
ABSTRACT
T cell trafficking into tumors depends on a “match” between chemokine receptors on effector cells (e.g., CXCR3 and CCR5) and tumor-secreted chemokines. There is often a chemokine/chemokine receptor “mismatch”, with tumors producing minute amounts of chemokines, resulting in inefficient targeting of effectors to tumors. We aimed to alter tumors to produce higher levels of CXCL11, a CXCR3 ligand, to attract more effector cells following immunotherapy. Mice bearing established subcutaneous tumors were studied. In our first approach, we used modified chimeric antigen receptor (CAR)-transduced human T cells to deliver CXCL11 (CAR/CXCL11) into tumors. In our second approach, we intravenously (iv) administered a modified oncolytic vaccinia virus (VV) engineered to produce CXCL11 (VV.CXCL11). The effect of these treatments on T cell trafficking into the tumors and anti-tumor efficacy after subsequent CAR T cell injections or anti-tumor vaccines was determined. CAR/CXCL11 and VV.CXCL11 significantly increased CXCL11 protein levels within tumors. For CAR/CXCL11, injection of a subsequent dose of CAR T cells did not result in increased intra-tumoral trafficking, and appeared to decrease the function of the injected CAR T cells. In contrast, VV.CXCL11 increased the number of total and antigen-specific T cells within tumors after CAR T cell injection or vaccination and significantly enhanced anti-tumor efficacy. Both approaches were successful in increasing CXCL11 levels within the tumors; however, only the vaccinia approach was successful in recruiting T cells and augmenting anti-tumor efficacy. VV.CXCL11 should be considered as a potential approach to augment adoptive T cell transfer or vaccine immunotherapy.
Introduction
Breaking tolerance to self-antigens has recently been achieved in some cancers with the use of antibodies to checkpoint inhibitors such as CTLA4 and PD-1.Citation1-3 Unfortunately, only a small subset of patients respond to checkpoint inhibitors, and many who initially respond eventually relapse. Additional tactics are thus needed, especially strategies to generate, rather than just revitalize, anti-tumor T cells. Two approaches that are being actively pursued are vaccines and adoptive cell immunotherapy (ACT). ACT is the transfer of immune cells for the treatment of cancer or infectious disease. It is now possible to genetically engineer T cells to express chimeric antigen receptors (CARs) or T cell receptors (TCRs) that target specific tumor cell surface antigens.Citation4
Although the adoptive transfer of CAR T cells has demonstrated remarkable success in treating hematologic tumors, prominently, the use of CD19 CARs in leukemias,Citation5-8 success in solid tumors has been much more limited. The reason for this difference is not yet known, and is likely multifactorial,Citation9,Citation10 but one major limitation appears to be limited “trafficking”; that is, once an effector T cell or CAR T cell is infused into a patient or generated endogenously by a vaccine, an immediate obstacle is the ability of the T cells to successfully infiltrate the tumor.
T cell trafficking into tumors is dependent on the appropriate expression of adhesion receptors on both T cells and the tumor endothelium and on a “match” between the chemokine receptors on the effector T cells and the chemokines secreted by the tumors.Citation11 The chemokine receptors expressed at the highest levels on central or effector memory cells (which includes CAR T cells or vaccine-induced T cells) are CXCR3 and CCR5.Citation12,Citation13 Effector T cells would thus be attracted to tumors expressing chemokines such as IFN-induced CXCR3 ligands (i.e., CXCL9 [MIG], CXCL10 [IP10], or CXCL11 [ITAC]) or a CCR5 ligand (i.e., CCL5 [RANTES]). Unfortunately, there is often a chemokine/chemokine receptor “mismatch”, with tumors producing very small amounts of RANTES or CXCR3 ligands, thus resulting in inefficient targeting of CXCR3high CD8+ effectors to tumor sites.Citation14 For example, using a human CAR T cell model, we found that less than 1% of injected T cells entered subcutaneous tumors by 5 days.Citation9
One proposed approach to overcome this problem in genetically engineered T cells is to design cells that co-express “better-matched” chemokine receptors. For example, using mesothelioma tumors that make large amounts of CCL2, we demonstrated enhanced intratumoral migration of CAR T cells when they were engineered to co-express the CCR2 gene, thus leading to subsequent tumor eradication.Citation12 Similarly, others have shown that the use of GD2-CAR T cells co-expressing CCR2b exhibited improved trafficking and tumor control compared to GD2-CAR alone.Citation15 We have also recently found that the genetic inhibition of protein kinase A (PKA) activation in CAR T cells increased their ability to infiltrate tumors in vivo, in part due to higher baseline expression of CXCR3.Citation10
There are some potential limitations of this approach, however. First, although tumors may secrete chemokines like CCL2 or IL-8, these chemokines are not tumor-specific. Second, some preliminary data suggest that chronic stimulation of T cells through a chemokine receptor like CCR2 leads to the continuous production of high levels of intracellular calcium which can result in accelerated T cell hypofunction.Citation16,Citation17 Third, genetic manipulation is not possible in immunotherapy approaches that do not use adoptive T cell transfer, such as vaccines.
An alternative approach to enhancing the chemokine/chemokine receptor “match” would be to alter the tumors to enable them to secrete chemokines like CCL5 or one of the IFN-induced CXCR3 ligands so that they could potentially attract effector T cells generated by vaccines or adoptive T cell transfer.Citation13 In this study, we explored two approaches to induce tumor secretion of the chemokine CXCL11 with the goal of augmenting immunotherapy. We chose CXCL11 since it potently attracts T cells into tumor sites, thereby enhancing anti-tumor effects via association with the receptor CXCR3.Citation18-20 CXCL11 expression is also strongly induced by interferons and CXCL11 has been shown to bind CXCR3 with a higher affinity than other ligands such as CXCL9 or CXCL10.Citation21
In our first approach, we used modified CAR T cells to serve as vehicles to deliver CXCL11 into the tumor. We have previously characterized a model in which human CAR T cells targeted to the tumor antigen mesothelin (mesoCAR T cells) injected intravenously into immunodeficient mice bearing treatment-resistant human tumors expressing mesothelin enter the tumors in only very small numbers.Citation9,Citation12 Once there, they proliferate and slow tumor growth. However, the tumors are not eradicated, largely due to the loss of antitumor activity of the T cells over time. We reasoned that CAR T cells that produced CXCL11 could accumulate within the tumors and “reprogram” the tumor microenvironment, even if they became hypofunctional with regard to tumor cell killing. This could potentially attract more T cells sequestered in other organs from the original CAR T cell injection or enhance trafficking from a second injection of mesoCAR T cells, given 2 weeks after the first dose.
In our second approach, we utilized a modified oncolytic vaccinia virus (VV) that was engineered to produce CXCL11. Oncolytic vaccinia viruses (VVs) have been shown to successfully and specifically infect tumor cells and lyse them.Citation22,Citation23 However, VVs “armed” with cytokines or chemokines have been shown to be even more effective by inducing anti-tumor immune responses.Citation24-27 As an example, we reported enhanced anti-tumor efficacy with a VV expressing β.Citation28,Citation29 A VV expressing CCL5 has been used to enhance the efficacy of vaccines and cytokine-superactivated T cells.Citation30 Similarly, the use of an oncolytic adenoviral vector expressing CCL5 combined with GD2-CAR T cells robustly controlled neuroblastoma progression in mice and improved CAR T cell influx.Citation31 Recently, a VV expressing CXCL11 was shown to increase the numbers of endogenous CD8 T cells in specific murine tumors.Citation32,Citation33 Accordingly, we combined the use of a VV producing CXCL11 with mesoCAR T cells and with an anti-tumor vaccine.
Results
CAR/CXCL11 T cell studies
Human T cells expressing either a CAR targeting mesothelin (mesoCAR) or the CAR plus the human CXCL11 gene (mesoCAR/CXCL11) were generated (Suppl. ). After 10 days, expression of the CAR by flow cytometry was 40–50% for both of the T cell types (Suppl ).
Figure 1. Effects of CXCL11 transduction into Human CAR T cells. A) CXCL11 secretion: Human CAR T cells were transduced with the CXCL11 gene and placed into culture for 24 hours. As measured by ELISA assay, the CXCL11 transduced T cells secreted significantly (p < 0.001) more CXCL11. B) Chemotaxis: Activated human T cells were placed in the top of a Boyden chamber. In the lower well was placed cell media (R10) as a negative control, 10 ng/ml of recombinant human CXCL11 protein as a positive control, or conditioned media from the CAR/CXCL11 cells. Both recombinant CXCL11 and the CXCL11-conditioned media significantly (p<0.001) enhanced migration of human T cells compared to media control. C) CAR T cell Killing: Equal numbers of CAR or CAR/CXCL11 T cells were added to mesothelioma cells not expressing mesothelin (EMParental) or to mesothelioma cells expressing human mesothelin (EMMeso) at various T cell to tumor ratios. After overnight incubation, their ability to kill the target cells was determined.
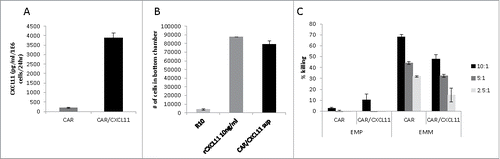
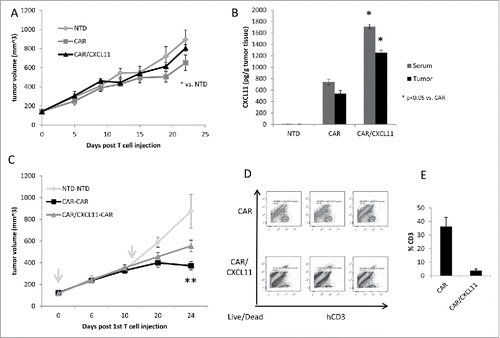
Figure 2. In vivo studies using CAR T cells. A) Tumor-bearing NSG mice were injected intravenously once with 10 million activated T cells that were either non-transduced (NTD), mesothelin-targeted CAR T cells (CAR-GFP), or CAR-CXCL11 T cells. Tumor size was followed over time. Data shown are means ± SEM, n = 5 mice per group. CAR-GFP T cells were significantly smaller than the mice receiving the NTD T cells (* = p < 0.05), with no effect elicited by he CAR-CXCL11 cells. B) On Day 22, tumors were harvested, homogenized and the amount of CXCL11 determined using an ELISA assay. C) In a second experiment, Group 1 animals received one dose of 10 million NTD T cells on Day 0 (arrow) and one dose on Day 11 (arrow). Group 2 mice received one dose of 10 million Meso-CAR T cells on Day 0 and one dose of Meso-CAR T cells on Day 11. Group 3 mice received 10 million CAR-CXCL11 CAR T cells on Day 0 and one dose of 10 million CAR T cells on Day 11. The growth of the tumors was followed until Day 24. Data shown are means ± SEM, n = 7 mice per group. The tumors in the Group 2 mice receiving of two doses of CAR T cells were significantly smaller than the NTD Group 1 mice (p < 0.001). The tumors in the Group 3 mice were significantly smaller than the NTD tumors (p < 0.01), however, the Group 3 tumors were significantly larger than the Group 2 tumors (p < 0.05). D). Tumors were harvested at the end of the study, digested, and the % of live human CD3 T cells measured by flow cytometry. E) Average percent of human CD3 T cells within the CAR vs CAR/CXCL11 groups is plotted. There were significantly more T cells in the CAR group (p < 0.001).
Supernatant from the T cell cultures were analyzed for CXCL11 concentrations by ELISA and showed high levels of secretion of CXCL11 by the CAR/CXCL11 T cells (). Supernatant from the T cells was also used to test the chemoattraction of activated human T cells in a migration assay. Compared to cell media as a negative control, conditioned media from the CAR/CXCL11 cells significantly enhanced migration of human T cells to a degree similar to that of 10 ng/ml of recombinant human CXCL11 protein used as a positive control ().
To evaluate the effect of CXCL11 expression on the function of the CAR T cells, equal numbers of mesoCAR or mesoCAR/CXCL11 T cells were added to parental, non-mesothelin-expressing cells (EMP) or to mesothelioma cells expressing human mesothelin (EMMeso) at various E:T ratios. After overnight incubation, their ability to kill the target cells was determined. As shown in , there was minimal killing of EMP cells by CAR T cells. In contrast, both types of CAR T cells were able to kill the EMMeso cells in a dose-responsive fashion. However, the killing efficiency of the same number of mesoCAR/CXCL11 T cells was significantly (P < 0.05 at all E:T ratios) reduced by 30–50% compared to MesoCAR T cells.
To test the efficacy of CAR T cells in vivo, EMMeso tumors were injected into the flanks of NSG mice and allowed to grow to ∼150 mm3. At this time point (Day 0), mice were injected intravenously with 10 million activated CAR T-positive cells that were either non-transduced (NTD), mesothelin CAR T cells (CAR/GFP), or CAR/CXCL11 T cells. As shown in , after 22 days, the tumors on CAR/GFP-treated mice were 28% smaller than NTD-treated mice (P < 0.05). In contrast, the tumors in the CAR/CXCL11-treated mice were not significantly different in size than the tumors in the NTD group. To confirm the effect of the transgene, we measured the levels of human CXCL11 in the serum and homogenized tumors from each group (). Mice receiving NTD T cells showed no detectable CXCL11 in either serum or tumor. Interestingly, clearly detectable levels of CXCL11 were seen in the serum and tumor of the mice receiving CAR T cells, suggesting that activated CAR T cells (perhaps through secretion of γ) stimulated the production of some CXCL11. However, both the serum and tumor levels were significantly (P < 0.05) higher in CAR/CXCL11-treated mice.
Despite their inability to effectively kill tumors, we hypothesized that since the CAR/CXCL11 T cells were producing CXCL11, they could potentially be used to alter the tumor microenvironment to attract other effector T cells. We therefore designed an experiment in which EMMeso tumor-bearing mice (n = 6 per group) received two intravenous doses of T cells (). Mice in Group 1 received one dose of 10 million NTD T cells on Day 0 and another dose on Day 11. Mice in Group 2 received one dose of 10 million mesoCAR T cells on Day 0 and another dose on Day 11. Mice in Group 3 received 10 million mesoCAR/CXCL11 CAR T cells on Day 0 and a dose of 10 million mesoCAR T cells on Day 11. The growth of the tumors was followed until Day 24. At this time point, the tumors in Group 2 that received two doses of CAR T cells were 68% smaller than the tumors in Group 1 that received NTD T cells (P < 0.001). The tumors in Group 3 that received CAR/CXCL11 followed by CAR T cells were 36% smaller than the tumors in Group 1 (P < 0.01). However, tumors in Group 3 tumors were significantly larger than the tumors in Group 2 (P < 0.05). On Day 24, tumors and spleens were harvested and analyzed by flow cytometry. The materials from 2 mice were pooled, therefore yielding three samples for each group for analysis. In the tumors from the mice receiving two doses of mesoCAR T cells ( and ), 36.2% of the digested cells were human CD3+ T cells. Surprisingly, in mice that received mesoCAR/CXCL11 followed by mesoCAR T cells, only 3.9% of the tumor digest was human CD3 T cells (P < 0.01). However, the percent of human T cells in the spleens of mice receiving two doses of CAR T cells was 80.3% versus 50.5% in the CAR/CXCL11 spleens (P < 0.02) (Suppl. ).
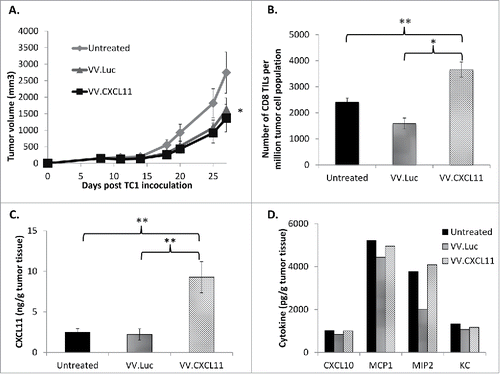
Figure 3. VV.CXCL11 administration into tumor-bearing mice led to robust CXCL11 generation, and tumor control. Wild type C57 Bl/6 mice were subcutaneously inoculated with TC1 murine lung cancer cells on the right flanks, and when tumors were established (approximately 200 mm3), they were treated with 108 pfu of VV.luc and VV.CXCL11, administered intravenously. A) Tumors were then monitored for the following 10–14 days. Data shown are means ± SEM, n = 5 mice per group. Tumors treated with both VV's were significantly smaller than control tumors (p < 0.05) but not different from one another. B) At Day 25, mice were euthanized, and tumors were excised for ex vivo analysis. Flow cytometry indicated a significant (** = p < 0.01) influx of CD8 tumor-infiltrating lymphocytes (TILs) in the VV.CXCL11-treated mice. C) Tumor tissues were processed and analyzed for CXCL11 production via ELISA. Values are reported in nanogram CXCL11 detected per gram of tumor tissue. CXCL11 levels were significantly (** = p < 0.01) higher in the VV.CXCL11 treated mice. D) The presence of other cytokines were also evaluated by ELISA to assess the specificity of CXCL11 production, and others that may be induced in this model. Values are reported in picogram of cytokines detected per gram of tumor tissue. There were no significant differences.
VV.CXCL11 Studies
Given the lack of enhanced anti-tumor efficacy by delivering CXCL11 via CAR T cells, we studied a second delivery approach using a recombinant oncolytic vaccinia virus (VV) modified to secrete CXCL11. We hypothesized that this approach might have some efficacy due to direct oncolytic effects, but would also serve to augment trafficking of adoptive transfer of T cells or endogenous generation of anti-tumor T cells using a vaccine. Thus, in these studies, we utilized a fully murine system and a VV that secreted murine CXCL11.
VV.CXCL11 particles are able to replicate and kill lung cancer cells in vitro while producing high levels of CXCL11
To evaluate the replicative potential and killing ability (and therefore, oncolytic capability) of VV.CXCL11 in culture, we infected murine TC1 lung cancer cells at a MOI of 1. Cell supernatants and lysates were collected at various time points, and titered using a standard plaque protocol. VV.CXCL11 replicated and killed TC1 cells efficiently in a time-dependent manner (Suppl. ). The in vitro cytolytic effect of our control (VV.luc) and CXCL11-producing vaccinia vectors 48 hours post-TC1 infection was identical (Suppl. ). Since this virus vector expresses CXCL11, we performed ELISA assays to detect CXCL11 secretion at differing MOIs. As expected, CXCL11 secretion intensified with increasing MOI (Suppl. ). The proliferation of two viruses in TC1 cells was also very similar (Suppl. ).
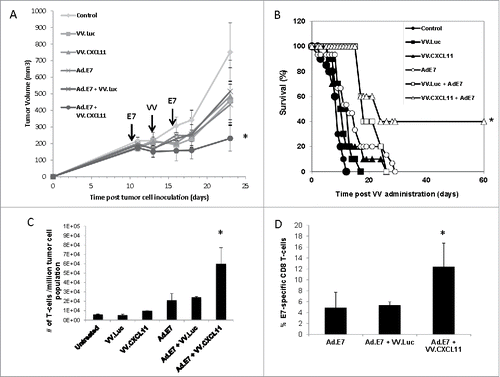
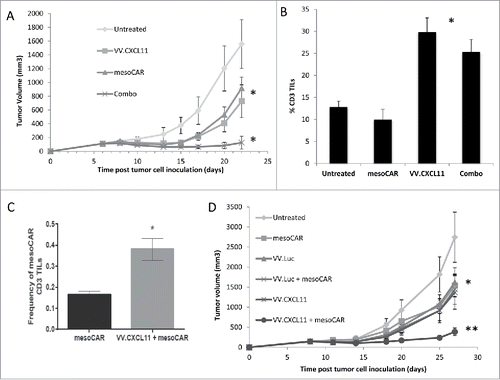
Figure 4. VV.CXCL11 significantly augmented an Ad.E7 cancer vaccine and increased T cell infiltration. A) TC1-bearing wild type mice were treated with Ad.E7 vaccine or vaccinia viruses – mice were vaccinated against E7 at Day 10 and again at Day 15, while vaccinia vectors were administered at Day 12. Tumor size was measured. Data shown are means ± SEM, n = 5 mice per group. A) Ad.E7 alone, VV.luc, VV.CXCL11, and Ad.E7 plus VV.luc all reduced the size of the TC1 tumors, but these changes did not reach statistical significance. In contrast, Ad.E7 plus VV.CXCL11 was the only group significantly smaller (p < 0.05) than control tumors. B) Survival curves from the same study, show that only the Ad.E7 plus VV.CXCL11 had prolonged survival (40%) (p < 0.05 using log-rank test). C) Tumors were harvested on Day 23. Ex vivo TIL analysis of tumors from each group revealed increased influxes of CD3 TILs in the Ad.E7 and Ad.E7 + VV.CXCL11 combination group. Data is expressed as means ± SEM, n = 5 mice per group (* = p < 0.05). D) The frequency of HPV-E7 positive CD8 TILs was determined. The percentage was significantly (p < 0.05) increased in the Ad.E7 + VV.CXCL11 combination group.
Intravenous administration of VV.CXCL11 induces generation of CXCL11 in vivo and results in modest anti-tumor control
We next determined if VV.CXCL11 was able to control tumor progression in vivo. Wild type C57 Bl/6 mice were inoculated with TC1 murine lung cancer cells and when tumors were approximately 200 mm3, 108 pfu of VV.luc or VV.CXCL11 were administered intravenously. Tumors were subsequently monitored for the next 2 weeks. At endpoint, we observed that both vectors slowed tumor progression modestly, but equally well, compared to untreated tumors (). Our group previously showed that TC1 tumors are generally sensitive to the oncolytic effectCitation29 and are thus susceptible to both vaccinia vectors in this study. We then performed ex vivo analysis of tumors and analyzed the number of CD8 tumor-infiltrating lymphocytes (TILs) by flow cytometry. Compared to the untreated and VV.luc-treated tumors, VV.CXCL11 tumors had significantly more CD8 TIL infiltration () and significantly higher levels of CXCL11 in homogenized tumor tissue (). However, we did not detect differences in the levels of other chemokines in treated TC1 tumors, indicating that CXCL11 was specifically produced in this model ().
The combinatorial administration of intravenous VV.CXCL11 and the E7 cancer vaccine elicits robust anti-tumor control and heightened influx of T cells into tumor sites
Although the intravenous administration of these vaccinia vectors had only a modest effect on tumor growth, presumably due largely to their oncolytic effects, we did observe a two-fold increase in endogenous CD8 TIL influx (). With this in mind, we next sought to augment the VV.CXCL11 effect by combining its use with an HPV-E7 cancer vaccine as delivered by an adenoviral vector (Ad.E7). Mice bearing established TC1 tumors (which expressed HPV-E7 antigen) were vaccinated subcutaneously twice, five days apart, with 109 pfu of the Ad.E7 vaccine. Two days after the first vaccination, 108 VV.luc or VV.CXCL11 viruses were intravenously administered. Each treatment had a small effect by itself, however the combinatorial therapy utilizing Ad.E7 vaccination and VV.CXCL11 controlled tumor progression to a superior and statistically significant (P < 0.05) extent compared to the other treatment groups (). Survival was also significantly longer in the animals treated with the combination (). Ex vivo TIL analysis using flow cytometry revealed a significantly higher influx of CD3+ T cells (). We also examined the % of T cells that were antigen specific by staining the CD8 T cells with an HPV-E7-specific tetramer. In tumors from mice that were untreated or treated with VV.luc or VV.CXCL11, the percent of tetramer+ cells was less than 2% (Suppl. ). Treatment with the vaccine alone or with VV.luc and vaccine resulted in an increase of intra-tumoral tetramer+ cells to about 4%, however, the percent of tetramer+ cells was significantly higher (12.4%; P < 0.05) in the tumors from mice treated with vaccine plus VV.CXCL11 ().
Figure 5. VV.CXCL11 significantly augmented mesoCAR immunotherapy. A) Mice bearing TC1-meso tumors were left untreated, injected iv with 108 pfu VV.CXCL11 on Day 5, injected iv with 107 mesothelin CAR-expressing T cells (iv) on day 8, or given both the iv VV.CXCL11 on Day 5 and the iv Meso-CAR T cells on Day 8. Tumor size was followed. Data is expressed as means ± SEM, n = 5 mice per group (* = p < 0.05). Both single treatments significantly (* = p < 0.05) reduced tumor size compared to control mice, however the combination was significantly (* = p < 0.05) more effective than either treatment alone. B) Tumors were digested and analyzed by flow cytometry on Day 22. The % of digested tumor cells that were CD3+ T cells was significantly higher in both groups receiving VV.CXCL11 (* = p < 0.05)). C) The % of adoptively transferred meso-CAR T cells present in the VV.CXCL11 treated tumors was more than double than that seen in the tumors treated with mesoCAR T cells alone (* = p < 0.05). D) In a separate experiment, groups received iv VV.luc alone and iv VV.luc plus iv Meso-CAR T cells in addition to the groups describe above in (A). Data is expressed as means ± SEM, n = 5 mice per group (* = p < 0.05). The combination of meso-CAR T cells plus VV.CXCL11 was again significantly better (p < 0.05) than all other treatments, however, administration of VV.luc did not augment the efficacy of Meso-CAR T cells. One way ANOVA with the appropriate post hoc testing, with * (P ≤ 0.05), ** (P ≤ 0.01). All experiments were replicated at least twice in an independent manner.
Intravenous VV.CXCL11 significantly augments mesoCAR immunotherapy
We next examined the ability of VV.CXCL11 to enhance the efficacy of mesoCAR adoptive immunotherapy in mice bearing mesothelin-transduced TC1 tumors (TC1-meso). Mice (n = 5 per group) bearing TC1-meso tumors were left untreated, intravenously injected with 108 pfu VV.CXCL11 on Day 6, intravenously injected with 107 mesothelin CAR-expressing T cells on day 8, or given both the VV.CXCL11 on Day 6 and mesoCAR T cells intravenously on Day 8. As shown in , both single treatments significantly (P < 0.05) reduced tumor size compared to control mice, however the combination was significantly (P < 0.05) more effective than either treatment alone. Tumors were digested and analyzed by flow cytometry on Day 22. Consistent with , the % of digested tumor cells that were CD3+ T cells was significantly higher in both groups receiving VV.CXCL11 (). Because the numbers of adoptively transferred mouse T cells peak at day 7 and then rapidly decline, we did not expect to detect mesoCAR T cells intratumorally at Day 22. However, we did observe a low, but detectable number of adoptively transferred mesoCAR T cells present in the VV.CXCL1- treated tumors that was more than double than that seen in the tumors treated with mesoCAR T cells alone (P < 0.05) ().
To demonstrate the specificity of the CXCL11 transgene in this effect, we repeated the animal experiment, but added groups that received iv VV.luc alone and iv VV.luc plus iv mesoCAR T cells. Whereas the combination of mesoCAR T cells plus VV.CXCL11 was again significantly better (P < 0.05) than all other treatments, administration of VV.luc did not augment the efficacy of mesoCAR T cells ().
Discussion
After intravenous injection, oncolytic VVs can enter tumors where they directly kill a proportion of tumor cells, and at the same time, alter the tumor microenvironment, inducing the production of many types of inflammatory cytokines.Citation24-26,Citation28,Citation29 Although this induction of inflammation is usually detrimental to viral spreading, it has become increasingly recognized that this activation of the immune system can also trigger anti-tumor immunologic responses.Citation22,Citation27 This VV-induced inflammation can be further manipulated by the introduction of specific cytokine genes into the VV genome. The most successful of the vaccinia vectors in clinical trials expresses the cytokine GM-CSF which is thought to attract dendritic cells and trigger anti-tumor immunity.Citation24-26 Our group has introduced the type 1 interferon, β, into the vaccinia vector for reasons of safety and increased efficacy.Citation28,Citation29
It is also possible to engineer VV's to express specific chemokine genes, such as CCL5 and CCL19.Citation30,Citation39,Citation40 The VV.CCL5 vector was able to increase attraction of lymphocytes and interestingly, for unclear reasons, also enhanced the persistence of the VV, raising potential safety concerns.Citation30 However, the virus enhanced the efficacy of a dendritic cell vaccine. A VV expressing CCL19 was produced with the goal of attracting dendritic cells and cytokine-induced killer cells (which unlike CARs, express the receptor of CCL19, CCR7).Citation40 The VV.CCL19 did not cause enhanced persistence of the virus and did appear to be more efficacious. It also augmented the efficacy of infused cytokine killer cells.
More recently, an oncolytic vaccinia virus that secreted CXCL11 was produced by the same group.Citation32,Citation33 As discussed above, CXCL11 was chosen as a ligand for CXCR3, the CCR most highly expressed on effector T cells. The effects of VV.CXCL11 in an intraperitoneal model of mesothelioma were studied. The results showed that compared to a control VV, endogenous T cells were attracted into intraperitoneal mesothelioma tumors in higher numbers and this was accompanied by enhanced anti-tumor efficacy.Citation32
In our study, which used intravenous injection of VV.CXCL11, we could also demonstrate marked increases in intratumoral levels of CXCL11 () along with increased number of total T cells (). In our subcutaneous model, the VV.CXCL11 was not more efficacious than the control VV (). The reasons for this difference with previous studies is not known for certain, however, since the two studies used different tumor lines, there are possible differences in the levels of endogenous immunogenicity of the tumors, the strains of mice used, and likely in the degree of replication of the virus. In the Liu paper,Citation32 the authors used the AB12 mesothelioma tumor which grows in Balb/C mice and is quite an immunogenic tumor. It appears that in that model that the tumors generate strong endogenous immunogenicity. By augmenting the trafficking of endogenously active anti-tumor T cells, they saw an increase in anti-tumor efficacy. We used a different cell line (TC1 lung cancer model) and a different mouse strain (C57B6). Since our study shows that the VV-CXCL11 induces an increase in the number of TILs, but no increase in efficacy, our data suggest the inherent immunogenicity in this line is likely lower- thus, increasing the number of T cells infiltrating the tumor has little effect. However, when the VV.CXCL11 vector was combined with immunotherapy, we did increase the trafficking of tumor-specific T cells into tumors that were generated endogenously by a vaccine or exogenously by infusion of CAR T cells targeted to the tumor. Importantly, this increased trafficking was accompanied by significantly augmented anti-tumor efficacy in both models ( and ).
Our other approach was based on the idea of using CAR T cells as vehicles to deliver a chemokine into the tumor.Citation41 This idea was pioneered in studies where production of the cytokine interleukin-12 (IL12) by CAR T cells appeared to enhance their efficacy in murine models.Citation41,Citation42 This approach resulted in some toxicity in the first human trial using TILs, but additional studies using CD19-IL-12 CARs are underway.Citation8 We postulated that secretion of CXCL11 by CAR T cells might also enhance efficacy. However, in contrast to our studies using VV.CXCL11, we were not able to enhance T cell trafficking or anti-tumor efficacy, and, in fact, when compared in a “head-to-head” fashion after one injection, the CAR/CXCL11 T cells performed much more poorly than the control CAR T cells (). We have not comprehensively explored the reasons for this, but have a number of possible explanations. It appears that the CAR-CXCL11 T cells were able to make relatively large amounts of CXCL11 (). Although that did not affect our ability to expand the cells after transduction (data not shown), when we studied the ability of the CAR-CXCL11 T cells in vitro, we noted a clear decrement (∼30%) in their ability to kill tumor cells compared to control CAR T cells (). Whereas normal chemokine/chemokine receptor signaling is relatively transient and accompanied by rapid downregulation of the CCR, we think we may have perturbed this situation by the unabated secretion of chemokine. Since CCR signaling involves increases in intracellular calcium and chronic calcium signaling can lead to T cell anergy,Citation16,Citation17 we think it is likely that the presence of chronic chemokine stimulation (or high levels of transduced CCRs (see above)) leads to enhanced T cell hypofunction, explaining their poor killing ability in vivo. It is possible that a lower level of CXCL11 secretion might have led to less hypofunction, however, since the expression of the CAR was linked to that of CXCL11, we did not have an easy way to “titrate” the level of CXCL11 secretion in our T cells.
We had hoped that the first “wave” of injected CAR T cells would start to produce CXCL11 which would then help to attract CAR T cells that were injected subsequently. Unfortunately, the opposite occurred, with fewer CAR T cells visualized in the mice that received CAR-CXCL11 followed by CAR T cells. In addition to inducing T cell hypofunction, another possible problem with having chronic, relatively high levels of CXCL11 in the tumor is the known ability of CXCL11 to inhibit angiogenesis.Citation43 Alterations in the tumor blood vessels might have inhibited the trafficking of the second dose of injected T cells explaining the low numbers of T cells we observed in the tumors (but not spleens).
Regardless of the reasons, our data clearly favor the approach using the vaccinia virus to deliver CXCL11. Our study suggests that the transient intra-tumoral pulse of CXCL11 as delivered by a vaccinia virus might avoid negative effects, like induction of T cell hypofunction or loss of blood vessels that might been seen with the chronic production of CXCL11 in the CAR T cells. Although we focused primarily on lung cancer tumor lines in this study, other murine tumor lines also appear to be similarly affected by the VV-CXCL11.Citation32,Citation33 It is also possible that vaccinia virus can work in other ways to alter the tumor microenvironment.Citation39
It may also be possible to use other viral vectors to deliver intratumoral chemokines or cytokines to enhance T cell therapy. Prior studies have shown that intratumoral injection of an adenovirus expressing CXCL10 can enhance adoptive T cell transferCitation44 or a vaccine therapy.Citation45 A recent study described the use of an intratumoral injection of oncolytic adenovirus producing CCL5 and IL15 that augmented the efficacy in intravenously injected GD2-directed CAR T cells to attack neuroblastoma.Citation46,Citation47 Other groups have proposed using oncolytic adenovirus enhance to secrete cytokines to enhance immunotherapy.Citation48 In our opinion, however, vaccinia has a major advantage over adenovirus in that can be delivered intravenously versus intra-tumorally and thus can target the multiple areas of tumor likely to be present in a patient with metastatic cancer.
This study may have important translational implications. Neither CAR T cell therapy nor cancer vaccines have shown significant effects in the treatment of solid tumors yet.Citation10 There are many possible reasons, but in both approaches, large number of anti-tumor T cells have been detected in blood, without effective trafficking to tumors. Oncolytic VV's have been tested in clinical trials and shown safety and some efficacy.Citation26 The VV-GM-CSF virus is in Phase 3 testing and may soon receive regulatory approval.Citation49 We propose that combining VV-CXCL11 with a vaccine or with adoptive T cell transfer could take advantage of the strengths of each approach with the exciting possibility of enhancing efficacy with tolerable toxicity. Enhancing T cell tumor trafficking with a VV-CXCL11 could also be used in other types of immunotherapies (dendritic cell vaccines, checkpoint inhibitors, etc.) to enhance efficacy.
Materials and methods
Creation of the CXCL11-expressing CAR
The CXCL11 gene was ordered from Integrated DNA Technologies with 5′ XbaI and 3′ BspeI flanking restriction enzyme sites. The CXCL11 gene was subcloned into a bicistronic lentiviral expression plasmid driven by the EF1α (eukaryotic translation elongation factor 1 alpha) promoter to be co-expressed with our mesoCAR (Suppl ). The SS1BBz mesoCAR design has been previously described.Citation34 Human CD4 and CD8 T cells acquired from the healthy donors were activated in vitro with anti-CD3/CD28 microbeads and were transduced with high-titer lentivirus encoding SS1BBz or SS1BBz/CXCL11. After 10 days, expression of SS1BBz by flow cytometry was 40–50% for each of the T cell types (Suppl ).
Cell lines and animals
The EMMeso human malignant mesothelioma cell line was generated from a mesothelioma pleurectomy specimen. Loss of mesothelin expression on the parental cell line (EMP) occurred in culture. Thus, EMP was transduced to stably express high levels of mesothelin to produce the EMMeso cell line. These lines were also transduced to stably express green fluorescent protein (GFP) and firefly luciferase (ff-luc).Citation12 TC1 murine lung cancer cells were established from murine lung epithelial cells immortalized with human papillomavirus-16 E6 and E7 proteins, and subsequently transformed with c-Ha-Ras.Citation35 TC1 cells stably transduced with human mesothelin (TC1-meso)Citation36 were used for immunotherapy studies. All animal protocols were approved and carried out in compliance with the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania. Pathogen-free wild-type C57 Bl/6 (strain CD45.2) mice were obtained from Charles River Laboratories. All test animals used were females used at 10–12 weeks of age.
Viruses
The vaccinia virus vector background is vvDD which as deletions in the VGF and TK viral genes.Citation32 The VV expressing firefly luciferase (VV.luc), or firefly luciferase and murine CXCL11 (VV.CXCL11) were generated as previously described.Citation32 The replicative potential of these viruses in vitro using TC1 murine lung cancer cells was assessed as using a standard plaque assay protocol.Citation29,Citation32 In vivo, TC1 tumors were dissected from mice treated with these virus vectors, and processed as described.Citation29 The generation of CXCL11 and other cytokines was measured using standard ELISA assays.Citation29 The use of the E7 cancer vaccine, as delivered by an adenoviral vector (Ad.E7) was previously established by our group.Citation37
Generation of CAR T cells
Human T cells were transduced with lentiviruses expressing the mesothelin CAR or mesothelin-CXCL11 CAR as previously described.Citation9,Citation12 Expression levels averaged between 40–70% (Supp. Figure 2). To produce murine cells expressing CAR construct targeting human mesothelin (mesoCAR), the mesothelin-CAR construct was subcloned into the retroviral MigR1 backbone also expressing GFP; primary murine T cells were isolated and transduced with these retroviral particles as previously described.Citation36 Expression levels averaged between 50–70%.
ELISA assays
Measurement of antigen-induced T cell IFN-γ secretion and transgene-induced CXCL11 secretion were measured by the Biolegend ELISA assay using the manufacturer's protocol. For IFNγ measurements, T cells were cocultured with target tumor cells at specified effector-to-target (E:T) ratios for 18 hours at 37oC and 5% CO2. For CXCL11 measurements, serum or homogenized tumor lysates were used. Supernatant was collected and analyzed for CXCL11 or IFNγ levels by ELISA.
Flow cytometry analysis
After harvesting, tumors were digested as previously described.Citation29,Citation37 Single cell suspensions were stained for surface and intracellular markers using fluorescently-labeled antibodies purchased from BD Biosciences based on the manufacturer's recommendations. The allophycocyanin-labelled H-2Db tetramer loaded with E7 peptide (RAHYNIVTF) was obtained from the National Institute of Allergy and Infectious Diseases Tetramer Core. Acquisition was performed on a CyAn-ADP Analyzer (Beckman Coulter) or a BD LSRFortessa (BD Biosciences). Data was analyzed using FlowJo (TreeStar).
MTT assays
5000 TC1 cells were plated per well in a 96-well tissue culture plate and infected with virus vectors of varying multiplicity of infection (MOI). Cell viability was assessed at several time points post-infection using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay per the manufacturer's instructions (Promega). Optical density was read at 570 nm and corrected against background control values.
In vivo tumor studies in immunodeficient mice
Animal studies were performed using NOD/SCID/γ -chain Knockout (NSG) mice that were bred at the University of Pennsylvania as described.Citation9,Citation12 Five million EMMeso tumor cells in a solution of X-Vivo media (Lonza) and Matrigel (BD Biosciences) were injected in the flanks of NSG mice. After moderately-sized tumors (150mmCitation3) were established, the mice were grouped to receive one of three treatments via intravenous tail-vein administration: 1) 10 million non-transduced (NTD) Dynabead®-activated T cells, 2) 10 million Dynabead®-activated T cells transduced with mesoCAR and GFP or 3) 10 million Dynabead®-activated T cells transduced with mesoCAR and CXCL11. Tumors were harvested 22–24 days after T cell administration and digested per a previously published protocol.Citation38 Digested tumor was then filtered through 70 um nylon mesh cell strainers and washed twice with PBS + 1% fetal bovine serum (FBS). If the digested tumor appeared contaminated with blood, red blood cell lysis with PharmLyse (BD Biosciences) was also performed. Three million cells were placed in standard flow cytometry (FACS) tubes and were stained with fluorochrome-conjugated anti-human antibodies.
In vivo tumor studies in immunocompetent mice
1.2 million TC1 or TC1-meso cells were subcutaneously inoculated on the right flanks of wild type mice, and after tumors were established (around 200 mm3), a single (unless otherwise indicated) intravenous dose of 108 pfu (100 µl) of viral vectors (VV.luc and VV.CXCL11) was administered. Tumors and mice were then monitored 2–3 times weekly, and tumor volumes were measured using digital calipers. Mice bearing tumors that exceeded 2000 mm3 and sickly mice were euthanized. For combinatorial studies with the Ad.E7 cancer vaccine, TC1-bearing mice were vaccinated subcutaneously on the left flanks (contralateral to the tumor site) with 109 pfu Ad.E7 after tumors were established (approximately 200 mm3). 2 days after vaccination, 108 pfu of VV.luc and VV.CXCL11 were intravenously administered. For combinatorial studies with mesoCAR T cells, 107 murine CAR-expressing T cells were intravenously injected 2 days after intratumoral VV.luc or VV.CXCL11 administration. At endpoint, tumors were harvested from mice and digested as described.Citation10,Citation29,Citation36,Citation37 Flow cytometry was conducted as described above.
Statistical analyses
All results were reported as means ± SEM. For studies comparing 2 groups, the Student's t test was used, while for studies comparing more than 2 groups, one- or two- way ANOVA with the appropriate post hoc testing, with * (P ≤ 0.05), ** (P ≤ 0.01). All experiments were replicated at least twice in an independent manner.
Disclosure of potential conflicts of interest
EM and SMA have received research funding from Novartis, Janssen Pharmaceuticals and Incyte Corporation; LSW is an employee and stockholder at Incyte Corporation; KB, RCL and AL have nothing to disclose; SHT is an employee and has stock equity at Western Oncolytics.
Combined_Figs_and_Suppl_Figs_for_OncoImmunology.pdf
Download PDF (645.8 KB)Suppl_Figs_for_OncoImmunology__1_.doc
Download MS Word (434.5 KB)Additional information
Funding
References
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39. doi:10.1056/NEJMoa1507643.
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi:10.1056/NEJMoa1501824.
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61. doi:10.1016/j.ccell.2015.03.001.
- June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117:1466–76. doi:10.1172/JCI32446.
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi:10.1056/NEJMoa1215134.
- Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi:10.1126/scitranslmed.aac5415.
- Sadelain M. CAR therapy: the CD19 paradigm. J Clin Invest. 2015;125:3392–400. doi:10.1172/JCI80010.
- Zhang T, Cao L, Xie J, Shi N, Zhang Z, Luo Z, Yue D, Zhang Z, Wang L, Han W, et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: a meta-analysis. Oncotarget. 2015;6:33961–71. doi:10.18632/oncotarget.5582.
- Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Puré E, Milone MC. et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20:4262–73. doi:10.1158/1078-0432.CCR-13-2627.
- Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, Puré E, Moon E, Albelda SM. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol Res. 2016;4:541–51. doi:10.1158/2326-6066.CIR-15-0263.
- Ager A, Watson HA, Wehenkel SC, Mohammed RN. Homing to solid cancers: a vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem Soc Trans. 2016;44:377–85. doi:10.1042/BST20150254.
- Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, Powell DJ Jr, Riley JL, June CH, Albelda SM.. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17:4719–30. doi:10.1158/1078-0432.CCR-11-0351.
- Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, Kato M, Prévost-Blondel A, Avril MF, Nardin A, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 2011;71:6997–7009. doi:10.1158/0008-5472.CAN-11-1466.
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85. doi:10.1158/0008-5472.CAN-08-2281.
- Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–8. doi:10.1097/CJI.0b013e3181ee6675.
- Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, Rao A. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–65. doi:10.1038/ni1047.
- Adachi Y, Hattori M, Yoshida T. Regulation of T cell activation and anergy by the intensity of the Ca(2)+ signal in cooperation with other signals. Biosci Biotechnol Biochem. 2010;74:1788–93. doi:10.1271/bbb.100107.
- Hensbergen PJ, Wijnands PG, Schreurs MW, Scheper RJ, Willemze R, Tensen CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J Immunother. 2005;28:343–51. doi:10.1097/01.cji.0000165355.26795.27.
- Berencsi K, Meropol NJ, Hoffman JP, Sigurdson E, Giles L, Rani P, Somasundaram R, Zhang T, Kalabis J, Caputo L, et al. Colon carcinoma cells induce CXCL11-dependent migration of CXCR3-expressing cytotoxic T lymphocytes in organotypic culture. Cancer Immunol Immunother. 2007;56:359–70. doi:10.1007/s00262-006-0190-2.
- Flier J, Boorsma DM, van Beek PJ, Nieboer C, Stoof TJ, Willemze R, Tensen CP. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J Pathol. 2001;194:398–405. doi:10.1002/1096-9896(200108)194:4%3c397::AID-PATH899%3e3.0.CO;2-S.
- Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, Lin W, Boyd JG, Moser B, Wood DE, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med 1998;187:2009–21. doi:10.1084/jem.187.12.2009.
- Thorne SH. Immunotherapeutic potential of oncolytic vaccinia virus. Front Oncol. 2014;4:155. doi:10.3389/fonc.2014.00155.
- Kim M. Replicating poxviruses for human cancer therapy. J Microbiol. 2015;53:209–18. doi:10.1007/s12275-015-5041-4.
- Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16:1637–42. doi:10.1038/mt.2008.143.
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, Oh SY, Han SY, Yoon JH, Hong SH, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–42. doi:10.1016/S1470-2045(08)70107-4.
- Heo J, Breitbach CJ, Moon A, Kim CW, Patt R, Kim MK, Lee YK, Oh SY, Woo HY, Parato K, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19:1170–9. doi:10.1038/mt.2011.39.
- Sampath P, Thorne SH. Arming viruses in multi-mechanistic oncolytic viral therapy: current research and future developments, with emphasis on poxviruses. Oncolytic Virother. 2014;3:1–9.
- Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi:10.1371/journal.pmed.0040353.
- Wang LC, Lynn RC, Cheng G, Alexander E, Kapoor V, Moon EK, Sun J, Fridlender ZG, Isaacs SN, Thorne SH, et al. Treating tumors with a vaccinia virus expressing IFNbeta illustrates the complex relationships between oncolytic ability and immunogenicity. Mol Ther. 2012;20:736–48. doi:10.1038/mt.2011.228.
- Li J, O'Malley M, Urban J, Sampath P, Guo ZS, Kalinski P, Thorne SH, Bartlett DL. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. 2011;19:650–7. doi:10.1038/mt.2010.312.
- Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74:5195–205. doi:10.1158/0008-5472.CAN-14-0697.
- Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology. 2016;5:e1091554. doi:10.1080/2162402X.2015.1091554.
- Francis L, Guo ZS, Liu Z, Ravindranathan R, Urban JA, Sathaiah M, Magge D, Kalinski P, Bartlett DL. Modulation of chemokines in the tumor microenvironment enhances oncolytic virotherapy for colorectal cancer. Oncotarget. 2016;7:22174–85. doi:10.18632/oncotarget.7907.
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi:10.1073/pnas.0813101106.
- Lin KY, Guarnieri FG, Staveley-O'Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res 1996;56:21–6.
- Riese MJ, Wang LC, Moon EK, Joshi RP, Ranganathan A, June CH, Koretzky GA, Albelda SM. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013;73:3566–77. doi:10.1158/0008-5472.CAN-12-3874.
- Haas AR, Sun J, Vachani A, Wallace AF, Silverberg M, Kapoor V, Albelda SM. Cycloxygenase-2 inhibition augments the efficacy of a cancer vaccine. Clin Cancer Res. 2006;12:214–22. doi:10.1158/1078-0432.CCR-05-1178.
- Quatromoni JG, Singhal S, Bhojnagarwala P, Hancock WW, Albelda SM, Eruslanov E. An optimized disaggregation method for human lung tumors that preserves the phenotype and function of the immune cells. J Leukoc Biol. 2015;97:201–9. doi:10.1189/jlb.5TA0814-373.
- Sampath P, Li J, Hou W, Chen H, Bartlett DL, Thorne SH. Crosstalk between immune cell and oncolytic vaccinia therapy enhances tumor trafficking and antitumor effects. Mol Ther. 2013;21:620–8. doi:10.1038/mt.2012.257.
- Li J, O'Malley M, Sampath P, Kalinski P, Bartlett DL, Thorne SH. Expression of CCL19 from oncolytic vaccinia enhances immunotherapeutic potential while maintaining oncolytic activity. Neoplasia. 2012;14:1115–21. doi:10.1593/neo.121272.
- Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257:83–90. doi:10.1111/imr.12125.
- Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, Rosenberg SA. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–83. doi:10.1158/1078-0432.CCR-11-3050.
- Santoni M, Bracarda S, Nabissi M, Massari F, Conti A, Bria E, Tortora G, Santoni G, Cascinu S. CXC and CC chemokines as angiogenic modulators in nonhaematological tumors. Biomed Res Int. 2014;2014:768758. doi:10.1155/2014/768758.
- Huang H, Liu Y, Xiang J. Synergistic effect of adoptive T-cell therapy and intratumoral interferon gamma-inducible protein-10 transgene expression in treatment of established tumors. Cellular Immunology. 2002; 217: 12–22. doi:10.1016/S0008-8749(02)00508-7.
- Rodriguez MMB, Ryu S-M, Qian C, Geissler M, Grimm C, Preito J, Blum H, Mohr L, Immunotherapy of murine hepatocellular carcinoma by DNA vaccination combined with adenovirus-mediated chemokine and cytokine expression. Human Gene Therapy. 2008; 19:753–759, 2008. doi:10.1089/hum.2007.130.
- Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–26. doi:10.1111/imr.12131.
- Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4:e988098. doi:10.4161/21505594.2014.988098.
- Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, Tähtinen S, Oksanen M, Heiskanen R, Pesonen S, et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res. 2013;19:2734–44. doi:10.1158/1078-0432.CCR-12-2546.
- Breitbach CJ, Moon A, Burke J, Hwang TH, Kirn DH. A phase 2, open-label, randomized study of pexa-vec (JX-594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol Biol. 2015;1317:343–57. doi:10.1007/978-1-4939-2727-2_19.