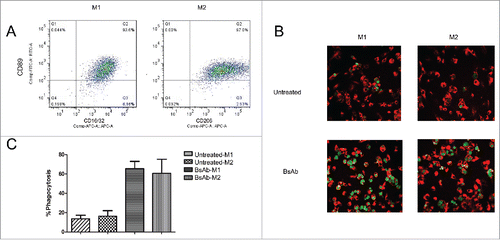
In tumor sections, myeloid cells, including tumor-associated macrophages, constitute the major component of the leukocyte tumor infiltrate.Citation21,22 Tumor-associated macrophages are divided into the tumor growth inhibiting M1 and the tumor promoting M2 phenotype.Citation23 A previous study demonstrated that IgA could activate the anti-tumor ability of macrophages following stimulation with IL-4 in vitro, suggesting that tumor-associated M2 macrophages could also be recruited against cancer cells activated by the IgA-CD89 interaction.Citation24 To assess the ability of our BsAb antibody (CD89-CD20) to activate tumor associated macrophages, macrophages within CD20-LLC tumors of CD89 Tg xenograft mice were characterized. Single-cell suspensions of freshly isolated CD20-LLC tumors were first labeled by CD11b-FITC and F4/80-PE antibodies (BD Pharmingen) and were selected by BD FACs AriaII (BD Bioscience). Then the CD11b+F4/80+macrophages were determine the CD16CD32+M1 and CD206+M2 macrophage profile using microbeads. Cell surface expression of FcαRI(CD89) was detected on both M1 and M2 macrophages in the analyzed tumors (). Further characterization of M1 and M2 macrophages was conducted by analyzing the cytokines secreted from the in vitro macrophage subsets. M1 macrophages secreted IFN-γ, TNF-α, while M2 macrophages secreted IL-13 and TGF-β (data not shown). Next, we analyzed whether polarized macrophages still possess the ability to mediate antibody-dependent cellular phagocytosis (ADCP) of human Raji tumor cells via the CD89-CD20 bispecific antibody. Interestingly, M1 and M2 macrophages showed similar killing efficiency with the bispecific antibody ( and ), demonstrating that M1 and M2 macrophages were effectively recruited for ADCP by the BsAb (CD89-CD20) bispecific antibody.
Figure 6. Tumor-associated macrophages trigger ADCP in Raji cells with BsAb (CD89-CD20). (A) Flow cytometric analysis of CD89 surface expression on TAMs. CD11b+F4/80+macrophage cells were selected by BD FACs AriaII and TAMs were further purified by microbeads and stained with anti-CD89-FITC. Cells were stained with CD16/32-APC or CD206-APC to identify M1 and M2 macrophages, respectively. The experiment was repeated at least three times, yielding essentially identical results. (B) Representative microscope images showing specific phagocytosis induced by the bispecific antibody. Raji cells (CFSE; green) incubated with macrophages (F4/80-Cy3; red) for 6 hours in the presence of the bispecific antibody (1 μg/ml) demonstrated substantial phagocytosis. (C) Phagocytosis of Raji cells was analyzed and quantified as the percentage of double-positive cells relative to total CFSE-positive cells and F4/80+cells.
Discussion
Therapeutic antibodies have become a major therapeutic option for patients with cancer, autoimmune, inflammatory, and various other diseases.Citation25 Over the past few years, numerous bispecific antibody formats have been reported; the approach of simultaneously modulating two molecular targets on a tumor cell or redirecting immune effector cells to kill tumor cells may develop into an important therapeutic alternative to monoclonal antibodies.
The most successful strategy to date, which involves blinatumomab, a bispecific antibody targeting CD3 on T cells and CD19 on B cells in patients with acute lymphoblastic leukemia, has demonstrated impressive clinical anti-tumor activity. Various other bispecific formats are currently under preclinical and clinical evaluation. Several BsAbs presently in clinical development are designed to redirect T cells to tumor cells.Citation26,27 In addition to T cells, other immune cells, such as macrophages, monocytes, granulocytes, and NK cells, can also exert tumor-killing effects. BsAbs with a non-IgG-like format are smaller in size, leading to enhanced tissue penetration, while BsAbs with an IgG-like format usually have a longer serum half-life due to their larger size and FcRn-mediated recycling.Citation28 The Fc region of BsAbs facilitates purification and improves solubility and stability.Citation29
Recently, we described a bispecific antibody format with a loss of ADCC activity to prevent the side effect of binding to FcγRIIIA on immune effector cells in vivo. Therefore, the antibodies in this study lack the core fucose at Asn-297 of the Fc region. The production of BsAbs with an Fc region poses some challenges such as the formation of undesirable homodimers and other product-related contaminants including mispaired molecules. Thus, we used the “knobs-into-holes” approach to tackle these problems by substituting a large amino acid for a small one in the CH3 domain (the “knob”) of one antibody and reciprocally in the other antibody (the “hole”).Citation30
A previous study has identified the myeloid receptor for IgA (FcαRI, CD89) as a promising trigger molecule for BsAb-based therapy.Citation31 The results reported here demonstrate that the FcαRI-directed BsAb could also mediate phagocytosis of HER2/neu-expressing tumor cells. Thus, the FcαRI directed BsAb could promote tumor cell destruction via the effector cells circulating in peripheral blood (PMNs and monocytes), as well as tissue macrophages (including tumor-associated macrophages).Citation32 However, in vivo evidence regarding the potential of FcαRI remains limited.
In this study, we demonstrated that the use of transgenic CD89 mice would induce enhanced tumor cell killing through the recruitment and activation of both FcαRI-effector cells (monocytes/macrophages). To this end, we generated a BsAb targeting CD89 and CD20 containing an IgG-Fc to allow for a prolonged half-life (73.6 h). We further examined the cytotoxicity induced by this BsAb both in vitro and in vivo. Moreover, we demonstrated that macrophages, in addition to PBMCs and PMNs, may constitute another potent effector cell population for anti-CD89 antibodies. In this transgenic mouse model, the CD89 bispecific antibody showed significant anti-tumor activities.
In this paper, the LLC-CD20 cells were used. At first we found that LLC-CD20 cells were immunogenic, and a certain percentage of mice injected with LLC-CD20 cells did not develop tumor. Thus the mice with tumors were sacrificed and the tumor cells were isolated, the CD20+cells were selected by BD FACs AriaII and again injected into C57 mice. Moreover, our previous report has proved that the peritoneal macrophages showed the same efficacy to the LLC and LLC-CD20 cells,Citation16 indicating there was almost no unwanted off-site engagement macrophage. A better approach was to generate transgenic mice both expressing human CD89 and CD20, however the present mouse model could already prove our CD89-CD20 Ab work well.
Interestingly, M1 and M2 macrophages from the CD89 transgenic mice displayed similar ADCP activity, suggesting that tumor-associated M2 macrophages could also be recruited against cancer cells by tumor-specific anti-CD89 antibodies. Moreover, we proved that TAMs in human tumors expressed CD89 by immunohistochemistry methods(data not shown), strengthening the argument for a clinical benefit of the CD89-targeted strategy. Recently, an in vivo study showed that depletion of macrophages resulted in decreased efficacy of anti-CD142 antibody therapy, thus demonstrating that TAMs could contribute an anti-tumor function.Citation33 And pro-tumor polarization or directly promoting macrophage activation cytotoxic therapy have been used successfully in preclinical models. On the other hand, immune checkpoint blockade antibody therapy was proved to be very efficient in antitumor activities. Macrophages could directly suppress anti-tumor T cell responses through programmed death ligand 1(PD-L1).Citation34 Could CD89-macrophage targeting combined checkpoint blockade therapy enhance antitumor response? Taken together, our findings indicate that the tumor microenvironment will become an important factor in the design of cancer therapies.
Monocytes and macrophages have been showed to induce ADCC and ADCP of tumor cells in the presence of CD89 anti-tumor mAb. Furthermore, neutrophils are the most abundant population of circulating blood cells, and when targeting neutrophils for tumor therapy, antibodies of the IgA subclass have been demonstrated to be more effective.Citation35 in conclusion, the results of our study underline the importance of CD89, which warrants further investigation of CD89 as a potential trigger molecule for cancer therapy.
Materials and methods
Ethical approval
All animal experiments were approved by the Animal Ethics Committee of Tongji University. All the mice were housed in a pathogen-free animal facility at the experimental animal center of Tongji University. Mice were fed standard chow and provided with distilled water ad libitum. Fresh cages were provided weekly. Pentobarbital sodium (60 mg/kg) was used as the anesthetic drug, and mice were euthanized using 100 mg/kg sodium pentobarbital. Appropriate efforts were made to minimize animal suffering.
Cell lines
Cell lines were obtained from the American Tissue Type Collection (ATCC); the human cell line Raji (ATCC) was used. The mouse cell line Lewis Lung Carcinoma (LLC) was stably transfected with human CD20 antigen using a lentivirus system as previously described.Citation16 All cell lines were maintained according to the cell bank recommendations or in normal growth medium consisting of RPMI-1640 (Gibco) supplemented with 2 mM glutamine and 10% (vol/vol) fetal calf serum (Gibco) at 37°C and 5% (vol/vol) CO2.
Bispecific antibody generation
BALB/c mice were injected in soluble CD89-his protein (Sino Biotech, Beijing, China). Four days after the final immunization, Spleens cells were harvested and fused with SP2/0 myeloma cells using 35% polyethylene glycol (Sigma). Hybridoma cell lines that produced specific antibodies against CD89 protein were identified by enzyme-linked immunosorbent assay (ELISA) and screened by flow cytometry using human neutrophils and mouse macrophages from CD89-transgenic mice generated previously in our lab.Citation18 All antibody gene segments were generated by gene synthesis and cloned using unique restriction sites into the pcDNA-3.1 expression vector. The anti-CD89 antibody sequence was derived from the hybridoma cell Line 2A9C11. Bispecific and control antibodies were expressed by transient transfection of suspended human embryonic kidney (HEK293F) cells. Antibodies were produced by co-transfection of the cells with three plasmids. Supernatants were harvested 7 days post transfection and purified by affinity chromatography using protein A-Sepharose ™ (GE Healthcare).
To promote heterodimerization of the CH3 domains, the bispecific antibody heavy chain was modified to encode the T366S:L368A:Y407V mutations and the Fc fragment was modified to contain the T366W mutation.Citation36 CD89-CD20 antibody containing the N297A mutation was generated to abrogate the unwanted FcγR function.
Cell-binding analysis
To examine CD20 binding, we incubated 5 × 105 Raji cells with CD89-CD20 at different concentrations for 1 hour at 4°C. To examine CD89 binding, human PMNs were isolated from the blood of healthy donors using a ficoll-histopaque density gradient and incubated with CD89-CD20 for 1 hour at 4°C, followed by two washes with PBS. The cells were then incubated with an anti-human Fc-FITC mAb (invitrogen) for 30 min on ice in the dark before two further washes with PBS. The cell-binding activity of purified CD89-CD20 bispecific antibody was determined by flow cytometry.
ADCC assay
Human PMNs and PBMCs were isolated from the blood of healthy donors using a ficoll-histopaque density gradient, ADCC was measured using a lactate dehydrogenase (LDH)-release assay as previously described;Citation16 Mouse PBMCs were isolated from freshly drawn peripheral blood of CD89 Tg and WT mice using mouse Percoll (Sigma) following the manufacturer's directions. Monocytes were isolated from the PBMC fraction using CD14-positive microbeads (Miltenyi Biotech). Effector cells were added with an effector-to-target (E: T) ratio of 40:1 in a final assay volume of 200 mL/well. The cells were incubated for 4 h/20 h at 37°C /5% CO2. Subsequently, the supernatant was transferred to a Deepwell Plate (Corning) and the (LDH) released into the medium was quantified by measuring the absorbance at 490 nM. Lysis was calculated using the following formula: % lysis = [(counts of sample – minimal release)/(maximum release – minimum release)]*100.
In vivo pharmacokinetics (PK) study
Four female CD89 Tg mice were injected intravenously (i.v.) with a single dose of control antibody (Rituximab, Roche) (n = 4) or CD89-CD20 bispecific antibody (n = 4) at 10 mg/kg. Blood was collected by tail vein cut or cheek pouch from alternating mice at various time points (0.25, 4, 8, 24, 48, 72, 96, 120 and 144 h post-dose), four mice per time point. The antibody concentrations in the sera were determined using human IgG ELISA Kits (Bethyl Laboratories) according to the manufacturer's instructions. PK parameters were calculated from the final dataset using the PK solver software, as previously described.Citation37
In vivo tumor killing studies
The anti-tumor properties of CD89-CD20 in vivo were determined using a Burkitt's lymphoma xenograft model in NOD/SCID mice reconstituted with human PMNs. Raji cells were s.c. inoculated into female NOD/SCID mice (2 × 106 cells/mouse). Animals were randomly assigned into treatment groups (6–8 per group), with the mean tumor volume for each group being 100–150 mm3. tumor volumes were determined according to the formula: tumor volume (mm3) = longer diameter × (shorter diameter)Citation2 × 0.5 mm3.
Tumor burden NOD/SCID mice were intravenously injected on day 0 with vehicle or CD89-CD20 antibody (10 mg/kg) pre-mixed with 1 × 107 IFN-γ (10 U/mL) activated human PMNs from healthy donors. PBS group without PMN effector cells served as the negative control. All animals in the experimental groups transplanted with tumor cells and PMNs received an i.v. bolus on days 0, 4, and 8 of CD89-CD20. Tumor size was monitored twice a week.
CD20-Positive LLC cells (1 × 106) were s.c. injected into wild type (WT) and FcαRI Tg C57BL/6 mice. The mice (n = 6–8) received twice a week i.v. injections of antibodies (10 mg/kg bispecific antibodies or rituximab [Roche]) starting on day 0, when tumor growth volume for each group reached 100–150 mm3.
Tumor-associated macrophage (TAM) phagocytosis
CD20-Positive LLC cells (1 × 106) were s.c. injected into FcαRI Tg C57BL/6 mice (n = 6). The mice were sacrificed when the tumors reached the desired diameter. Following resection, the tumors were shipped in RPMI-1640 on wet ice. Tumors were digested to single-cell suspensions using a human tumor dissociation kit (Miltenyi Biotec). The cell suspensions were first labeled by CD11b-FITC and F4/80-PE antibodies (BD Pharmingen) and were selected by BD FACs AriaII (BD Bioscience). Then the CD11b+F4/80+macrophages were incubated with APC rat anti-mouse CD16/CD32or APC rat anti-mouse CD206 (BioLegend) and then with anti-APC magnetic microbeads (Miltenyi Biotech) according to the manufacturer's instructions. To determine CD89 expression levels on TAMs, M1or M2 macrophages were stained with a fluorescein isothiocyanate (FITC)-conjugated anti-CD89 antibody (CD89-FITC; Miltenyi Biotech) for 30 min at 4°C and then evaluated by FACS using a FACSVerse system (BD Bioscience).
For microscopy experiments, macrophages and CFSE-Raji cells (1 μM CFSE; Life Technologies) were incubated on cover slips in 24-well plates at the appropriate effector / tumor cell ratio (5:1) together with 1.0 µg/mL of the CD89-CD20 antibody at 37°C for 6 h. Subsequently, cells were fixed after wash twice, permeabilized, and stained for macrophages, using a rabbit anti-mouse F4/80 antibody and a goat anti-rabbit IgG-Cy3 antibody (Boster Biotech, Wuhan, China). Confocal microscopy was performed according to routine procedures.
Statistical analysis
Data were graphed and analyzed using GraphPad Prism 6.0 (Graph Pad Software). Data are expressed as the median and range or mean ± standard error of the mean (SEM). Quantitative data between groups were compared using Student's t test with unpaired two-tailed t tests or two-way analysis of variance (ANOVA). Differences with P values of < 0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Abbreviations
| ADCC | = | antibody-dependent cell-mediated cytotoxicity |
| ADCP | = | antibody-dependent cellular phagocytosis |
| BsAb | = | bispecific antibody |
| CD20 | = | tumor antigen |
| CD89 | = | FcαRI receptor |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| ELISA | = | enzyme-linked immunosorbent assay |
| E:T | = | effector-to-target |
| FITC | = | fluorescein isothiocyanate |
| i.v. | = | intravenously |
| LDH | = | lactate dehydrogenase |
| mAbs | = | monoclonal antibodies |
| NK | = | natural killer |
| PBMCs | = | peripheral blood mononuclear cells |
| PK | = | polymorphonuclear |
| PMN | = | pharmacokinetics |
| s.c | = | subcutaneous |
| TAM | = | tumor-associated macrophage |
| WT | = | wild-type |
Acknowledgments
We wish to thank all members of our laboratory for their helpful suggestions and support. This study was supported by grants from the National Natural Science Foundation of China (NSFC31270987, NSFC31470896, and NSFC81402399), National Basic Research Program of China (973 Program) 2015CB553706.
References
- Valerius T, Stockmeyer B, van Spriel AB, Graziano RF, van den Herik-Oudijk IE, Repp R, Deo YM, Lund J, Kalden JR, Gramatzki M, et al. FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood. 1997;90:4485-92. PMID:9373259
- Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. Journal of hematology & Oncology. 2015;8:130. doi:10.1186/s13045-015-0227-0
- Chen S, Li J, Li Q, Wang Z. Bispecific antibodies in cancer immunotherapy. Human vaccines & immunotherapeutics. 2016;12:2491-500. doi:10.1080/21645515.2016.1187802
- Kiefer JD, Neri D. Immunocytokines and bispecific antibodies: two complementary strategies for the selective activation of immune cells at the tumor site. Immunol Rev. 2016;270:178-92. doi:10.1111/imr.12391. PMID:26864112
- Compte M, Alvarez-Cienfuegos A, Nunez-Prado N, Sainz-Pastor N, Blanco-Toribio A, Pescador N, Sanz L, Alvarez-Vallina L. Functional comparison of single-chain and two-chain anti-CD3-based bispecific antibodies in gene immunotherapy applications. Oncoimmunology. 2014;3:e28810. doi:10.4161/onci.28810. PMID:25057445
- Deo YM, Graziano RF, Repp R, van de Winkel JG. Clinical significance of IgG Fc receptors and Fc gamma R-directed immunotherapies. Immunol Today. 1997;18:127-35. doi:10.1016/S0167-5699(97)01007-4. PMID:9078685
- Guyre PM, Graziano RF, Vance BA, Morganelli PM, Fanger MW. Monoclonal antibodies that bind to distinct epitopes on Fc gamma RI are able to trigger receptor function. J Immunol. 1989;143:1650-5.
- Morton HC, van Egmond M, van de Winkel JG. Structure and function of human IgA Fc receptors (Fc alpha R). Critical Rev Immunol. 1996;16:423-40. PMID:8954257
- Stockmeyer B, Dechant M, van Egmond M, Tutt AL, Sundarapandiyan K, Graziano RF, Repp R, Kalden JR, Gramatzki M, Glennie MJ, et al. Triggering Fc alpha-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J Immunol. 2000;165:5954-61. doi:10.4049/jimmunol.165.10.5954. PMID:11067958
- van Egmond M, van Spriel AB, Vermeulen H, Huls G, van Garderen E, van de Winkel JG. Enhancement of polymorphonuclear cell-mediated tumor cell killing on simultaneous engagement of fcgammaRI (CD64) and fcalphaRI (CD89). Cancer Res. 2001;61:4055-60. PMID:11358825
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49-61. doi:10.1016/j.immuni.2014.06.010. PMID:25035953
- Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286-91. doi:10.1126/science.1232227. PMID:23329041
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39-51. doi:10.1016/j.cell.2010.03.014. PMID:20371344
- Fukuda K, Kobayashi A, Watabe K. The role of tumor-associated macrophage in tumor progression. Frontiers in bioscience. 2012;4:787-98.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Prot Eng. 1996;9:617-21. doi:10.1093/protein/9.7.617. PMID:8844834
- Li B, Xu L, Tao F, Xie K, Wu Z, Li Y, Li J, Chen K, Pi C, Mendelsohn A, et al. Simultaneous exposure to FcγR and FcαR on monocytes and macrophages enhances antitumor activity in vivo. Oncotarget. 2017;8:39356-39366. doi: 10.18632/oncotarget.17000. PMID:28454118
- Boross P, Lohse S, Nederend M, Jansen JH, van Tetering G, Dechant M, Peipp M, Royle L, Liew LP, Boon L, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. 2013;5:1213-26. doi:10.1002/emmm.201201929. PMID:23918228
- Xu L, Li B, Huang M, Xie K, Li D, Li Y, Gu H, Fang J. Critical Role of Kupffer Cell CD89 Expression in Experimental IgA Nephropathy. PloS one. 2016;11:e0159426. doi:10.1371/journal.pone.0159426. PMID:27437939
- Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47:115-23. doi:10.1053/j.seminhematol.2010.01.011. PMID:20350658
- Haas C, Krinner E, Brischwein K, Hoffmann P, Lutterbuse R, Schlereth B, Kufer P, Baeuerle PA. Mode of cytotoxic action of T cell-engaging BiTE antibody MT110. Immunobiology. 2009;214:441-53. doi:10.1016/j.imbio.2008.11.014. PMID:19157637
- Algars A, Irjala H, Vaittinen S, Huhtinen H, Sundstrom J, Salmi M, Ristamäki R, Jalkanen S. Type and location of tumor-infiltrating macrophages and lymphatic vessels predict survival of colorectal cancer patients. Int J Cancer. 2012;131:864-73. doi:10.1002/ijc.26457. PMID:21952788
- Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, Van Ginderachter JA. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol. 2011;55:861-7. doi:10.1387/ijdb.113371dl. PMID:22161841
- Guo Q, Jin Z, Yuan Y, Liu R, Xu T, Wei H, Xu X, He S, Chen S, Shi Z, et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J Immunol Res. 2016;2016:9720912. doi:10.1155/2016/9720912. PMID:27975071
- Lohse S, Brunke C, Derer S, Peipp M, Boross P, Kellner C, Beyer T, Dechant M, van der Winkel JG, Leusen JH, et al. Characterization of a mutated IgA2 antibody of the m(1) allotype against the epidermal growth factor receptor for the recruitment of monocytes and macrophages. J Biol Chem. 2012;287:25139-50. doi:10.1074/jbc.M112.353060. PMID:22679018
- Kontermann RE. Dual targeting strategies with bispecific antibodies. mAbs. 2012;4:182-97. doi:10.4161/mabs.4.2.19000. PMID:22453100
- Satta A, Mezzanzanica D, Turatti F, Canevari S, Figini M. Redirection of T-cell effector functions for cancer therapy: bispecific antibodies and chimeric antigen receptors. Future Oncology. 2013;9:527-39. doi:10.2217/fon.12.203. PMID:23560375
- Zugmaier G, Klinger M, Schmidt M, Subklewe M. Clinical overview of anti-CD19 BiTE((R)) and ex vivo data from anti-CD33 BiTE((R)) as examples for retargeting T cells in hematologic malignancies. Mol Immunol. 2015;67:58-66. doi:10.1016/j.molimm.2015.02.033. PMID:25883042
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9:182-212. doi:10.1080/19420862.2016.1268307. PMID:28071970
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95-106. doi:10.1016/j.molimm.2015.01.003. PMID:25637431
- Shatz W, Chung S, Li B, Marshall B, Tejada M, Phung W, Sandoval W, Kelley RF, Scheer JM. Knobs-into-holes antibody production in mammalian cell lines reveals that asymmetric afucosylation is sufficient for full antibody-dependent cellular cytotoxicity. mAbs. 2013;5:872-81. doi:10.4161/mabs.26307. PMID:23995614
- Stockmeyer B, Valerius T, Repp R, Heijnen IA, Buhring HJ, Deo YM, Kalden JR, Gramatzki M, van de Winkel JG. Preclinical studies with Fc(gamma)R bispecific antibodies and granulocyte colony-stimulating factor-primed neutrophils as effector cells against HER-2/neu overexpressing breast cancer. Cancer Res. 1997;57:696-701. PMID:9044847
- Deo YM, Sundarapandiyan K, Keler T, Wallace PK, Graziano RF. Bispecific molecules directed to the Fc receptor for IgA (Fc alpha RI, CD89) and tumor antigens efficiently promote cell-mediated cytotoxicity of tumor targets in whole blood. J Immunol. 1998;160:1677-86.
- Gul N, van Egmond M. Antibody-Dependent Phagocytosis of Tumor Cells by Macrophages: A Potent Effector Mechanism of Monoclonal Antibody Therapy of Cancer. Cancer Res. 2015;75:5008-13. doi:10.1158/0008-5472.CAN-15-1330. PMID:26573795
- Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462-72. doi:10.1016/j.ccell.2015.02.015. PMID:25858805
- Guettinger Y, Barbin K, Peipp M, Bruenke J, Dechant M, Horner H, Thierschmidt D, Valerius T, Repp R, Fey GH, et al. A recombinant bispecific single-chain fragment variable specific for HLA class II and Fc alpha RI (CD89) recruits polymorphonuclear neutrophils for efficient lysis of malignant B lymphoid cells. J Immunol. 2010;184:1210-7. doi:10.4049/jimmunol.0902033. PMID:20042573
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16:677-81. doi:10.1038/nbt0798-677. PMID:9661204
- Kreuer S, Hauschild A, Fink T, Baumbach JI, Maddula S, Volk T. Two different approaches for pharmacokinetic modeling of exhaled drug concentrations. Sci Rep. 2014;4:5423. doi:10.1038/srep05423. PMID:24957852