
ABSTRACT
Antibody-drug conjugates (ADCs) are emerging as effective tools in cancer therapy, combining the antibody's exquisite specificity for the target antigen-expressing cancer cell together with the cytotoxic potency of the payload. Much success stems from the rational design of “toxic warheads”, chemically linked to antibodies, and from fine-tuning the intricate properties of chemical linkers. Here, we focus on the antibody moiety of ADCs, dissecting the impact of Fab, linkers, isotype and Fc structure on the anti-tumoral and immune-activating functions of ADCs. Novel design approaches informed by antibody structural attributes present opportunities that may contribute to the success of next generation ADCs.
Introduction
Antibody-Drug Conjugates and known mechanisms of action in oncology
The treatment of cancer remains a formidable challenge.Citation1 For many tumors, chemotherapy achieves significant clinical benefit, however these agents suffer from lack of specificity for cancer cells and high toxicity, often resulting in adverse effects, poor quality of life, early discontinuation and reduced clinical efficacy.Citation1 Targeted treatments in the form of tumor antigen-specific and checkpoint inhibitor antibodies, envisioned for over 100 years since Ehrlich proposed the concept of “the magic bullet”, have now been established in clinical oncology and have earned their place alongside chemotherapeutic agents and small molecule inhibitors in the care of cancer patients.Citation1 However, antibodies targeting tumor-associated antigens also suffer from limitations. These include limited tissue penetrance and blocking target-associated pathways due to intrinsic and acquired resistance.Citation2
Antibody-drug conjugates (ADCs) are designed to combine the selectivity of monoclonal antibodies (mAbs) with the cytotoxic potential of chemotherapeutic drugs.Citation3 ADCs are tripartite drugs, comprising of a tumor antigen-specific mAb conjugated to a potent cytotoxin via a stable chemical linker.Citation3 The three components together give rise to a powerful oncolytic agent, capable of delivering normally-intolerable cytotoxic drugs directly and specifically to cancer cells, guided by the exquisite specificity and high affinity of antibodies for their targets in tumors ().Citation3
Figure 1. Schematic of ADC components and their role in ADC design, engineering and functions. The Fab region (A) is responsible for antigen recognition and binding, and can lead to ADC internalization. Therefore, the Fab region needs to be targeted to tumor-associated antigens that are homogenously expressed on tumor cells, ideally with little or no expression on normal cells. The payload is attached to the antibody via a cleavable or non-cleavable linker (B). Non-cleavable linkers rely on the complete degradation of the antibody after internalization of the ADC, whereas most cleavable linkers are cleaved by different mechanisms depending on the linker (i.e. proteases, reduction) and some cleavable linkers do not depend on ADC internalization for payload release and can result in higher off-target cytotoxicities. The hydrophobicity of linkers can play a vital role in the biodistribution of an ADC. Linkers can be attached non-selectively via lysines or the hinge thiols of cysteines, or antibody engineering can be performed for site-specific linking. The payload (C) is responsible for ADC toxicity and is usually a small hydrophobic molecule, able to cross cell membranes and cause cell death by targeting the cytoskeleton or DNA. Once cleaved from the antibody payloads can enter other (tumor) cells, resulting in further tumor killing (i.e. bystander effect) as well as off-target cytotoxicity when entering normal cells. The Fc region of the antibody (D) can trigger immune effector functions such as Antibody-Dependent Cytotoxicity through binding to Fcγ-receptors. However, if the ADC is internalized into non-malignant cells, it can cause off-target cytotoxicity. Antibody engineering can enhance or impair immune effector functions through, for example, single point mutations, Thiomabs, glycoengineering or incorporation of unnatural amino acids.
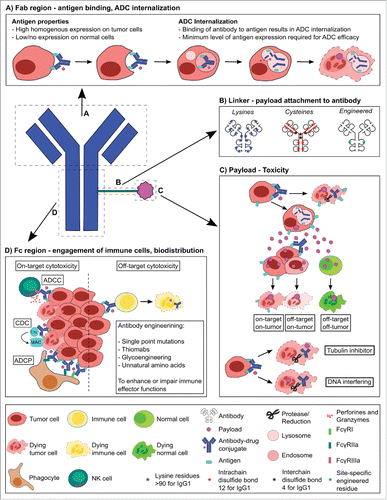
ADCs have a well-described mechanism of action, namely binding of the mAb to the target antigen resulting in complex internalization through receptor-mediated endocytosis.Citation4 Upon fusion of two internalized vesicles, an early endosome is formed whereby cargo is sent through two pathways: recycling which results in trafficking back to the plasma membrane,Citation5 or endolysosomal degradation.Citation4
The mechanism and location of toxin release depends on the type of linker. Non-cleavable linkers depend on degradation of the antibody with or without a portion of the linker to liberate the toxin from the ADC.Citation6 However, cleavable linkers can release toxins through acidic conditions in the lysosome, reduction of the linker in the cytoplasm or cleavage by specific proteases.Citation6 For ADCs containing cleavable linkers, the antibody-part of the ADC is either degraded once the toxin is cleaved or is recycled and released outside the cell in vesicles.Citation4 Once the toxin is cleaved from the ADC, it enters the cytoplasm and can either bind to its molecular target in the cytoplasm (usually tubulin) or can cross into the nucleus and cause cell cycle arrest and apoptosis by interfering with DNA.Citation7 Almost all payloads in clinical development are small hydrophobic molecules, that are able to cross biomembranes once cleaved from the ADC.Citation8 Therefore, nuclear DNA as well as the cytoskeleton in the cytoplasm are suitable locations for the payload to interfere with critical cellular mechanisms resulting in cell death.
The majority of ADCs have clinical efficacy through on-target on-tumor effects; however some ADCs also cause off-target on-tumor cell killing, referred to as the bystander-effect.Citation9 This effect exploits the ability of payloads to cross cell membranes and exert their cytotoxic effect on neighbouring cancer cells that lack target antigen expression. The payload may be cleaved from the antibody in the tumor microenvironment due to local acidic pH conditions and the presence of proteases released locally by tumor and dead cells.Citation10 The bystander effect is especially advantageous for heterogeneous tumors consisting of tumor cells with low or absent target antigen expression that cannot be recognized through the antibody variable regions of the ADC.Citation7,11 The majority of ADCs internalize, however there are also non-internalizing ADCs that target tumor cells in the same way but can exert off-target on-tumor effects, making use of the bystander mechanism as one of their primary modes of action. However, this type of ADCs come with a perceived risk of also killing immune cells, such as antigen-presenting cells, that may be important in tumor growth restriction, in an off-target fashion. Since non-internalizing ADCs have had restricted success in the clinic, internalizing ADCs are still at the forefront of the development of targeted anti-cancer therapeutics.Citation3
ADCs in clinical use
After decades of failure with the first approved ADCs being withdrawn from the market due to fatal cytotoxicity,Citation12 ADCs are now one of the fastest growing fields of oncology therapeutics. There are currently four FDA- and EMA- approved ADCs within clinical practice with durable clinical responses reported: gentuzumab ozogamicin (Mylotarg™) targeting CD33 expressed by acute myeloid leukemia (AML) has just been re-approved in the US, after being withdrawn in 2010,Citation13,14 brentuximab vedotin (Adcetris™) which targets CD30 expressed by Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (ALCL),Citation15 trastuzumab emtansine (Kadcyla™, also known as T-DM1) targeting HER2 expressed by 20–25% of breast carcinomas and other solid tumor typesCitation16 and the recently-approved inotuzumab ozogamicin (Besponsa™) targeting the B-cell lineage marker CD22, expressed on acute lymphoblastic leukemia cells.Citation17 (). Since their approval, there has been significant growth in this class of oncology therapeutics, with over 65 agents in early and late stage clinical trials (see for ADCs in Phase I/II, II and III clinical trials).
Table 1. Approved ADCs in clinical use.
Table 2. ADCs in clinical development in Phase I/II, II or III (U.S. National Institutes of Health. Clinical trials. at https://clinicaltrials.gov).
Two of the approved ADCs, brentuximab vedotin and trastuzumab emtansine, use auristatin- and maytansinoid-based warheads, respectively. These microtubule-targeting agents represent two-thirds of all clinical stage ADC payloads.Citation18 Mirvetuximab soravtansine (IMGN853, Phase III) and Depatuximab mafodotin (ABT-414, Phase IIb/III) also fall into this category line, consisting of the maytansinoid warhead DM4, similar to DM1, the drug component of trastuzumab emtansineCitation19 and MMAF, respectively. The other three ADCs in Phase III clinical trials (Sacituzumab Govitecan, Rovaltuzumab tesirine, Vadastuximab talirine) consist of drugs that target DNA and are therefore cytotoxic to both proliferating and non-proliferating cells (). SGN-CD33A (recently discontinued)Citation20 and rovalpituzumab tesirine (Rova-T™) consist of pyrrolobenzodiazepines (PBDs),Citation21,22 payloads designed to interfere with the minor groove of the DNA. The drug component of IMMU-132 is SN38a camptothean analogue, which inhibits DNA Topoisomerase I.Citation23,24 Generally, there is a trend towards payloads with higher potency featuring IC50s in the nanomolar and sub-nanomolar scale.Citation1 Despite many advances, finding novel payloads with optimal in vivo tolerability and high therapeutic index is still an ongoing challenge. New classes of toxins, evaluated in pre-clinical settings, such as the DNA alkylating agent indolinobenzodiazepine pseudodimers (IGNs), resulted in promising ADCs characterized by both high in vivo tolerability and high therapeutic indices and has been now moved forward to clinical evaluation.Citation25
Over the past decade, much research has been conducted focusing on linker chemistry and optimizing cytotoxic payloads. However, while the antibody is often viewed as a mechanism to deliver the cytotoxic payload, other potential effects pertaining to the antibody's unique structural characteristics, remain less-well elucidated. In this review, we consider the ADC as a tripartite drug, specifically focusing on the underexplored antibody rather than the toxic payload portion or the linker.
The Fab regions: Selection of, and specificity to, target expression of antigen
The successful development of an ADC is dependent on the premise of selecting an appropriate target antigen, with its suitability forming a key determinant of the efficacy of the ADC. Targeted therapies such as ADCs exploit the difference in protein expression between cancer cells and normal cells in order to select a suitable target antigen. Current targets being exploited include HER2 (i.e. trastuzumab emtansine)Citation26 and CD30 (i.e. brentuximab vedotin).Citation27 It is widely accepted that the target antigen should be homogeneously and selectively expressed on the surface of tumor cells with little or no expression on normal tissues in order to limit on-target off-tumor toxicity.Citation28
Target validation for ADCs must be based upon the reliable identification of target antigens. Genomic studies have highlighted the complex heterogeneity in tumors with different cell sub-populations harboring distinct phenotypic diversity resulting from the integration of genetic and non-genetic influences that define intra-tumor and inter-tumor heterogeneity.Citation29 This represents a profound challenge for antigen selection and, combined with sampling bias, may negatively impact drug discovery and validation of suitable antigen targets for ADCs.Citation29 While homogeneous target antigen expression is not an absolute requirement for ADC efficacy, since heterogeneous tumors may benefit from bystander killing, the most advanced ADCs within clinical development are for hematological indications which have a largely consistent expression of lineage specific markers (i.e. CD22, CD30)Citation30, such as CD79b (RG-7596, DCDS4501A). These lineage specific markers of hematologic cells are also targets for three out of four approved ADCs: CD22 (Besponsa™) for the treatment of acute lymphoblastic leukemia, CD30 (Acetris™) for the treatment of relapsed Hodgkin Lymphoma and systemic anaplastic large cell lymphoma and CD33 (Mylotarg™) for the treatment of acute myeloid leukemia ().Citation31
Traditionally, interest in target antigens has largely focused on those expressed on tumor cells. However, there has been growing interest in targeting antigens present within the tumor microenvironment, including those within the neovasculature, sub-endothelial extracellular matrix and the tumor stroma.Citation32–34 ADCs that target the stroma cause tumor cell death by reducing the concentration of growth factors produced by the stroma.Citation35 Since all tumor cells are dependent on angiogenesis and stromal factors for their survival, ADCs that target such tissues may have a broader efficacy. This is particularly attractive due to the fact that, unlike cancer cells, these cells are genomically stable, and are less likely to develop mutation-related drug resistance.Citation3
The target antigen should be well internalized by receptor-mediated endocytosis and should not be down-regulated by endocytosis or by the effects of repeated stimulation during treatment.Citation1,36 In order for an ADC to generate a clinical effect, antibody recognition of its epitope on the antigen must result in endocytosis.Citation37 In general, antigens that internalize well, with low expression on normal tissue and high expression on tumors are preferred for an ADC approach as they minimize potential toxicity through unwanted on-target, off-tumor expression. However the results of clinical trials indicate it may be difficult to predict the toxicity based on target expression in healthy tissue and toxicities due to off-tumor on-target expression can occur.Citation38 In the case of glembatumumab vedotin that targets the transmembrane glycoprotein NMB (gpNMB) on cancer cells, development of skin rash was one of the observed dose-limiting toxicities, which is likely due to membrane expression of gpNMB in epithelial cells of the skin. Previously the development of an ADC directed against CD44v6 was discontinued due to severe skin toxicity linked to high CD44v6 expression in the skin.Citation39,40 Interestingly, unlike findings with unconjugated antibody functions, current experimental evidence generally suggests that the antigen density does not directly correlate with the efficacy of the ADC.Citation41 Studies of lymphoma, melanoma and prostate cancer have demonstrated no direct correlation between antigen density and therapeutic response for ADCs, and findings suggest that a minimum antigen expression threshold is required for ADC efficacy.Citation31,42,43
The location and roles of the linker: Considerations for conjugation strategies
The linker plays a central role in connecting the cytotoxic agent to the antibody structure. One of the key functions of the linker is to maintain complex stability in the blood circulation, while allowing toxin release upon ADC internalization by target cells.
The first generation of ADCs mostly relied on linking via the antibody's lysine or cysteine residues.Citation6 IgG1 isotype antibodies consist of approximately 90 lysine residues, however, due to structural constrains, only 30 can be modified.Citation8 Therefore, linking the antibody to the toxin via the lysine residues results in heterogeneous ADCs with regards to both the number of toxins conjugated per antibody (drug-to-antibody ratio, DAR) and the positions of conjugation within the structure. Furthermore, monitoring changes on the antibody scaffold may be challenging, since, in addition to batch-to-batch variations, ADCs within one batch could differ significantly.Citation44 Therefore, novel methods are being developed to improve protein characterization and control protein modification processes.Citation45 Nevertheless, Kadcycla™ would not be a product unless regulatory authorities were satisfied that the conjugate product could be made consistently.
When conjugating to cysteine residues, disulfide bonds within the antibody must be reduced in order to produce free thiols for conjugation.Citation46 IgG1 antibodies consist of 4 inter-chain disulfide bonds – two connecting heavy and light chains and two connecting the two heavy chains in the hinge region, keeping the two half-antibodies together.Citation47 There are also 12 intra-chain disulfide bonds, however it has been shown that mild reduction of the antibody with DTT (Dithiothreitol) or TCEP (Tris(2-Carboxyethyl)phosphine) will result in inter-chain bonds being reduced without having an impact on the intra-chain disulfide bonds of the antibody.Citation48 However, one of the pitfalls of partial antibody reduction may be loss of the light chain, which can impair the binding properties of the ADC as well as its antigen cross-linking properties which may be key to facilitating internalization.
The location of the linker on the antibody structure could also negatively impact ADC functions. If the linker is conjugated near the Fab antigen binding site, this may interfere with or completely block antigen recognition and hence targeting of the ADC to tumors. Conjugation at or near the Fc regions, may also hinder binding to the neonatal Fc receptor (FcRn) and/or Fc receptors on immune effector cells, or alter antibody folding and structure. Either individually or collectively, these could substantially modify pharmacokinetics, pharmacodynamic properties, target recognition and engagement, effector functions and consequently bioavalability and efficacy. Quantifying the impact of these effects may be particularly complex for heterogeneous ADC preparations designed from lysine or cysteine bound linkers.
Despite drawbacks, cysteine and lysine conjugated heterogeneous ADC products with complex structural characteristics and variable DARs have facilitated ADC design and clinical translation. Current trends are focused on: designing homogenous ADC products with defined DARs via site-specific conjugation;Citation8 increasing the polarity and decreasing hydrophobicity of linkers which can provide improved pharmacokinetics, solubility and a larger therapeutic window.Citation49 Furthermore, ADCs with higher DARs can have improved potency because of greater delivery of toxin per antibody bound and may have the potential to eradicate off-target tumor cells, but can suffer from drawbacks including increased plasma clearance and off-target cytotoxicity.Citation50
Site-specific engineering may be achieved by integrating additional cysteines or non-natural amino acids with reactive groups for linking payloads.Citation8 Thiomab-ADCs, for example, contain engineered cysteines for site-specific conjugation giving a controlled DAR of 2, and have improved homogeneity as well as improved efficacy and toxicity profiles as demonstrated in vivo in cynomolgus monkey studies.Citation51–53
The trend towards homogenous ADC design enables the emergence of novel bio-orthogonal chemistries that utilize reactive moieties other than thiols or amines, and is broadening the diversity of linking methods.Citation8 Shifting the focus by investigating the impact of the antibody scaffold for ADC design might result in future ADCs with: defined DARs, higher solubility, lower off-target toxicity, better-characterized structural and functional attributes and improved efficacy. Therefore, the challenge is to design linking strategies that retain antibody structural integrity and stability, recognition and affinity for the target, Fc-mediated attributes that complement and enhance bioavailability and anti-tumor functions.
The Fc regions: Antibody scaffold and potential influence on function
The main role of the antibody moiety of an ADC is to deliver the cytotoxic drug selectively to the target cells due to its specificity and high affinity for an antigen expressed on the surface of target cells. Therefore, in designing ADCs, much attention has been paid to the Fab portion of the antibody, responsible for antigen recognition. The antibodies used to develop ADCs are mainly full length recombinant monoclonal antibodies, almost exclusively of the IgG class. Yet the Fc domains of such agents have received less consideration.
Contributions to efficacy
The Fc portion of IgG antibodies contains the binding domain to the neonatal Fc receptor (FcRn) that regulates serum half-life, and recognition of different activating and inhibitory Fc receptors on immune effector cells that can influence bioavailability, sequestration to tissues, trafficking to tumors, antigen-targeting and immune functions.
An intact IgG ADC might be able to recruit and activate complement components and immune effector cells into the tumor site, mediating secondary immune functions such as complement dependent cytotoxicity (CDC), antibody dependent cell-mediated cytotoxicity (ADCC) and antibody dependent cell-mediated phagocytosis (ADCP).Citation54 The ability of ADCs to trigger immune effector functions could offer an advantage through anti-tumor activity, or a disadvantage by sequestering ADCs through immune cells in the circulation and affecting the localization and target cell internalization of ADCs at the tumor site, or by being internalized by immune cells resulting in off-target toxicity. Studies have demonstrated similar antibody-mediated effector functions between the naked antibody and the corresponding ADC. For instance, the capacity of trastuzumab to induce ADCC of breast cancer cells was not affected by conjugation to DM1,Citation55 while brentuximab has been described to induce ADCP in vivo, believed to contribute to the potent anti-tumor efficacy observed for the brentuximab vedotin ADC.Citation56,57 While Fc receptor binding can be advantageous, other studies have identified Fc receptor engagement as a possible cause of side effects of ADC therapeutics. T-DM1 has been demonstrated to be internalized by megakaryocytes in vivo via FcγRIIa binding. This has been proposed to be involved in the development of thrombocytopenia induced by T-DM1.Citation58,59 However, there are other mechanisms besides FcγRIIa binding, such as macropinocytosis, which could also account for sufficient non-receptor/non-target mediated uptake by megakaryocytes to cause thrombocytopenia.Citation59
For these reasons, depending on the type of tumor, the expression of the antigen and the affinity of the antibody for its antigen, it might be appropriate to design an ADC with defined Fc-mediated functions either able or unable to engage with the immune system. ADCs can therefore be designed by selecting an appropriate subclass of IgG or by engineering the Fc portion.
The impact of antibody subclass
There are 4 subclasses of IgG (IgG1, IgG2, IgG3 and IgG4), and so far all the sub-classes apart from IgG3 have been used to develop ADCs that are currently in clinical trials.Citation60
IgG1 is the most commonly used subclass for ADC design. It has comparable serum stability (21 days) to IgG2 and IgG4 but has a greater ability to fix complement and a higher affinity for activating FcγRs expressed on effector cells such as monocytes and macrophages (FcγRI, FcγRIIa, FcγRIIIa) and natural killer (NK) cells (FcγRIIIa).Citation61 Therefore, this sub-class has a superior ability to engage the immune system and trigger CDC, ADCC and ADCP.
IgG3 has a superior ability to fix complement and to bind activating FcγRsCitation61 but it has so far been avoided for the development of ADCs because of its low half-life in serum compared to the other classes (e.g., 7 days instead of 21 days for IgG1, 2 and 4), its long hinge region that is subject to proteolysis and also evidence of potential immunogenicity.Citation62
IgG2 and IgG4 have low or no capacity to fix complement and have lower affinity for the activating FcγRs compared with IgG1. IgG2 and IgG4 are used in the design of therapeutic antibodies when the recruitment of the immune system is not desired. IgG2 has four disulfide bridges, while IgG1 and IgG4 have only two, so it seems more suitable for the use of malemide-linkers allowing a higher DAR.Citation63 IgG2 is also able to form dimers, but the impact of dimerization on its therapeutic effects would require more in-depth study. IgG2 ADCs, such as AGS-16M8F (anti-ENPP3 IgG2-MMAF),Citation64 are currently under evaluation in clinical trials.Citation60,65 IgG4 is a sub-class with very unusual characteristics, such as the ability to undergo Fab arm exchange (FAE) with other IgG4 antibodies, and can thus result in bispecific and functionally monovalent forms with reduced anti-target functions.Citation66 Therefore, specific single point mutations (S228P or S228P/R209K)Citation67,68 are usually introduced to stabilize the antibody and prevent FAE. Despite lower affinity for the activating FcγRs compared with IgG1, the affinity of IgG4 for FcγRI form is sufficient for functional activationCitation66 and in its non-fucosylated form IgG4 is able to bind FcγRIIIa.Citation69 Therefore, immune cell activation needs to be considered, when designing ADCs with IgG4 antibodies. The first ADC approved by the FDA in 2000 was an IgG4 antibody (Gemtuzumab ozogamicin, anti-CD33 IgG4-Calicheamicin), withdrawn from the market voluntarily in 2010, since a post approval study showed no improvements in survival and fatal toxicities. However the ADC has been re-approved this year for the treatment of acute myeloid leukemia.Citation12 More IgG4 ADCs are currently being evaluated in clinical trials.Citation60,65
Fc domain engineering strategies
Another method to modulate the ability of ADCs to engage the immune system is to engineer their Fc domains. A widely used method to enhance their ability to recruit the immune system and to trigger effector functions is antibody glycoengineering, such as the production of afucosylated IgGs. J6M0-mcMMAF (anti-BCMA IgG1-MMAF) is the first afucosylated-ADC that entered a clinical trial for the treatment of multiple myeloma.Citation70
If the sub-class of choice is IgG1, the Fc portion can be engineered to introduce a single point mutation, or a combination of mutations, to enhance or impair IgG1 binding to FcγRs or complement (C1q), and consequently to enhance or impair ADCC, ADCP or CDC.Citation71–73 An example is the Phase I clinical trial of MEDI4276 (anti-HER2 IgG1-tubulysin analogue) which has been engineered with three single point mutations (E234F, S239C and S442C) to reduce FcγR binding with the aim to minimize thrombocytopenia seen with T-DM1.Citation23
Fc engineering could also be used to improve the pharmacokinetics of ADCs. An example is the humanized IgG1 MEDI-524-YTE engineered with three single point mutations (M252Y, S254T and T256E) to enhance IgG1 binding to the neonatal Fc receptor (FcRn). MEDI-524-YTE has been shown to have a four-fold increase in serum half-life in cynomolgus monkeys compared with the wild type antibody.Citation74 It is worth investigating if increased FcRn binding and longer half-life might result in increased activity and decreased toxicity for an ADC.Citation75
ADC design based on biodistribution considerations
Stand-alone small molecule therapeutic agents such as those conjugated to ADCs can be widely distributed in the body. In contrast, antibodies are restricted primarily to plasma and extracellular fluids, and have been shown to target tissues that express the relevant antigen(s). ADCs typically retain the pharmacokinetic properties of their antibody componentCitation3,76 as opposed to the attached drug, and thus exhibit relatively low clearance and longer half-lives.Citation77
Despite the optimization of therapeutic antibodies and the availability of antibodies with higher affinity for the tumor than for normal tissues, the amount of antibody that reaches a tumor is only a small percentage of that administered (e.g., approximately 1–2%).Citation1,78,79 For this reason the use of recombinant antibody fragments for ADC production has been evaluated. Antibody fragments such as diabodies are much smaller then IgGs (around 50 kDa versus 150 kDa) and thus have superior tissue penetration abilities. However, due to their smaller size and the lack of the Fc portion that usually binds to FcRn, diabodies are cleared much faster than whole IgG isotypes.Citation80 A promising anti-CD30 diabody-drug conjugate has already demonstrated high anti-tumor activity,Citation81 but the use of diabodies for ADC design needs further study and optimization with a view to striking a balance between optimum tissue penetration and low clearance rates.
The Fc portion of an ADC is responsible for ADC-mediated effector functions and pharmacokinetics. Therefore, these biological properties can be optimized according to therapeutic requirements via engineering and glycoengineering.
Conclusion
Over the past decade, research efforts have focused on linker chemistry and optimizing cytotoxic payloads for ADCs. The antibody scaffolds of ADCs provide the required specificity for tumor-associated antigens and can be used to transport prohibitively-cytotoxic agents to cancer tissues. However, the potential clinical effects of the choice of antibody and its structural characteristics remain less-well explored, and perhaps under-exploited. Emerging evidence and novel technologies now provide good reason for a closer consideration of antibody structure, how this is influenced by linking to toxic payloads, and how both the antigen-recognizing Fab regions, as well as the immune-engaging Fc domains, can affect the functions and potency of ADCs ().
In designing the next generation of more effective, specific and efficacious ADCs, we postulate that translational opportunities that can be harnessed by considering the attributes of the antibody component. Careful selection of linking approaches could preserve antibody stability and high target affinities may define and optimize immune cell engagement functions when desired. In the future, ADCs with Fc regions of specific antibody isotypes, and with engineered Fc structure scaffolds may offer better control of biodistribution, and improved immune cell engagement and activation.
Each component of the tripartite complex can be evaluated and optimized to deliver pharmacokinetic and oncolytic properties whilst minimizing off-target toxicities. Novel design approaches informed by antibody structural attributes may thus present opportunities to contribute to the success of the next generation of optimized ADCs.
Conflicts of interest
SNK and JFS are founders and shareholders of IGEM Therapeutics Ltd. All other authors declare no conflicts of interest.
Author contributions
SNK, SC, RMH and BGTC conceived and designed the study. RHM, BGTC, DHJ, SM, KMI, AC, ANT, JFS, DET, SC and SNK discussed and edited the manuscript. SNK and SC supervised the study and coordinated the project. RMH, BGTC, SNK, DET and SC discussed and wrote the manuscript.
Acknowledgements
The research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) based at Guy's and St Thomas' NHS Foundation Trust and King's College London (IS-BRC-1215-20006). The authors acknowledge support by the Medical Research Council (MR/L023091/1); Breast Cancer Now (147); Cancer Research UK (C30122/A11527; C30122/A15774); The Academy of Medical Sciences; BBSRC IBCarb Network (Proof-of-Concept award IBCarb-PoC-0616-040); CRUK/NIHR in England/DoH for Scotland, Wales and Northern Ireland Experimental Cancer Medicine Centre (C10355/A15587). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Additional information
Notes on contributors
Silvia Crescioli
Silvia Crescioli, PhD, St. John's Institute of Dermatology, School of Basic & Medical Biosciences, King's College London & NIHR Biomedical Research Centre at Guy's and St. Thomas's Hospitals and King's College London, Guy's Hospital, Tower Wing, 9th Floor, London, SE1 9RT, United Kingdom. E-mail: [email protected]
References
- Diamantis N, Banerji U. Antibody-drug conjugates–an emerging class of cancer treatment. Br J Cancer. 2016;114:362–7. doi:10.1038/bjc.2015.435.
- McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi:10.1016/j.ccell.2014.12.001.
- Peters C, Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015;35:e00225. doi:10.1042/BSR20150089. PMID:26182432
- Ritchie M, Tchistiakova L, Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs. 2013;5:13–21. doi:10.4161/mabs.22854.
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–25. doi:10.1038/nrm2728.
- Lu J, Jiang F, Lu A, Zhang G. Linkers Having a Crucial Role in Antibody-Drug Conjugates. Int J Mol Sci. 2016;17:561. doi:10.3390/ijms17040561. PMID:26182432
- Kim EG, Kim KM. Strategies and Advancement in Antibody-Drug Conjugate Optimization for Targeted Cancer Therapeutics. Biomol Ther (Seoul). 2015;23:493–509. doi:10.4062/biomolther.2015.116.
- Jain N, Smith SW, Ghone S, Tomczuk B. Current ADC linker chemistry. Pharm Res. 2015;32:3526–40. doi:10.1007/s11095-015-1657-7.
- Singh AP, Sharma S, Shah DK. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J Pharmacokinet Pharmacodyn. 2016;43:567–82. doi:10.1007/s10928-016-9495-8.
- Perrino E, Steiner M, Krall N, Bernardes GJL, Pretto F, Casi G, Neri D. Curative properties of noninternalizing antibody-drug conjugates based on maytansinoids. Cancer Res. 2014;74:2569–78. doi:10.1158/0008-5472.CAN-13-2990.
- Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, Senter PD, Alley SC. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16:888–97. doi:10.1158/1078-0432.CCR-09-2069.
- Beck A, Haeuw J-F, Wurch T, Goetsch L, Bailly C, Corvaïa N. The Next Generation of Antibody-drug Conjugates Comes of Age. Discov Med. 2010;10:329–39. PMID:26182432
- Pfizer. PFIZER RECEIVES FDA APPROVAL FOR MYLOTARG™ (GEMTUZUMAB OZOGAMICIN) [Internet]. 2017 [cited 2017 Sep 9]; Available from: http://www.pfizer.com/news/press-release/press-release-detail/pfizer_receives_fda_approval_for_mylotarg_gemtuzumab_ozogamicin
- Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, Estey EH, Dombret H, Chevret S, Ifrah N, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15:986–96. doi:10.1016/S1470-2045(14)70281-5.
- Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363:1812–21. doi:10.1056/NEJMoa1002965.
- Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Diéras V, Guardino E, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91. doi:10.1056/NEJMoa1209124.
- Pfizer. BESPONSA® Approved in the EU for Adult Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia [Internet]. 2017 [cited 2017 Aug 4]; Available from: http://press.pfizer.com/press-release/besponsa-approved-eu-adult-patients-relapsed-or-refractory-b-cell-precursor-acute-lymp
- Beck A, Reichert JM. Antibody-drug conjugates. MAbs. 2014;6:15–7. doi:10.4161/mabs.27436.
- Ponte JF, Ab O, Lanieri L, Lee J, Coccia J, Bartle LM, Themeles M, Zhou Y, Pinkas J, Ruiz-Soto R. Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, Potentiates the Activity of Standard of Care Therapeutics in Ovarian Cancer Models. Neoplasia. 2016;18:775–84. doi:10.1016/j.neo.2016.11.002.
- Seattle Genetics. Vadastuximab Talirine discontinued [Internet]. VADASTUXIMAB TALIRINE2017 [cited 2017 Aug 15]; Available from: http://www.seattlegenetics.com/pipeline/vadastuximab-talirine
- Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, Stone I, Ryan MC, Sussman D, Lyon RP, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122:1455–63. doi:10.1182/blood-2013-03-491506.
- Rossi A. Rovalpituzumab tesirine and DLL3: a new challenge for small-cell lung cancer. Lancet Oncol. 2017;18:3–5. doi:10.1016/S1470-2045(16)30575-7.
- Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–37. doi:10.1038/nrd.2016.268.
- Cardillo TM, Govindan SV, Sharkey RM, Trisal P, Arrojo R, Liu D, Rossi EA, Chang C-H, Goldenberg DM. Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody-Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. Bioconjug Chem. 2015;26:919–31. doi:10.1021/acs.bioconjchem.5b00223.
- Miller ML, Fishkin NE, Li W, Whiteman KR, Kovtun Y, Reid EE, Archer KE, Maloney EK, Audette CA, Mayo MF, et al. A New Class of Antibody-Drug Conjugates with Potent DNA Alkylating Activity. Mol Cancer Ther. 2016;15:1870–8. doi:10.1158/1535-7163.MCT-16-0184.
- Lambert JM, Chari RVJ. Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. J Med Chem. 2014;57:6949–64. doi:10.1021/jm500766w.
- Haddley K. Brentuximab vedotin: its role in the treatment of anaplastic large cell and Hodgkin's lymphoma. Drugs Today. 2012;48:259–70. doi:10.1358/dot.2012.48.4.1788435.
- Damelin M, Zhong W, Myers J, Sapra P. Evolving Strategies for Target Selection for Antibody-Drug Conjugates. Pharm Res. 2015;32:3494–507. doi:10.1007/s11095-015-1624-3.
- Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108:479–85. doi:10.1038/bjc.2012.581.
- Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, Leece BA, Chittenden T, Blättler WA, Goldmacher VS. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66:3214–21. doi:10.1158/0008-5472.CAN-05-3973.
- Dornan D, Bennett F, Chen Y, Dennis M, Eaton D, Elkins K, French D, Go MAT, Jack A, Junutula JR, et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood. 2009;114:2721–9. doi:10.1182/blood-2009-02-205500.
- Boyiadzis M, Foon KA. Approved monoclonal antibodies for cancer therapy. Expert Opin Biol Ther. 2008;8:1151–8. doi:10.1517/14712598.8.8.1151.
- Palumbo A, Hauler F, Dziunycz P, Schwager K, Soltermann A, Pretto F, Alonso C, Hofbauer GF, Boyle RW, Neri D. A chemically modified antibody mediates complete eradication of tumours by selective disruption of tumour blood vessels. Br J Cancer. 2011;104:1106–15. doi:10.1038/bjc.2011.78.
- Mukherjee S, Richardson AM, Rodriguez-Canales J, Ylaya K, Erickson HS, Player A, Kawasaki ES, Pinto PA, Choyke PL, Merino MJ, et al. Identification of EpCAM as a molecular target of prostate cancer stroma. Am J Pathol. 2009;175:2277–87. doi:10.2353/ajpath.2009.090013.
- Hofmeister V, Schrama D, Becker JC. Anti-cancer therapies targeting the tumor stroma. Cancer Immunol Immunother. 2008;57:1–17. doi:10.1007/s00262-007-0365-5.
- Mack F, Ritchie M, Sapra P. The next generation of antibody drug conjugates. Semin Oncol. 2014;41:637–52. doi:10.1053/j.seminoncol.2014.08.001.
- Hurwitz E, Stancovski I, Sela M, Yarden Y. Suppression and promotion of tumor growth by monoclonal antibodies to ErbB-2 differentially correlate with cellular uptake. Proc Natl Acad Sci U S A. 1995;92:3353–7. doi:10.1073/pnas.92.8.3353.
- de Goeij BE, Lambert JM. New developments for antibody-drug conjugate-based therapeutic approaches. Curr Opin Immunol. 2016;40:14–23. doi:10.1016/j.coi.2016.02.008.
- Sauter A, Kloft C, Gronau S, Bogeschdorfer F, Erhardt T, Golze W, Schroen C, Staab A, Riechelmann H, Hoermann K. Pharmacokinetics, immunogenicity and safety of bivatuzumab mertansine, a novel CD44v6-targeting immunoconjugate, in patients with squamous cell carcinoma of the head and neck. Int J Oncol. 2007;30:927–35. doi:10.3892/ijo.30.4.927.
- Rupp U, Schoendorf-Holland E, Eichbaum M, Schuetz F, Lauschner I, Schmidt P, Staab A, Hanft G, Huober J, Sinn H-P, et al. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6-positive metastatic breast cancer: final results of a phase I study. Anticancer Drugs. 2007;18:477–85. doi:10.1097/CAD.0b013e32801403f4.
- Polson AG, Ho WY, Ramakrishnan V. Investigational antibody-drug conjugates for hematological malignancies. Expert Opin Investig Drugs. 2011;20:75–85. doi:10.1517/13543784.2011.539557.
- Smith LM, Nesterova A, Alley SC, Torgov MY, Carter PJ. Potent cytotoxicity of an auristatin-containing antibody-drug conjugate targeting melanoma cells expressing melanotransferrin/p97. Mol Cancer Ther. 2006;5:1474–82. doi:10.1158/1535-7163.MCT-06-0026.
- Wang X, Ma D, Olson WC, Heston WDW. In vitro and in vivo responses of advanced prostate tumors to PSMA ADC, an auristatin-conjugated antibody to prostate-specific membrane antigen. Mol Cancer Ther. 2011;10:1728–39. doi:10.1158/1535-7163.MCT-11-0191.
- Panowksi S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6:34–45. doi:10.4161/mabs.27022.
- Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs. 2011;3:161–72. doi:10.4161/mabs.3.2.14960.
- Agarwal P, Bertozzi CR. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug Chem. 2015;26:176–92. doi:10.1021/bc5004982.
- Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs. 2012;4:17–23. doi:10.4161/mabs.4.1.18347.
- Schumacher FF, Nunes JPM, Maruani A, Chudasama V, Smith MEB, Chester KA, Baker JR, Caddick S. Next generation maleimides enable the controlled assembly of antibody-drug conjugates via native disulfide bond bridging. Org Biomol Chem. 2014;12:7261–9. doi:10.1039/C4OB01550A.
- Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, Jonas M, Anderson ME, Setter JR, Senter PD. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33:733–5. doi:10.1038/nbt.3212.
- Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi:10.1158/1078-0432.CCR-04-0789.
- Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–32. doi:10.1038/nbt.1480.
- Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, Bhakta S, Nguyen T, Dugger DL, Li G, et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin Cancer Res. 2010;16:4769–78. doi:10.1158/1078-0432.CCR-10-0987.
- Shen B-Q, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu S-F, Mai E, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30:184–9. doi:10.1038/nbt.2108.
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–87. doi:10.1038/nrc3236.
- Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128:347–56. doi:10.1007/s10549-010-1090-x.
- Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7:303–10. doi:10.1080/19420862.2015.1011450.
- Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, Lin WY, Hu Z, Lu Y, Chen Y, et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. 2005;174:817–26. doi:10.4049/jimmunol.174.2.817.
- Uppal H, Doudement E, Mahapatra K, Darbonne WC, Bumbaca D, Shen B-Q, Du X, Saad O, Bowles K, Olsen S, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res. 2015;21:123–33. doi:10.1158/1078-0432.CCR-14-2093.
- Zhao H, Gulesserian S, Ganesan SK, Ou J, Morrison K, Zeng Z, Robles V, Snyder J, Do L, Aviña H, et al. Inhibition of Megakaryocyte Differentiation by Antibody-Drug Conjugates (ADCs) is Mediated by Macropinocytosis: Implications for ADC-induced Thrombocytopenia. Mol Cancer Ther. 2017;16:1877–86. doi:10.1158/1535-7163.MCT-16-0710.
- Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi:10.3389/fimmu.2014.00520. PMID:26182432
- Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daëron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–25. doi:10.1182/blood-2008-09-179754.
- Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi:10.1038/nri1837.
- Wiggins B, Liu-Shin L, Yamaguchi H, Ratnaswamy G. Characterization of cysteine-linked conjugation profiles of immunoglobulin G1 and immunoglobulin G2 antibody-drug conjugates. J Pharm Sci. 2015;104:1362–72. doi:10.1002/jps.24338.
- Doñate F, Raitano A, Morrison K, An Z, Capo L, Aviña H, Karki S, Morrison K, Yang P, Ou J, et al. AGS16F Is a Novel Antibody Drug Conjugate Directed against ENPP3 for the Treatment of Renal Cell Carcinoma. Clin Cancer Res. 2016;22:1989–99. doi:10.1158/1078-0432.CCR-15-1542.
- Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J. 2008;14:154–69. doi:10.1097/PPO.0b013e318172d704.
- Crescioli S, Correa I, Karagiannis P, Davies AM, Sutton BJ, Nestle FO, Karagiannis SN. IgG4 Characteristics and Functions in Cancer Immunity. Curr Allergy Asthma Rep. 2016;16:7. doi:10.1007/s11882-015-0580-7. PMID:26182432
- Labrijn AF, Rispens T, Meesters J, Rose RJ, Bleker TH den, Loverix S, van den Bremer ETJ, Neijssen J, Vink T, Lasters I, et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3-CH3 interaction strength. J Immunol. 2011;187:3238–46. doi:10.4049/jimmunol.1003336.
- Davies AM, Rispens T, Bleker TH den, McDonnell JM, Gould HJ, Aalberse RC, Sutton BJ. Crystal structure of the human IgG4C(H)3 dimer reveals the role of Arg409 in the mechanism of Fab-arm exchange. Mol Immunol. 2013;54:1–7. doi:10.1016/j.molimm.2012.10.029.
- Bruggeman CW, Dekkers G, Bentlage AEH, Treffers LW, Nagelkerke SQ, Lissenberg-Thunnissen S, Koeleman CAM, Wuhrer M, van den Berg TK, Rispens T, et al. Enhanced effector functions due to antibody defucosylation depend on the effector cell fcγ receptor profile. J Immunol. 2017;199:204–11. doi:10.4049/jimmunol.1700116.
- Tai Y-T, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, Craigen J, Yates J, Gliddon L, Fieles W, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128–38. doi:10.1182/blood-2013-10-535088.
- Stopforth RJ, Cleary KLS, Cragg MS. Regulation of monoclonal antibody immunotherapy by fcγriib. J Clin Immunol. 2016;36 Suppl 1:88–94. doi:10.1007/s10875-016-0247-8.
- Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2017;. doi:10.1007/s13238-017-0457-8. PMID:26182432
- Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, Scallon B, Teplyakov A, Malia TJ, Strohl WR. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65:114–26. doi:10.1016/j.ymeth.2013.06.035.
- Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281:23514–24. doi:10.1074/jbc.M604292200.
- Hamblett KJ, Le T, Rock BM, Rock DA, Siu S, Huard JN, Conner KP, Milburn RR, O'Neill JW, Tometsko ME, et al. Altering Antibody-Drug Conjugate Binding to the Neonatal Fc Receptor Impacts Efficacy and Tolerability. Mol Pharm. 2016;13:2387–96. doi:10.1021/acs.molpharmaceut.6b00153.
- Han TH, Zhao B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab Dispos. 2014;42:1914–20. doi:10.1124/dmd.114.058586.
- Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM. Antibody-drug conjugates: current status and future directions. Drug Discov Today. 2014;19:869–81. doi:10.1016/j.drudis.2013.11.004.
- Scott AM, Lee F-T, Tebbutt N, Herbertson R, Gill SS, Liu Z, Skrinos E, Murone C, Saunder TH, Chappell B, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A. 2007;104:4071–6. doi:10.1073/pnas.0611693104.
- Teicher BA, Chari RVJ. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res. 2011;17:6389–97. doi:10.1158/1078-0432.CCR-11-1417.
- Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther. 2009;8:2861–71. doi:10.1158/1535-7163.MCT-09-0195.
- Kim KM, McDonagh CF, Westendorf L, Brown LL, Sussman D, Feist T, Lyon R, Alley SC, Okeley NM, Zhang X, et al. Anti-CD30 diabody-drug conjugates with potent antitumor activity. Mol Cancer Ther. 2008;7:2486–97. doi:10.1158/1535-7163.MCT-08-0388.