
ABSTRACT
Autologous patient-derived dendritic cells (DCs) modified ex vivo to present tumor-associated antigens (TAAs) are frequently used as cancer vaccines. However, apart from the stringent logistics in producing DCs on a patient basis, accumulating evidence indicate that ex vivo engineered DCs are poor in migration and in fact do not directly present TAA epitopes to naïve T cells in vivo. Instead, it is proposed that bystander host DCs take up material from vaccine-DCs, migrate and subsequently initiate antitumor T-cell responses. We used mouse models to examine the possibility of using pro-inflammatory allogeneic DCs (alloDCs) to activate host DCs and enable them to promote antigen-specific T-cell immunity. We found that alloDCs were able to initiate host DC activation and migration to draining lymph node leading to T-cell activation. The pro-inflammatory milieu created by alloDCs also led to recruitment of NK cells and neutrophils at the site of injection. Vaccination with alloDCs combined with Ad5M(gp100), an infection-enhanced adenovirus encoding the human melanoma-associated antigen gp100 resulted in generation of CD8+ T cells with a T-cell receptor (TCR) specific for the gp10025-33 epitope (gp100-TCR+). Ad5M(gp100)-alloDC vaccination in combination with transfer of gp100-specific pmel-1 T cells resulted in prolonged survival of B16-F10 melanoma-bearing mice and altered the composition of the tumor microenvironment (TME). We hereby propose that alloDCs together with TAA- or neoepitope-encoding Ad5M can become an “off-the-shelf” cancer vaccine, which can reverse the TME-induced immunosuppression and induce host cellular anti-tumor immune responses in patients without the need of a time-consuming preparation step of autologous DCs.
Introduction
Cancer immunotherapy has made enormous progress recently, thanks to the introduction of checkpoint blockade antibodies, in particular antibodies blocking programmed cell death protein 1 (PD-1) and PD-1 ligand (PD-L1) interactions.Citation1 However, only a fraction of cancer patients benefits from this treatment and it is only effective against certain forms of cancer. As there is still room for improvement in the field, cancer immunotherapies evolve to be more efficient and robust.
Cancer vaccines aim to educate patients' immune system to recognize and eradicate cancer cells,Citation2 in particular to activate CD8+ T cells against peptides from tumor-associated antigens (TAAs) or patient-specific neoepitopes expressed on MHC class I by the cancer cells.Citation3 This is particularly important since clinical studies have demonstrated that therapeutic benefit has a strong correlation with the presence of tumor-infiltrating CD8+ T cells.Citation4 Dendritic cells (DCs) are professional antigen-presenting cells that are often used as cancer vaccines due to their educative roles in immunity. DCs have the ability to secrete pro-inflammatory cytokines and play a key role by bridging innate and adaptive immune responses. Immature DCs (imDCs) capture antigens at the site of inflammation, mature and migrate to draining lymph nodes (dLNs), process the antigens into peptides and present the peptides on major histocompatibility complex (MHC) class II molecules to activate CD4+ helper T cells. They can also cross-present exogenously captured antigens on MHC class I molecules to activate CD8+ cytolytic T cells. In addition, they communicate with a gamut of innate immune cells, specifically NK cells which secret interferons (IFNs) when stimulated.
DC-based cancer vaccines have been tested extensively in clinical trials.Citation5 DC vaccines are typically produced from patient's blood monocytes and loaded ex vivo with TAAs. Lately, selection of myeloid or plasmacytoid DC-subsets have been used for DC vaccines in order to take advantage of certain characteristics these subtypes possess.Citation6,7 However, recent findings indicate that ex vivo-generated DCs cannot migrate efficiently after re-administration.Citation8,9 Moreover, it appears that endogenous DCs are required for the induction of potent CD8+ T-cell responses, as bystander host DCs have been found to be essential in priming specific CD8+ T-cell immunity after DC vaccination.Citation10,11 This can be an explanation for the limited therapeutic clinical success of DC vaccines as the tumor microenvironment (TME) negatively affects the endogenous host DCs, not only in the vicinity of the tumor site but at distant sites, such as the dLN.Citation12 TME-mediated immune suppression is achieved mainly by the accumulation of myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), anergic imDCs and regulatory T cells (Tregs), together with high levels of immune-suppressing cytokines, notably TGF-β and IL-10.Citation13–16 Anti-tumor immune reactions are further hindered by poor infiltration of immune effector cells into the tumor site and expression of PD-L1 by tumor cells.Citation17 Thus, novel effective immunotherapeutic strategies should aim to reverse this state of immune suppression and achieve expansion of cytotoxic TAA-directed T cells.Citation18
This led us to develop TAA-modified, pro-inflammatory allogeneic DCs (alloDCs) as a stimuli for host bystander DCs. AlloDCs as vaccine cells are designed to release substantial amounts of immune stimulating chemokines and cytokines. The local secretion of these factors within the tumor will recruit and activate endogenous immune cells, including NK cells, T cells and imDCs. Such scenario is in line with several experimental in vivo studies where autologous vaccine DCs have been shown to function as pure immune enhancers, generating antigen-specific T-cells indirectly through recruiting and activating endogenous immune cells, including bystander DCs.Citation10,11,19 AlloDCs can be further regarded as MHC-incompatible invaders, which can potentiate an inflammatory allogeneic mixed leucocyte reaction (MLR) at the vaccination site that further promotes the recruitment and activation of bystander DCs.Citation20 Thus, we hypothesize that alloDCs can function as strong adjuvants to attract innate effector immune cells and break the immunosuppressive TME in order to promote effector T-cell responses against TAAs released from the destroyed tumor cells.
Intratumoral administration of pro-inflammatory alloDCs (Ilixadencel) was recently shown to induce an anti-tumor immune response that may prolong survival in unfavorable risk metastatic renal cell carcinoma (mRCC)-patients given subsequent standard of care.Citation21 Additionally, we have shown that pro-inflammatory, adenoviral-transduced alloDCs created an immunostimulatory environment in vitro, which promoted the maturation of bystander DCs able to cross-present antigens to antigen-specific effector T cells (co-published data). Here, we evaluate alloDCs in combination with an infection-enhanced adenoviral vector in order to load alloDCs with TAAs. We found that Ad5M(gp100)-alloDCs stimulated the proliferation of both endogenous gp100-TCR+ T cells and adoptively transferred pmel-1 (gp-100-directed) CD8+ T cells and altered the immunosuppressive TME, resulting in delayed tumor growth and prolonged survival. The present findings indicate that the combination of pro-inflammatory alloDCs and a TAA-carrying infection-enhanced adenoviral vector facilitate a cost-effective immunotherapy strategy, flexible to be combined with other established tumor immunotherapies.
Results
Vaccination with Ad5M(gp100)-alloDCs resulted in host DC maturation and stable gp100-specific T-cell responses
Murine DCs were generated from the bone marrow of Balb/C (H-2Dd) mice and matured with polyI:C, R848 and IFN-γ,Citation22 alone or with Ad5M(gp100). Their maturation status was verified by surface marker staining and their secretion profile was verified by analysis of CXCL-10 and IL-12 secretion (Supplementary Fig. S1 A-C). Notably, addition of the Ad5M(gp100) vector does not negatively affect the DCs in terms of maturation status or cytokine secretion. Therefore, Ad5M can be used for antigen delivery to DCs. Since those DCs are allogeneic to the recipient C57BL/6NRj (H-2Db) mice, from here on they will be referred to as alloDCs.
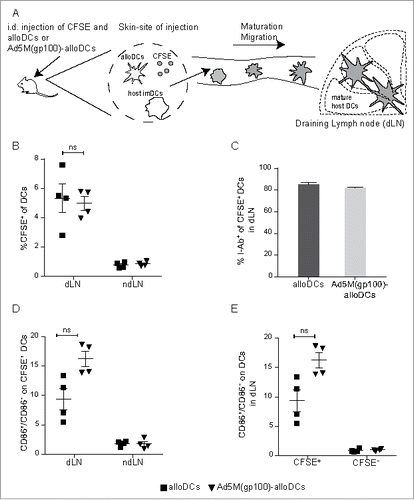
The inflammatory environment created by MLR and alloDCs has been found able to mature bystander DCs in vitro (20 and co-published data). It is also known that Th1-polarizing immune triggering factors secret by infected DCs have major importance in the activation of bystander DCs.Citation19 We have already shown that Ad5M vector injection by itself is not sufficient to activate resident DC and induce them to migrate to draining lymph nodes, unless Ad5M is encoding an immune-activating protein.Citation23 In order to evaluate if alloDCs can provide immune-activating signals in vivo and potentiate the migration of bystander DCs to dLN, C57BL/6NRj mice were injected intradermally (i.d.) with alloDCs or Ad5M(gp100)-alloDCs, together with the fluorescent cell-membrane dye CFSE (). We hypothesized that the co-injected CFSE will label host cells at the site of injection including host DCs, which are expected to get activated due to the emerging pro-inflammatory milieu and subsequently migrate into the dLNCitation23,24 (). CFSE-labeled (CFSE+) CD11chighCD11b+B220− DCs, were observed in the dLN 48 hours after injection (). The gating strategy can be found in Supplementary Fig. S2 A. Staining with the MHC-II marker I-Ab revealed that more than 80% of the CFSE+ DCs in the dLN are host C57BL/6NRj DCs (). These CFSE+ host DCs were activated, as observed by the expression of CD86 both after alloDC and Ad5M(gp100)-alloDC injections (). No activation was observed for the CFSE− LN-resident host DCs (), indicating that activation was specific for the CFSE+ LN-migrating host DCs. Evaluation of the CFSE+CD86− non-activated population of DCs helped us exclude that CFSE was diffusing to the draining lymph nodes (Supplementary Fig. S2B). As a control for the specificity of the migratory-effect, we used the non-draining lymph node (ndLN) obtained from the opposite side of the same mice where no CFSE+ cells were detected (, , Supplementary Fig. S2B).
Figure 1. AlloDCs injection results in migration of host DCs to the draining lymph node. (A) C57BL/6NRj (H2-Db) mice were injected intradermally (i.d.) on the right hind flank with 1×106 alloDCs or Ad5M(gp100)-alloDCs, derived from BALB/c (H2-Dd) mice, and 20 μg CFSE (fluorescent cell-membrane dye). Activation and migration of host DCs to the draining lymph node was analyzed 48 h post-injection by flow cytometry. (B) The presence of migrated DCs was evaluated as % CFSE+ cells of CD11chighCD11b+B220− DCs in the draining (dLN) and non-draining (ndLN) lymph node. (C) Co-staining the CFSE+ DCs in the dLN with I-Ab, a C57BL/6NRj haplotype-specific antibody labelling only host DCs. Shown is the % of I-Ab+ cells of all CFSE+CD11b+B220− DCs. (D) The activation status (expression of the co-stimulatory molecule CD86) of the migrated CFSE-labelled DCs (CD11chighCD11b+B220−) in dLN and ndLN was evaluated and presented as the ratio of CD86+/CD86− cells. (E) The activation status (expression of CD86) of migrated (CFSE+) and resident (CFSE−) DCs (CD11chighCD11b+B220−) in the dLN was also evaluated and presented as the ratio of CD86+/CD86− cells. Data are shown as mean±SEM (n.s. p≥0.05).
We speculated that host bystander DCs activated by the emerged milieu can take up antigens from the site of injection and educate T cells in the dLN. In order to evaluate this hypothesis, C57BL/6NRj mice were vaccinated subcutaneously (s.c.) on day 0 and 10 with 5×109 evg of Ad5M(gp100) either alone or after transduction of 1×106 alloDCs (). Ad5M(MOCK) was used as negative control. On day 17 blood samples were drawn and analyzed and gp100-TCR+ CD8a+ T cells were identified in 10/15 mice vaccinated with Ad5M(gp100) alone and in 14/15 mice vaccinated with Ad5M(gp100)-alloDCs (). No gp100-TCR+ T cells were observed after vaccination with control Ad5M(MOCK) and Ad5M(MOCK)-alloDCs (data not shown). Mice were subsequently injected with Hcmel12 cells, derived from a primary HGF-CDK4(R24C) melanoma,Citation25 and tumor size was monitored. In this prophylactic setting, presence of gp100-TCR+ T cells was correlated with a lower tumor burden (). In a more specific analysis, only mice that received vaccination with Ad5M(gp100)-alloDCs had a significant correlation for smaller tumors than mice that received Ad5M(gp100) alone (, ).
Figure 2. Combination treatment with alloDCs and an infection-enhanced adenoviral vector providing TAA results in efficient induction of endogenous TAA-specific T cells. (A) C57BL/6NRj mice were vaccinated s.c. in a prime (day 0) and boost (day 10) prophylactic setup with 5×109 evg of Ad5M(gp100) either alone or after transduction of 1×106 alloDCs. (B) Blood was collected one week later (day 17) and the presence of T cells (CD8a+CD3+) with a TCR for the human gp10025-33 epitope (gp100-TCR+) were analyzed with an H-2Db/hgp10025-33-PE tetramer in flow cytometry. Presence of gp100-TCR+ T cells was determined as positive if a distinct population above 0.5% of the parental CD8a+CD3 a+ population (cut off value for Ad5M(MOCK)) was observed. Shown is a chart with the number of mice with gp100-TCR+ T cells. (C, D, E) The vaccinated mice were then inoculated (day 18) with 2×105 Hcmel12 cells, derived from a primary HGF-CDK4(R24C) melanomaCitation25 and tumor growth was monitored. Shown is a scatter plot representing the correlation of tumor size and % of gp100-TCR+ T cells at the time of the first recorded tumor-related death (day 40) for (C) all controls and treatments pooled (r = −0.3271, p = 0.0450), (D) mice treated with Ad5M(gp100) and control (r = −0.1256, p = 0.5681) and (E) mice treated with Ad5M(gp100)-alloDC and control (r = −0.5434, p = 0.0074). (F) For a therapeutic setting, C57BL/6NRj mice were inoculated with 5 ×105 Hcmel3 cells, another primary HGF-CDK4(R24C) melanoma with slower growth kinetics,Citation26 and received three consecutive weekly i.t injections with 5×109 evg Ad5M(gp100) alone or after transduction of 1×106 alloDCs. Control mice were treated with 5×109 evg Ad5M(MOCK) or 1×106 alloDCs. (G) The presence of gp100-TCR+ T cells was evaluated by tetramer flow cytometry, one month after inoculation of tumors. (H) Scatter plot representing the correlation of tumor size and % of gp100-TCR+ T cells of CD3+ T cells (r = −0.5837, p = 0.0155) at the time of sacrifice.
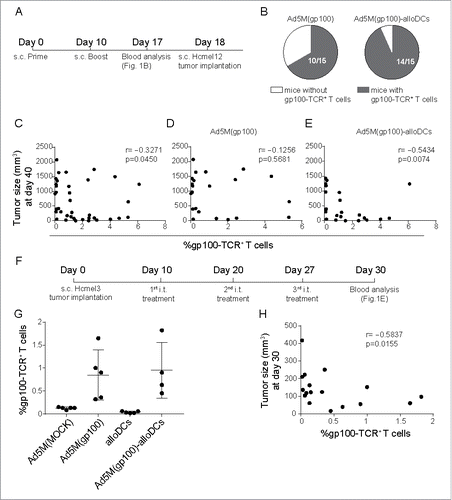
To validate our findings in the therapeutic setting, mice were inoculated with Hcmel3 cells, followed by three consecutive weekly intratumoral (i.t.) injections with 5×109 evg Ad5M(gp100) alone or after transduction of with 1×106 alloDCs (). Hcmel3 is a cell line derived from primary HGF-CDK4(R24C) melanoma that has slower growth kinetics than Hcmel12.Citation26 Control mice were treated with 5×109 evg Ad5M(MOCK) or 1×106 alloDCs. Mice were sacrificed one month after inoculation of tumor cells and the presence of gp100-specific immunity was confirmed (). A correlation between lower tumor burden and presence of gp100-TCR+ effector T cells was verified also in this setting (). Overall, vaccination with Ad5M(gp100)-alloDCs resulted in more frequent gp100-TCR+ T-cell responses in comparison to vaccination with Ad5M(gp100) alone, an important feature given the correlation between gp100-TCR+ T-cell presence and lower tumor burden.
Injection of alloDCs induced local infiltration of immune cells and altered the tumor microenvironment
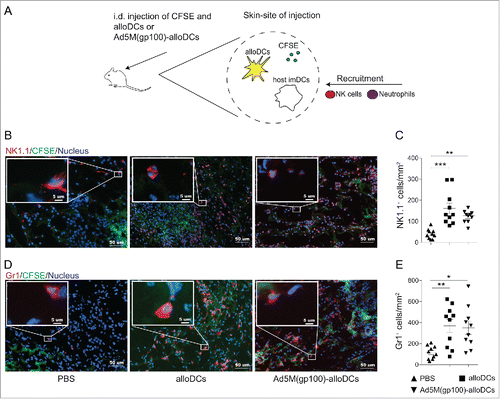
Besides inducing specific T-cell responses, an anti-cancer treatment is desirable to have a general immune-boosting effect, engaging several effector immune cells and altering the immunosuppressive TME.Citation18 In order to evaluate this potential of alloDCs, C57BL/6NRj mice were injected i.d. in the right hind limb with alloDCs or Ad5M(gp100)-alloDCs together with the fluorescent cell-membrane dye CFSE () and the injected sites were collected and examined for cell infiltration 48 hours post-injection. The skin CFSE staining guided us to identify the site of injection when analyzing the sections. Significantly higher populations of NK1.1+ and Gr1+ cells were found at the site of injection for both groups (, , and ), showing substantial infiltration of NK cells and neutrophils.
Figure 3. The pro-inflammatory milieu induced by alloDCs recruits innate immune cells to the site of injection. (A) C57BL/6NRj mice were injected i.d. with 1×106 alloDCs, along with 20 μg CFSE (green fluorescent cell-membrane dye) to be able to identify the injection site. Recruitment of innate immune cells at the site of injection was analyzed after 48 hours. (B-E) Recruitment of NK1.1+ NK cells (B, C) and Gr1+ neutrophils (D, E) to the site of injection in response to injected alloDCs was analyzed by immunofluorescent staining. Representative staining of one mouse per group is shown in B and D (green = injected area, blue = nuclei, red = NK1.1 or Gr1). Quantification of infiltrated NK-cells and neurophils (C, E) was determined as number of positive cells per mm2. Data are shown as mean±SEM (* P<0.05; ** P<0.01; *** P<0.001).
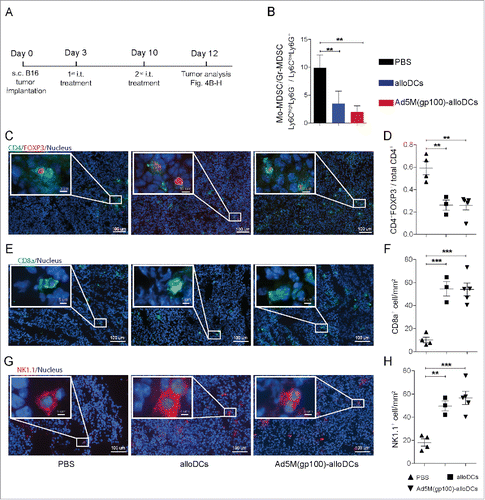
It is a common observation that tumor responses and increased survival are strongly correlated with a shift of the TME from immunosuppressive to immunogenic.Citation27,28 To examine if the aforementioned immune activating effect occurs in the tumor setting, alloDCs were injected i.t. in B16-F10 melanoma-carrying C57BL/6NRj mice (). B16-F10 melanoma tumors are known for having a very aggressive growth and are poorly immunogenic, therefore are a good model to evaluate the strength of potential immunotherapies. Myeloid-derived suppressor cells (MDSCs) have a major role in tumor immunosuppression and in the murine system they can be subcategorized based on Ly6C and Ly6G expression into “monocytic” or “granulocytic”, with the monocytic-MDSCs (Mo-MDSCs) having more potent immunosuppressive functions.Citation29 We sought to observe changes in these populations upon the different treatments, identifying granulocytic MDSCs (Gr-MDSCs) as Ly6ClowLy6G+ and Mo-MDSCs as Ly6ChighLy6G− (Supplementary Fig. S3 A). A lower frequency of Gr-MDSCs was found in tumors receiving control treatment (PBS) than alloDCs or Ad5M(gp100)-alloDCs, as indicated by the higher ratio of Mo-MDSC to Gr-MDSC (). The lower ratio of Mo-MDSC/Gr-MDSC was actually a result of higher Gr-MDSC infiltration (Supplementary Fig. S3B, C). Regulatory CD4+FOXP3+ T- cells (Tregs) are suppressive in the TME and capable to dismiss immune responses against tumors.Citation30 We therefore evaluated if the reduction in the ratio of Mo-MDSCs/Gr-MDSCs was accompanied by a decrease in the frequency of Tregs. Both alloDC-treated and Ad5M(gp100)-alloDC-treated tumors exhibited less CD4+FOXP3+ T-cell infiltration relative to total CD4+ T cells per tumor area, compared to the PBS-treated tumors, as illustrated in representative figures () and quantitative analysis (). In contrast, CD8+ T-cell infiltration, which is often a beneficial factor, was significantly higher in both alloDC-treated and Ad5M(gp100)-alloDC-treated tumors compared to PBS (, ). Moreover, higher NK-cell infiltration was observed in alloDC-treated and Ad5M(gp100)-alloDC-treated tumors (, ).
Figure 4. Intratumoral injection with Ad5M(gp100)-alloDCs reduces the immunosuppressive melanoma tumor microenvironment. (A) C57BL/6NRj mice were injected s.c. into the right hind flank with 1×105 B16-F10 cells (day 0) and received two consecutive i.t. injections of 1×106 alloDCs, Ad5M(gp100)-alloDCs or PBS (negative control) on days 3 and 10. (B) The effect on the tumor microenvironment (TME) was investigated two days later (day 12) as changes in the ratio of monocytic to granulocytic myeloid-derived suppressor cells, i.e., Mo-MDSC(Ly6ChighLy6G−)/Gr-MDSC(Ly6ClowLy6G+). Mean values from 5 individual mice per group are shown. (C) The CD4+FOXP3+ regulatory T cells were determined in the tumor by immunofluorescence staining and representative pictures for one mouse per group of each treatment are shown (green = CD4, red = FOXP3, blue = nuclei). (D) Evaluation was exhibited as changes in the ratio of CD4+FOXP3+/total CD4+ T cells. (E) Representative pictures of tumor infiltrating CD8a+ T cells (green = CD8a, blue = nuclei) and (F) quantification are shown. (G) Representative pictures of tumor infiltrating NK1.1+ NK cells (red = NK1.1, blue = nuclei) and quantification (H) are shown. Data are shown as mean±SEM ** P<0.01; *** P<0.001).
Ad5M(gp100)-alloDC vaccination augmented the efficacy of adoptively transferred pmel-1 T cells and delayed tumor growth
We proceed examining if Ad5M(gp100)-alloDCs in combination with adoptive transfer of gp100-specific T cells from splenocytes of pmel-1 mice has a positive survival effect in melanoma-bearing mice. Initially, naïve C57BL/6NRj mice were vaccinated s.c. on the right hind limb with 1×106 alloDCs or Ad5M(gp100)-alloDCs, followed by intravenous (i.v.) adoptive transfer of 10×107 pmel-1 splenocytes (). In contrast to alloDCs, Ad5M(gp100)-alloDCs were able to stimulate the adoptively transferred gp100-specific Thy1.1+ CD8+ T cells and sustain their presence five days after transfer ().
Figure 5. The combination of intratumoral administration of Ad5M(gp100)-alloDCs and adoptive transfer of pmel-1 T cells leads to prolonged survival of B16-F10 tumor-bearing mice. (A) The ability of Ad5M(gp100)-alloDCs to indirectly stimulate gp100-specific T cells in non-tumor-bearing mice was assessed by combining s.c. injection of Ad5M(gp100)-alloDCs (day 0) with i.v. adoptive cell transfer of gp100-specific Thy1.1+ pmel-1 splenocytes (day 2). T-cell read out was performed five days later by Thy1.1 staining (day 7). (B) Representative scatter plots of the specific proliferation of Thy1.1+ pmel-1 T cells (Thy1.1+) in response to alloDCs or Ad5M(gp100)-alloDCs from two mice are shown. (C) In a therapeutic setting, C57BL/6NRj mice were first injected s.c. into the hind flank with 1×105 B16-F10 cells (day 0) and received 1×107 pmel-1 splenocytes i.v. (day 8). The treatment was combined with i.t. injections of 1×106 Ad5M(gp100)-alloDCs (day 8 and 14) or PBS as negative control. Tumor growth was monitored by caliper measurement. (D) Average tumor growth is presented for each treatment (n = 8 per group). (E) Mice survival is shown by the Kaplan-Meier survival curve and compared by log-rank test (n = 8 per group) (* P<0.05). (F-K) Mice were sacrificed two days after the last treatment (day 16) for immunological systemic and tumor analyses. Systemic presence of pmel-1 T cells was verified as % Thy1.1+Vβ13+ of CD8a+ T cells in (F) spleen, (G) dLN and (H) blood. (I) The effect of each treatment on the TME was evaluated day 16 post B16-F10 inoculation. It is presented as the ratio of Mo-MDSC/Gr-MDSC; mean values from 5 individual mice per group are shown. Infiltration of CD8a+ T cells in the tumor is shown as (J) % CD8a+ of CD3+ T cells and exhausted CD8a+ T cells as (K) % PD1+Tim3+ double positive cells of CD8a+ T cells. Data are shown as mean±SEM (* P<0.05; ** P<0.01; *** P<0.001; **** P<0.0001).
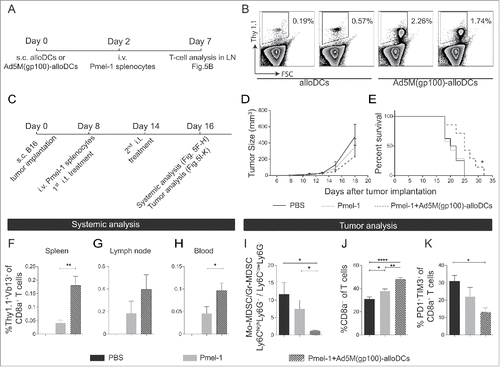
As pmel-1 T cells persist after vaccination with Ad5M(gp100)-alloDCs (), we next evaluated the combinatorial effect of adoptive pmel-1 splenocyte transfer with i.t. injection of Ad5M(gp100)-alloDCs () in a therapeutic setting. The combined treatment delayed tumor growth () and significantly prolonged survival of tumor-bearing mice (). An altered TME was also observed with a lower ratio of Mo-MDSCs/Gr-MDSCs () and increased infiltration of CD8+ T cells in the tumor samples (), in accordance with previous findings (, ). These T cells expressed a less exhausted phenotype, as assessed by double staining of Tim-3 and PD-1 (). The amount of pmel-1 T cells was higher in spleen (), dLN () and blood () eight days post pmel-1 splenocyte transfer when combined with i.t. Ad5M(gp100)-alloDCs treatments compared to pmel-1 splenocyte transfer alone.
Discussion
In the present study, we demonstrated the use of alloDCs as a cancer-immunotherapeutic adjuvant that can potentiate T-cell responses and reverse the immunosuppressive TME in a therapeutically favorable manner.
Effective priming of T-cell responses requires DCs to take up antigens, mature and migrate to dLNs where they can productively interact with naïve T cells.Citation31,32 AlloDCs could efficiently stimulate host DC activation and migration to the dLN (). Murine DCs activated by inflammatory stimuli have been reported to upregulate co-stimulatory molecules and migrate to the dLN.Citation23,33 In line with these findings, alloDC administration also induced migrating host DCs in the dLN (I-Ab+CFSE+) (, ) which had an activated phenotype identified by upregulated CD86 expression (, ). Unspecific diffusion of the CFSE dye to dLN or ndLN was not observed, as evaluated by the absence of a non-activated CFSE+CD86− population (Supplementary Fig. S2B).
Furthermore, we confirmed the ability of these migratory host DCs to take up the gp100 melanoma-associated antigen from the injected Ad5M(gp100)-alloDCs and cross-present the antigen to expand both endogenous CD8+ gp100-TCR+ T cells (, ) and adoptively transferred T cells from pmel-1 mice (). Both in the prophylactic and therapeutic setting, the presence of gp100-TCR+ T cells correlated with lower tumor burden (, and ). However, it is generally observedd that therapeutic vaccination aiming for the treatments of established tumors is ineffective, though capable in inducing antigen-specific T cells.Citation34 We observe the same effect, where gp100-specific T cells were evoked and could control early onset of tumors (, ) but eventually did not result in prolonged survival of tumor bearing mice (data not shown). The same inefficiency to induce tumor regression applies to TAA-specific T-cell transfer, however epitope-specific re-stimulation along with repeated IL-2 administration can efficiently prevent tumor growth, as has been observed before for pmel-1 T-cell transfer to mice with “hard-to-treat” B16-F10 tumors.Citation35,36 In combination with adoptive pmel-1 T-cell transfer, even in the absence of IL-2 support, Ad5M(gp100)-alloDCs were able to stimulate and expand pmel-1 T cells in vivo (, , , ). This resulted not only in tumor-growth control, but also to a small but significant survival benefit of B16-F10 melanoma-bearing mice (, ).
The alloDC-induced pro-inflammatory milieu was efficient in altering the immunosuppressive TME by recruiting innate immune cells, such as NK cells and neutrophils, to the site of injection (, and , ). This is in line with the latest findings regarding favorable prognosis and optimal therapeutic efficacy of immunotherapy suggesting that one should aim for a TME infiltrated by heterogeneous populations of effector immune cells.Citation18 In our setting, B16-F10 melanoma tumors receiving i.t. injections of alloDCs and Ad5M(gp100)-alloDCs exhibited an altered TME as revealed by higher infiltration of CD8+ T cells (, and ) and NK cells (, ), accompanied by relative reductions of suppressive types of MDSCs ( and ) and Tregs (, ). Interestingly, such findings were not observed when Ad5M(gp100) was injected i.t. alone (data not shown) indicating that the combination of alloDCs and the Ad5M vector is more favorable.
The adjuvant alloDCs can be used intratumorally in a therapeutic setting, which is beneficial, as intratumorally injected alloDCs led to recruitment and activation of innate and adaptive immune cells, which can aid in tumor cell killing and antigen release. We speculate that, apart from the release of TAAs (encoded by the adenovirus vector) from the alloDCs, bulk antigen release from the dying tumor cells may initiate neoepitope-specific T-cell responses.Citation10 This is in line with current paradigm shifts indicating that for modalities targeting immune subsets within the tumor, it might be preferable to deliver them locally rather than systemically.Citation37 Our research findings are also pertinent to the clinical setting, as the alloDCs approach using i.t. injected mature pro-inflammatory alloDCs has already been successfully tested in a phase I/II clinical trial in patients with mRCCCitation21 and is ongoing in a randomized multicenter phase II study in metastatic RCC patients (NCT02432846).
Concluding, we present a novel approach where an infection-enhanced adenovirus can be used to load pro-inflammatory alloDCs with high amounts of TAAs. We propose that this approach can be used in a clinical setting in order to elicit antigen-specific adaptive immune responses and in addition alter the immunosuppression in the TME. With this approach, one could from a single leukapheresis product create an “off-the-shelf” vaccine, which can readily be used for many patients, irrespectively of their MHC haplotype. Uniformity in the production of such a vaccine can eliminate the variability of responses due to the technical differences of creating individual-based autologous DC vaccines.
Materials and methods
Production of the recombinant virus
The Ad5M(MOCK) and Ad5M(gp100) vectors were constructed and produced as previously described.Citation38 They are E1 deleted adenovirus serotype 5 (Ad5) vectors having the fiber shaft and knob replaced with the serotype-35 counterpart. They also contain a cell-penetrating peptide from HIV Tat in the hypervariable loop of their hexon proteins (major capsid protein). They are infection-enhanced vectors with improved ability to transduce most hematopoietic cells including DCs.Citation39 Ad5M(MOCK) does not encode any transgene, while Ad5M(gp100) encodes the human melanoma antigen glycoprotein 100 (gp100, amino acids 25-33). Titers were determined by quantitative polymerase chain reaction as encapsidated virus genomes (evg) per ml.Citation39
Cell lines
The 911 cell line (Crucell, Leiden, The Netherlands) used for adenovirus production was cultured in DMEM supplemented with 10% heat inactivated FBS, 1% penicillin/streptomycin (PeSt) and 1% sodium pyruvate. The mouse melanoma cell line B16-F10 (ATCC, Rockville, MD) were cultured in DMEM supplemented with 10% heat inactivated FBS, 1% PeSt and 1% sodium pyruvate. The mouse melanoma cell lines Hcmel3 and Hcmel12 are derived from a primary HGF-CDK4(R24C) melanoma modelCitation25,26 and are a kind gift from Prof. Thomas Tüting, University Hospital Magdeburg. Both Hcmel3 and Hcmel12 were cultured in RPMI-1640 supplemented with 10% heat inactivated FBS. All components and culture media were from Thermo Fisher Scientific (Carlsbad, CA). All cells were cultured in a humidified incubator with a 5% CO2 atmosphere at 37°C.
Isolation and maturation of mouse bone marrow-derived allogeneic DCs
Bone marrow-derived DCs were generated from the femur and tibia of female 7-8 week old wild-type (wt) BALB/c mice (H-2Dd) (The Janvier Labs, France) by exposing the bone marrow and flushing out the cells with a sterile syringe. The harvested cells were cultured in IMDM supplemented with 10% heat inactivated FBS, 1% PeSt, 1% HEPES, 50 μM β-mercaptoethanol. Culture medium was supplemented with 20 ng/mL recombinant murine IL-4 and 20 ng/mL recombinant murine GM-CSF (Nordic BioSite, Stockholm, Sweden). Bone marrow cells were plated on non-treated Petri dishes (Sarstedt, Nümbrecht, Germany). Medium was replaced every 3 days. On day 7 the non-adherent imDCs were harvested and treated for 18 h with a cocktail of combined Toll-like receptor ligands with IFN-γ (COMBIG), consisting of 2.5 μg/mL R848 (InvivoGen, Sam Diego, CA), 20 μg/mL polyinosinic:polycytidylic acid (polyI:C) (Sigma-Aldrich, St. Louis, MO) and 1000 IU/mL IFN-γ (Nordic Biosite)Citation22 in order to obtain mature alloDCs. Ad5M(gp100)-alloDCs were obtained by transducing pelleted imDCs for 2 h, at 37oC with 2000 evg/cell Ad5M(gp100) and matured as above. Cells were cultured in a humidified incubator with a 5% CO2 atmosphere at 37°C.
Murine immune cell phenotyping
Flow cytometry was used to phenotype:
Murine DCs: anti-B220-Pacific Blue, anti-CD11c-PE, anti-CD11b-PerCP, anti-CD86-APC, anti-I-Ab-PE.
Murine T-cells: anti-CD3-PB, anti-CD8a-APC, anti-Thy1.1-PE, anti-Vβ-13-FITC, H-2Db/hgp10025-33-PE tetramer (Beckman Coulter, San Diego, CA).
Murine MDSCs: anti-B220-APC/Cy7, anti-Gr1-PE/Cy7, anti-Ly6C-BV421, anti-Ly6G-FITC, anti-CD11c-PE, anti-CD11b-PerCP.
All antibodies were purchased from BioLegend (San Diego, CA). Data acquisition was performed using a FACSCanto II (BD Biosciences) flow cytometer, and the analysis was performed using FlowJo software (version 7.6.5; Tree Star, Ashland, OR).
Animal experiments
The Uppsala Research Animal Ethics Committee (C215/12) and the Northern Stockholm Research Animal Ethics Committee (N170/13, N164/15) have approved the animal studies. All in vivo experiments were performed with female 6-7 weeks old C57BL/6NRj (H-2Db) mice receiving vaccinations with treated allogeneic DCs (vaccine cells) generated from the bone marrow of 7-8 weeks old female BALB/c (H-2Dd) mice and treated as described above. All mice were obtained from Janvier Labs.
In vivo maturation and migration of endogenous DC
Mice were injected i.d. with 6 × 106 vaccine cells and 20μg CellTrace CFSE fluorescent dye (Thermo Fisher Scientific). Draining (inguinal) lymph nodes were harvested 48 h post injections and digested into single-cell suspensions. CFSE+ DCs migrating from the site of injection to the lymph node were quantified after gating for CD11chighCD11b+B220− cells in flow cytometric analysis. Activation status of migrated DCs was analyzed as percentage of CFSE+CD86+ and CFSE+CD86− cells of CD11chighCD11b+B220− cells. The percentage of CFSE+ I-Ab + (detecting host cells of C57BL/6NRj origin) DCs in the lymph node was evaluated to exclude migration of non-host DC (vaccine cells of BALB/c origin). Non-draining lymph nodes from the same animals were used as control. The skin piece at the injection site was dissected and cryopreserved. The piece was sectioned (6 µm) and stained with anti-NK1.1-AF647 and anti-Gr-1-AF647 (Biolegend). Sections were then counterstained with Hoechst 33342 (Sigma-Aldrich), mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL, USA) and imaged using Zeiss Axioimager microscope (Oberkochen, Germany). Image J software was used for staining quantification, manual cell counting of NK1.1 and Gr1 positive cells.
Prophylactic and therapeutic vaccination with Ad5M(gp100) and Ad5M(gp100)-alloDCs
For the prophylactic setting, mice were vaccinated s.c. on the left hind flank on day 0 and 10 with 5×109 evg of Ad5M(MOCK) or Ad5M(gp100) alone or after transduction of 1×106 alloDCs. On day 17, blood was sampled and analyzed for the presence of gp100-TCR+ T- cells by staining with the H-2Db/hgp10025-33-PE tetramer. Mice were subsequently injected with 2×105 Hcmel12 tumor cells s.c. on the right hind flank and tumor growth was followed. Tumors were measured with caliper and the tumor size was calculated by the formula: Volume = length × widthCitation2 × π/6. Mice were sacrificed when the tumor volume exceeded 1 cm3 or if bleeding ulcers developed.
For the therapeutic setting, mice were injected s.c. on the right hind flank with 5×105 Hcmel3 tumor cells. Mice received 3 i.t. injections of 5×109 evg of Ad5M(MOCK) or Ad5M(gp100) alone or after transduction of 1×106 alloDCs on days 10, 20 and 27 after tumor implantation. On day 30 all mice were sacrificed and analyzed for the presence of gp100-TCR+ T cells and tumor size was calculated by the formula: Volume = length × widthCitation2 × π/6.
Adoptive transfer of pmel-1 (gp100-specific) splenocytes
Mice were injected s.c. with 1×106 vaccine cells. Pmel-1 splenocytes (10×107 per mouse) were injected i.v. 48 hours later. The transgenic pmel-1 mice have a C57BL/6NRj (H-2Db) background and around 20% of their splenocytes are Thy1.1+ CD8+ T cells that carry a gp10025-33-specific TCR. Pmel-1 mice were originally obtained from the Jackson Laboratory (Bar Harbor, Maine, USA) and kept in breeding by our group. Draining (inguinal) lymph nodes were harvested 7 days post pmel-1 adoptive splenocyte transfer and digested into single-cell suspensions.
In vivo assessment of TME and therapeutic efficacy in a B16-F10 melanoma model after intratumoral injection with alloDCs
Mice were injected s.c. into the hind flank with 1×105 B16-F10 tumor cells. Mice received i.t. injections of 1×106 vaccine cells on day 3 and 10 post tumor inoculation. On day 12 post tumor inoculation mice were sacrificed and tumors were excised. Half of each excised tumor was cryopreserved and sectioned, while the other half was enzymatically digested into single-cell suspensions. FACS analysis was used to evaluate the TME. Cryosections were stained with CD4-PE, FoxP3-AF647, NK1.1-AF647, unconjugated-CD8a (clone 53-6.7) and goat anti-rat-AF555 (Life Technologies). Tile-scan images from whole tumor sections were taken on a Zeiss Axioimager microscope. Image J software was used for manual cell counting in the total tumor area.
For the pmel-1 T-cell transfer experiment, mice received i.v. injection of 1×108 pmel-1 splenocytes on day 8 and i.t. injections of 106 alloDCs on day 8 and 14 post B16-F10 tumor cell inoculation. Tumors were measured with caliper and the tumor size was calculated as the formula: Volume = length × widthCitation2 × π/6. Mice were sacrificed when the tumor volume exceeded 1 cm3 or if bleeding ulcers developed. Cells were obtained from spleen, blood and lymph node of the sacrificed mice on day 15 post B16-F10 inoculation and stained for expression of the Thy1.1 and Vβ13 pmel-1 T-cell markers. Staining for TME analysis was performed as described above.
Statistics
The data are reported as mean and SEM. Statistical analysis was performed by GraphPad prism software version 6.01 (La Jolla, CA, USA). Statistical analyses were performed using parametric One way ANOVA test (> 2 experimental groups) with Holm-Sidak test for multiple comparison correction and Mann-Whitney U test (only 2 experimental groups). Association of gp100-TCR+ T-cells and tumor burden was evaluated by a linear regression model computing Spearman correlation. Statistical comparison of the Kaplan-Meier survival curve was performed using log-rank test. Values with P<0.05 were considered to be statistically significant.
Disclosure of potential conflicts of interest
AKP is founder of Immunicum AB, a company in the field of allogeneic dendritic cell-based immunotherapeutic of cancer. The other authors have no conflicting financial interests.
supp_data.zip
Download Zip (1.4 MB)Acknowledgments
The authors would like to thank Jing Ma and Berith Nilsson for technical assistance. The authors also wish to thank the BioVis platform, Science for Life Laboratory, Uppsala University for assisting with microscopy. This work is supported by The Swedish Cancer Society (CAN 2013/373; CAN 2016/318), The Swedish Children Cancer Foundation (PR2015-0049), The Swedish Research Council (2015-03688) and Immunicum AB. Conceived and designed the experiments: GF, CJ, MR, AKP, DY, ME. Performed the experiments and analyzed the data: GF, CJ, IK, M-MM. Wrote the paper: GF, CJ, DY, ME. All authors read and approved the final version of the manuscript. AKP is founder of Immunicum AB. The other authors have no conflicting financial interests.
Additional information
Funding
References
- Morrissey KM, Yuraszeck TM, Li CC, Zhang Y, Kasichayanula S. Immunotherapy and novel combinations in oncology: Current landscape, challenges, and opportunities. Clin Transl Sci. 2016;9:89–104. doi:10.1111/cts.12391. PMID:26924066.
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. doi:10.1038/nature10673. PMID:22193102.
- Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi:10.1146/annurev.immunol.24.021605.090733. PMID:16551247.
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi:10.1126/science.1129139. PMID:17008531.
- Bloy N, Pol J, Aranda F, Eggermont A, Cremer I, Fridman WH, Fučíková J, Galon J, Tartour E, Spisek R, et al. Trial watch: Dendritic cell-based anticancer therapy. Oncoimmunology. 2014;3:e963424. doi:10.4161/21624011.2014.963424. PMID:25941593.
- Schreibelt G, Bol KF, Westdorp H, Wimmers F, Aarntzen EH, Duiveman-de Boer T, van de Rakt MW, Scharenborg NM, de Boer AJ, Pots JM, et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin Cancer Res. 2016;22:2155–66. doi:10.1158/1078-0432.CCR-15-2205. PMID:26712687.
- Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, Boerman OC, Croockewit S, Oyen WJ, van Rossum M, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013;73:1063–75. doi:10.1158/0008-5472.CAN-12-2583. PMID:23345163.
- Fong L, Brockstedt D, Benike C, Wu L, Engleman EG. Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol. 2001;166:4254–9. doi:10.4049/jimmunol.166.6.4254. PMID:11238679.
- Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JF, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009;15:2531–40. doi:10.1158/1078-0432.CCR-08-2729. PMID:19318472.
- Liu C, Lou Y, Lizee G, Qin H, Liu S, Rabinovich B, Kim GJ, Wang YH, Ye Y, Sikora AG, et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J Clin Invest. 2008;118:1165–75. PMID:18259609.
- Yewdall AW, Drutman SB, Jinwala F, Bahjat KS, Bhardwaj N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One. 2010;5:e11144. doi:10.1371/journal.pone.0011144. PMID:20585396.
- Benencia F, Sprague L, McGinty J, Pate M, Muccioli M. Dendritic cells the tumor microenvironment and the challenges for an effective antitumor vaccination. J Biomed Biotechnol. 2012;2012:425476. doi:10.1155/2012/425476. PMID:22505809.
- Ghirelli C, Hagemann T. Targeting immunosuppression for cancer therapy. J Clin Invest. 2013;123:2355–7. doi:10.1172/JCI69999. PMID:23728169.
- Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:2447–63. doi:10.1172/JCI64859. PMID:23728179.
- Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol. 2015;34:22–7. doi:10.1016/j.coi.2014.12.009. PMID:25597442.
- Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi:10.1016/j.immuni.2013.07.005. PMID:23890064.
- Draghiciu O, Nijman HW, Daemen T. From tumor immunosuppression to eradication: targeting homing and activity of immune effector cells to tumors. Clin Dev Immunol. 2011;2011:439053. doi:10.1155/2011/439053. PMID:22190971.
- Jeanbart L, Swartz MA. Engineering opportunities in cancer immunotherapy. Proc Natl Acad Sci U S A. 2015;112:14467–72. doi:10.1073/pnas.1508516112. PMID:26598681.
- Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol. 2013;14:246–53. doi:10.1038/ni.2514. PMID:23314004.
- Wallgren AC, Andersson B, Backer A, Karlsson-Parra A. Direct allorecognition promotes activation of bystander dendritic cells and licenses them for Th1 priming: a functional link between direct and indirect allosensitization. Scand J Immunol. 2005;62:234–42. doi:10.1111/j.1365-3083.2005.01663.x. PMID:16179010.
- Laurell A, Lönnemark M, Brekkan E, Magnusson A, Tolf A, Wallgren AC, Andersson B, Adamson L, Kiessling R, Karlsson-Parra A. Intratumorally injected pro-inflammatory allogeneic dendritic cells as immune enhancers: a first-in-human study in unfavourable risk patients with metastatic renal cell carcinoma. J ImmunoTher Cancer. 2017;5:52. doi:10.1186/s40425-017-0255-0. PMID:28642820.
- Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–76. doi:10.1038/ni1223. PMID:15995707.
- Ramachandran M, Jin C, Yu D, Eriksson F, Essand M. Vector-encoded Helicobacter pylori neutrophil-activating protein promotes maturation of dendritic cells with Th1 polarization and improved migration. J Immunol. 2014;193:2287–96. doi:10.4049/jimmunol.1400339. PMID:25049358.
- Miller MJ, Hejazi AS, Wei SH, Cahalan MD, Parker I. T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node. Proc Natl Acad Sci U S A. 2004;101:998–1003. doi:10.1073/pnas.0306407101. PMID:14722354.
- Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn-Konijnenberg D, Hömig-Hölzel C, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014;507:109–13. doi:10.1038/nature13111. PMID:24572365.
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wölfel T, Hölzel M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490:412–6. doi:10.1038/nature11538. PMID:23051752.
- Chew V, Toh HC, Abastado JP. Immune microenvironment in tumor progression: characteristics and challenges for therapy. J Oncol. 2012;2012:608406. doi:10.1155/2012/608406. PMID:22927846.
- Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356–64. doi:10.1172/JCI80005. PMID:26168215.
- Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G, Koss B, Kim Y, Bronte V, Herold MJ, et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity. 2014;41:947–59. doi:10.1016/j.immuni.2014.10.020. PMID:25500368.
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–5. doi:10.1038/nature05543. PMID:17220874.
- Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014;35:571–80. doi:10.1016/j.it.2014.09.006. PMID:25443495.
- Kleindienst P, Brocker T. Endogenous dendritic cells are required for amplification of T cell responses induced by dendritic cell vaccines in vivo. J Immunol. 2003;170:2817–23. doi:10.4049/jimmunol.170.6.2817. PMID:12626531.
- Maroof A, Beattie L, Kirby A, Coles M, Kaye PM. Dendritic cells matured by inflammation induce CD86-dependent priming of naive CD8+ T cells in the absence of their cognate peptide antigen. J Immunol. 2009;183:7095–103. doi:10.4049/jimmunol.0901330. PMID:19917700.
- Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15:e257–67. doi:10.1016/S1470-2045(13)70585-0. PMID:24872109.
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi:10.1084/jem.20030590. PMID:12925674.
- Palmer DC, Balasubramaniam S, Hanada K, Wrzesinski C, Yu Z, Farid S, Theoret MR, Hwang LN, Klebanoff CA, Gattinoni L, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–16. doi:10.4049/jimmunol.173.12.7209. PMID:15585842.
- Marabelle A, Kohrt H, Caux C, Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin Cancer Res. 2014;20:1747–56. doi:10.1158/1078-0432.CCR-13-2116. PMID:24691639.
- Yu D, Jin C, Leja J, Majdalani N, Nilsson B, Eriksson F, Essand M. Adenovirus with hexon Tat-protein transduction domain modification exhibits increased therapeutic effect in experimental neuroblastoma and neuroendocrine tumors. J Virol. 2011;85:13114–23. doi:10.1128/JVI.05759-11. PMID:21957304.
- Yu D, Jin C, Ramachandran M, Xu J, Nilsson B, Korsgren O, Le Blanc K, Uhrbom L, Forsberg-Nilsson K, Westermark B, et al. Adenovirus serotype 5 vectors with Tat-PTD modified hexon and serotype 35 fiber show greatly enhanced transduction capacity of primary cell cultures. PLoS One. 2013;8:e54952. doi:10.1371/journal.pone.0054952. PMID:23372800.