
ABSTRACT
Regulatory T-cells (Tregs) are crucial for the maintenance of immune tolerance and homeostasis as well as for preventing autoimmune diseases, but their impact on the survival of cancer patients remains controversial. In the TC-1 mouse model of human papillomavirus (HPV)-related carcinoma, we have previously demonstrated that the therapeutic efficacy of the CyaA-E7-vaccine, targeting the HPV-E7 antigen, progressively declines with tumor growth, in correlation with increased intratumoral recruitment of Tregs. In the present study, we demonstrated that these TC-1 tumor-infiltrating Tregs were highly activated, with increased expression of immunosuppressive molecules. Both intratumoral effector CD4+ T-cells (Teffs) and Tregs expressed high levels of PD-1, but anti-PD-1 antibody treatment did not impact the growth of the TC-1 tumor nor restore the therapeutic effect of the CyaA-E7 vaccine. To analyze the mechanisms by which Tregs are recruited to the tumor site, we used MHC-II KO mice with drastically reduced numbers of CD4+ effector T-cells. We demonstrated that these mice still had significant numbers of Tregs in their lymphoid organs which were recruited to the tumor. In MHC-II KO mice, the growth of the TC-1 tumor was delayed in correlation with a strong increase in the intratumoral recruitment of CD8+ T-cells. In addition, in mice that spontaneously rejected their tumors, the infiltration of E7-specific CD8+ T-cells was significantly higher than in MHC-II KO mice with a growing tumor. These results demonstrate that tumor-specific CD8+ T-cells can be efficiently activated and recruited in the absence of MHC class II molecules and of CD4+ T-cell help.
Introduction
The role of immunity and immunosurveillance in cancer was suspected in the beginning of the 20th century and established in recent decades. Three phases of tumor interaction with the immune system have been proposed: the elimination phase, the equilibrium phase and the escape phase, in which the tumor grows uncontrollably, establishes an immunosuppressive microenvironment and escapes the immune system.Citation1 Several mechanisms are involved in tumor escape, including the activation and accumulation of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T-cells (Tregs) at the tumor site.Citation1,2
Tregs are a subset of CD4+ T-cells expressing the Foxp3 transcription factor and exerting immunosuppressive functions: they are crucial for the maintenance of immune tolerance to self-antigens, preventing the development of autoimmune diseases in healthy individuals.Citation3 They exert immune suppression through the expression of surface molecules, such as CTLA-4, CD39 and CD73, or through the secretion of suppressive cytokines such as IL-10, TGF-ß or IL-35.Citation4,5 These properties are currently being manipulated to improve patient health and survival after organ transplant and to prevent graft rejection. Furthermore, their involvement in cancer is being intensively investigated as they also dampen the immune response to tumor. However, their impact on prognosis and tumor progression is still controversial depending on the tumor type.Citation6 Treg numbers are indeed increased in peripheral blood, in tumor-draining lymph nodes (dLN) and at tumor sites in patients and in mouse models of cancer. Their increased levels have a positive impact in B cell lymphomas and colorectal cancers,Citation7–9 while they are associated with a poor prognosis in patients with lung, gastric, pancreatic, ovarian or hepatocellular carcinoma.Citation10–14 They are also associated with a poor prognosis in cervical carcinoma.Citation6,15
Cervical carcinoma is the second most frequent gynecologic cancer worldwide. Human papilloma virus (HPV) infections cause proliferative lesions in infected skin and squamous mucosa, resulting in hyperplasia, papillomas and condylomas. Although most HPV infections are transient, in some patients, high-risk HPV can induce persistent mucosal lesions in the ano-genital tract and the oropharynx, leading to in situ or invasive carcinomas.Citation16
Our team has recently developed a new immunotherapeutic vaccine candidate, CyaA-E7, that is currently undergoing clinical trials: the detoxified adenylate cyclase (CyaA) from B. pertussis, carrying the E7 oncoprotein from HPV-16. This vaccine is able to elicit an E7-specific CD8+ T-cell response, to reduce Treg recruitment to the tumor siteCitation17 and to induce complete tumor eradication in the TC-1 mouse model of HPV-related carcinoma. However, its therapeutic efficacy against larger tumors is reduced.Citation18 As observed in cancer patients, we found a massive accumulation of Tregs at the tumor site very early after tumor inoculation. They represented up to 80% of tumor-infiltrating CD4+ T-cells, thus likely contributing to the impaired anti-tumor immune response induced by the CyaA-E7 vaccine in mice with large tumors.Citation18
Understanding the mechanisms by which Tregs migrate and accumulate in tumors is important in order to develop strategies to overcome their recruitment and immunosuppressive properties. In a previous study,Citation19 we investigated whether tumor infiltration by Tregs is due to inflammation promoting the migration and conversion of CD4+ T-cells into tumor tissue, or to antigen-driven activation and expansion of Treg cells. This study established that TC-1 tumor-infiltrating Tregs displayed CDR3 spectratyping profiles characteristic of biased and strongly perturbed T-cell repertoires, typical of clonal expansions, suggesting that strong T-cell responses have occurred within the tumor tissue. In addition, we demonstrated that a large proportion of tumor-infiltrating Treg sequences, especially those encoding public sequences, are found in the repertoire of Treg cells obtained from the draining lymph node (dLN), but were absent in the repertoire of Tregs isolated from naïve mice. These observations suggest that Treg cells may be activated in the dLN and then migrate to the tumor site where they continue to proliferate and accumulate.
In the present study, we aimed to further analyze the mechanisms by which Tregs accumulate in the tumor tissue and to determine whether their activation and recruitment is dependent upon antigen presentation by MHC class II molecules.
We first characterized their phenotype and demonstrated that tumor-infiltrating Tregs were highly activated with increased expression of suppressive molecules, such as ICOS, CD39, CD103 but also PD-1, but exhibited suppressive functions similar to naive Tregs. However, treatment of TC-1-bearing mice with an anti-PD-1 antibody had no effect on the growth of the tumor, or on the therapeutic effect of the CyaA-E7 vaccine.
MHC-II KO mice were used to determine whether Tregs require antigen presentation by MHC class II molecules to be recruited to the tumor site. These mice lack five genes involved in antigen presentation to CD4+ T-cells, and have drastically reduced numbers of CD4+ T-cells. However, CD4+ Foxp3+ T cells are found in secondary lymphoid tissues, but not in the thymus, of these MHC class II deficient mice and can exert regulatory functions.Citation20–22 In these mice, Tregs were still recruited to the tumor site, although their activation state and their suppressive phenotype were altered compared with their counterparts from wild-type (WT) mice. TC-1 tumor growth was delayed in MHC-II KO mice, correlating with increased numbers of tumor-infiltrating CD8+ T-cells with a more activated phenotype. In addition, surprisingly, the MHC-II KO mice developed significantly increased numbers of E7-specific CD8+ T-cells after vaccination with CyaA-E7. An increased percentage of E7-specific CD8+ T-cells was also observed in the tumors of MHC-II KO mice, which spontaneously rejected the TC-1 tumor compared to mice with progressing tumor.
Our results clearly show that in tumor-bearing mice, Tregs alter the activation and intratumoral recruitment of CD8+ T-cells. These suppressive properties must be overcome to restore an efficient anti-tumor immune response.
Results
Tumor-infiltrating CD4+ Foxp3+ T-cells are highly activated with increased expression of immunosuppressive molecules
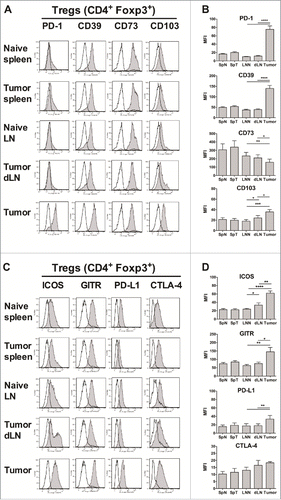
First, we analyzed the phenotype of tumor-infiltrating CD4+ Tregs and Teffs in the TC-1 mouse model of HPV-related carcinoma. TC-1 tumor cells were s.c. injected into Foxp3-GFP mice, and the infiltration of lymphocyte subsets and the phenotype of Tregs and effector T cells (Teffs) in tumors, dLN and spleens were analyzed twenty-five days later (Fig. S1 A-C). As previously described,Citation18,19 Tregs were highly accumulated at the tumor site (Fig. S1D-G). We have also previously shown that both tumor-infiltrating Tregs and Teffs are highly activated with an enhanced memory phenotype as shown by increased expression of CD69 and CD44 and decreased expression of CD62L.Citation19 Next, we analyzed the expression of other markers characterizing Treg phenotype and function (). We found that tumor-infiltrating Tregs had significantly increased expression of PD-1 and CD39 compared with naive Tregs, while their CD73 expression was significantly decreased. Recent studies have identified highly suppressive Treg subsets in tumors characterized by enhanced expression of CD103 or ICOS.Citation2–25 Indeed, the expression of these molecules was significantly increased on Tregs from both dLN and tumors, showing that Treg activation had already occurred in the dLN. Tumor Tregs also showed an increased level of GITR and PD-L1,Citation26,27 whereas the expression of CTLA-4 did not vary significantly (). The expression of PD-1, CD39, CD73, CD103, GITR, ICOS, PD-L1 and CTLA-4 was also significantly increased on tumor-infiltrating Teffs compared to naive Teffs (Fig. S2). These data clearly showed that both Treg and Teff tumor-infiltrating CD4+ T-cells were highly activated with an enhanced immunosuppressive phenotype.
Figure 1. Increased suppressive phenotype of tumor-infiltrating Tregs. Foxp3-GFP mice were injected on day 0 with 1 × 106 TC-1 cells, and 25 days later, the mice were sacrificed and cell suspensions were prepared from the spleens (SpT), tumor-dLN (dLN) and tumors, stained and analyzed by flow cytometry, and compared with the LN (LNN) and spleen (SpN) from naive mice. (A-D) The surface expression of PD-1, CD39, CD73, CD103, ICOS, GITR, PD-L1 and CTLA-4 (filled grey line) by CD4+ Foxp3+ Tregs purified from the indicated organs is shown. Labeling with isotype controls is represented by black lines. Data from one representative experiment are shown in A and C, whereas the geometric mean fluorescence intensity (MFI) ± SEM is shown in B and D. The results represent the cumulative data from 4 independent experiments (n = 8–9 mice per group). * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001 as determined by the Mann-Whitney test.
Recent data have established that neuropilin-1 (Nrp1) expression distinguishes natural Tregs (nTregs) from induced Tregs (iTregs).Citation28,29 We observed that a high percentage of Tregs isolated from the spleen and LN from naive or tumor-bearing mice, as well as from the tumor, expressed Nrp1 (Fig. S3A-C), clearly showing that tumor-infiltrating Tregs are nTregs and not Teffs that have been converted to iTregs by the tumor immunosuppressive microenvironment. In contrast to Tregs, Teffs expressed a low level of Nrp1 in the lymphoid tissues. However, the tumor-infiltrating Teffs had significantly increased expression of this molecule (Fig. S3D-F), showing that Nrp1 expression on Teffs is a marker of their activation as previously suggested.Citation30
Anti-PD-1 treatment does not improve CyaA-E7-induced tumor regression in mice
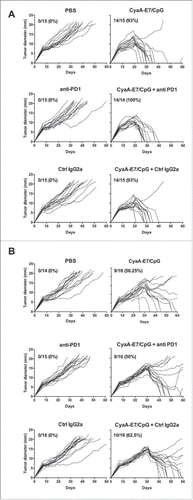
Treatment with anti-PD-1 mAb was shown to induce tumor regression in mouse models and in patients.Citation31 Since the expression of PD-1 was significantly increased on both CD4+ and CD8+ intratumoral T-cells, we then analyzed whether treatment by an anti-PD1 antibody would enhance the effect of the CyaA-E7 vaccine on tumor regression. Tumor-bearing mice were vaccinated with CyaA-E7/CpG either 14 or 24 days after tumor injection and treated chronically with anti-PD-1 or control mAb.This anti-PD-1 treatment alone had no impact on tumor growth (). As shown in , vaccination with CyaA-E7/CpG at day 14 induced rejection of the tumor in 93% of mice. The combination with anti-PD-1 mAb did not enhance this effect. When administered 24 days after tumor graft, the CyaA-E7/CpG induced complete tumor regression in only 56% of mice, and the combination with anti-PD1 did not improve this effect ().
Figure 2. Anti-PD-1 antibody treatment does not enhance the therapeutic efficacy of the CyaA-E7 vaccine. C57 BL/6 J mice were injected with 6 × 105 TC-1 cells on day 0, and after 14 (A) or 24 (B) days were i.v. immunized with CyaA-E7 (50 µg/mouse) and CpG-B-DOTAP (left panels) or received PBS (right panels). On day 13 (A) or 23 (B) and then every 3 days after vaccination, the mice were left untreated (upper panels) or received either i.p. injection of anti-PD-1 (middle panels) or isotype control antibodies (lower panels) for a total of 4 injections (250 µg/mouse/injection on days 13, 17, 20 and 23 in A or days 23, 27, 30 and 33 in B). The results represent the cumulative data from 2 independent experiments (n = 14–15 mice per group).
Tumor-infiltrating Tregs are functional
To more precisely characterize the phenotype and properties of the tumor-infiltrating CD4+ T-cells, we then analyzed the mRNA expression by purified Tregs and Teffs of transcription factors involved in the differentiation of naive CD4+ T-cells into effector T-cells. As expected, only Tregs expressed Foxp3 (), while RORγc expression was barely detectable in Teffs and Tregs (). T-bet was expressed by Teffs of lymphoid organs and was significantly increased in Teffs purified from tumors (). The expression of GATA-3 by tumor-infiltrating Teffs was similar to that of Teffs from lymphoid tissues (), showing that CD4+Foxp3− Teffs infiltrating the TC-1 tumor were mainly Th1 cells. The T-bet expression by Tregs purified from lymphoid organs was very low but was significantly increased in tumors (). In contrast, GATA-3 expression was greatly increased in Tregs purified from tumor. The increased intracellular expression of the T-bet protein by tumor Teffs and Tregs was confirmed by flow cytometry ().
Figure 3. Functional analysis of tumor-infiltrating T-cells. Foxp3-GFP mice were injected on day 0 with 1 × 106 TC-1 cells, and the spleens, dLN and tumors were processed 25 days later to obtain cell suspensions. The spleens and lymph nodes from naive mice were used as controls. CD4+ Foxp3-GFP+ Tregs and Foxp3-GFP− Teffs were FACS sorted, and then total RNA was purified from the sorted cells and reverse-transcribed. The corresponding cDNA material was then subjected to quantitative PCR micro-arrays with primers specific to Foxp3 (A), RORγc (B), T-bet (C) and GATA-3 (D). The results represent the mRNA expression normalized to house-keeping genes and are expressed as the mean arbitrary units ± SD from 2 independent experiments in which cells from each organ were pooled from 5–6 mice per group. One-way ANOVA was performed separately for each subset to compare its gene expression in the different tissues. *p<0.05 was obtained for T-bet expression in both Tregs and Teffs (C) and **p<0.01 for GATA-3 expression in Tregs (D). (E-F) The cell suspensions were stained and analyzed by flow cytometry. The intracellular expression of T-bet in Tregs and Teffs is shown in E, while F shows the mean MFI ± SEM of cumulative data from 3 independent experiments (n = 5–6 mice per group). * p< 0.05 as determined by the Mann-Whitney test in F. (G-H) After AutoMacs CD90+ cell enrichment, CD4+ Foxp3-GFP+ Tregs and Foxp3-GFP− Teffs were FACS sorted, mixed together at different ratios, and cultured in anti-CD3-coated plates (5 µg/mL) for 72 hours. Cell proliferation was determined by [3H]-thymidine incorporation (G), and IFNγ was measured in culture supernatants (H). The results are expressed as the mean ± SEM from 2 independent experiments in which cells from 8 mice were pooled.
![Figure 3. Functional analysis of tumor-infiltrating T-cells. Foxp3-GFP mice were injected on day 0 with 1 × 106 TC-1 cells, and the spleens, dLN and tumors were processed 25 days later to obtain cell suspensions. The spleens and lymph nodes from naive mice were used as controls. CD4+ Foxp3-GFP+ Tregs and Foxp3-GFP− Teffs were FACS sorted, and then total RNA was purified from the sorted cells and reverse-transcribed. The corresponding cDNA material was then subjected to quantitative PCR micro-arrays with primers specific to Foxp3 (A), RORγc (B), T-bet (C) and GATA-3 (D). The results represent the mRNA expression normalized to house-keeping genes and are expressed as the mean arbitrary units ± SD from 2 independent experiments in which cells from each organ were pooled from 5–6 mice per group. One-way ANOVA was performed separately for each subset to compare its gene expression in the different tissues. *p<0.05 was obtained for T-bet expression in both Tregs and Teffs (C) and **p<0.01 for GATA-3 expression in Tregs (D). (E-F) The cell suspensions were stained and analyzed by flow cytometry. The intracellular expression of T-bet in Tregs and Teffs is shown in E, while F shows the mean MFI ± SEM of cumulative data from 3 independent experiments (n = 5–6 mice per group). * p< 0.05 as determined by the Mann-Whitney test in F. (G-H) After AutoMacs CD90+ cell enrichment, CD4+ Foxp3-GFP+ Tregs and Foxp3-GFP− Teffs were FACS sorted, mixed together at different ratios, and cultured in anti-CD3-coated plates (5 µg/mL) for 72 hours. Cell proliferation was determined by [3H]-thymidine incorporation (G), and IFNγ was measured in culture supernatants (H). The results are expressed as the mean ± SEM from 2 independent experiments in which cells from 8 mice were pooled.](/cms/asset/93eb3049-31f3-4ff2-a633-900a8b8e0ef7/koni_a_1404213_f0003_b.gif)
Since tumor-infiltrating Tregs displayed increased expression of immunosuppressive molecules, we determined whether they were endowed with increased immunosuppressive activity. However, both Tregs isolated from either tumor or normal spleen similarly suppressed the proliferation of Teffs and their production of IFN-γ after polyclonal stimulation with an anti-CD3 mAb ().
The transcriptomic analysis of genes involved in apoptosis demonstrated a significant increase in TRAIL in tumor Tregs (Fig. S4A). Analysis of the expression of genes of the BCL2 family showed that both pro- and anti-apoptotic genes were activated in tumor-infiltrating T-cells, but no clear trend was observed (data not shown). Of the caspase genes analyzed, only caspase-14 was significantly increased in tumor Tregs (Fig. S4B). A strong increase in the expression of Redd (for Regulated in Development and DNA damage responses) and HIF1a was also observed in Tregs (Fig. S4C-D), strongly suggesting that the TC-1 tumor microenvironment is hypoxic.
We also analyzed the mRNA expression of cytokine genes and found that neither IL-10 nor TGF-ß genes were increased in tumor-infiltrating Tregs compared with Tregs from naive mice (data not shown).
Treg activation and recruitment to tumors is independent of Ag presentation by MHC-II molecules
We have previously demonstrated that the TCR repertoire of the Tregs infiltrating the TC-1 tumors is skewed towards public sequences that are only shared by Tregs from dLN but not by those of naive mice.Citation19 In addition, the present results suggest that tumor-infiltrating Tregs are nTregs. Thus, we hypothesized that tumor Tregs are activated in dLN by antigen-presenting cells (APCs) through the presentation of tumor antigen(s) by MHC-II molecules and then migration to the tumor. To test this hypothesis, we used MHC-II KO mice, which have a dramatically reduced CD4+ T-cell compartment, mainly composed of nTregs, due to the inability of their APCs to present antigens to CD4+ T-cells.Citation21,32
TC-1 tumor cells were injected into MHC-II KO mice and C57BL/6J wild-type (WT) mice as a control, and tumor growth was followed and T-cell infiltration in tumors analyzed on day 25 (the gating strategy is shown in Fig. S5A-D). TC-1 tumor cells did not express MHC-II molecules, as determined either after in vitro culture or ex vivo on MHC-II KO or C57 BL/6J WT mice grafted with TC-1 cells (Fig. S6).
The growth of the TC-1 tumor was clearly delayed in MHC-II KO mice compared to WT mice, with 13% of mice rejecting the tumor (). As expected, strongly reduced numbers of CD4+ T-cells were found in the spleen and LN of MHC-II KO compared to WT mice (), while the CD8+ T-cell compartment was enlarged, especially in the LNN. B cell numbers were also significantly increased, especially in tumor-bearing mice.
Figure 4. The intratumoral recruitment of CD8+ T lymphocytes is increased in MHC class II-deficient mice. (A) Wild-type C57BL/6J (WT; black lines) and MHC-II KO mice (green lines) were injected on day 0 with 6 × 105 TC-1 cells, and tumor growth was followed every 2–3 days. The number and percentage of tumor-free mice on day 70 compared with the total number of animals injected are shown. (B-E) Wild-type C57BL/6J and MHC-II KO mice were injected on day 0 with 6 × 105 TC-1 cells, and on day 25, cell suspensions were prepared from spleens, dLN and tumors and analyzed by flow cytometry. The spleens and lymph nodes from naive mice were used as controls. The numbers of lymphocyte subsets and their percentages within the total CD45+ in spleen (B and C), in LN (D and E), and in tumors (F and G), respectively are shown. B-G show the mean ± SEM of cumulative results from 3 independent experiments (n = 6–7 mice per group). *p < 0.05, ** p < 0.01 and ***p < 0.001 as determined by Mann-Whitney's test between each lymphoid subset in WT vs MHC-II KO mice for each organ.
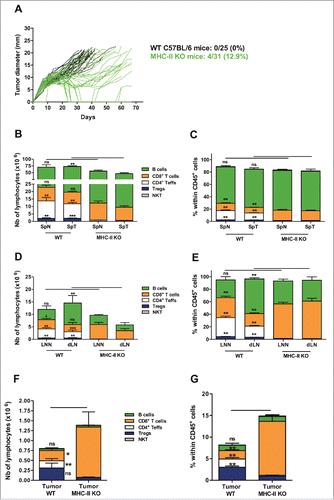
The few remaining CD4+ T-cells observed in the spleen of naive or tumor-bearing MHC-II KO mice consisted of conventional Teffs (40%, CD4+ NK1.1− Foxp3−), Tregs (20%, CD4+ NK1.1− Foxp3+) and NKT-cells (40%, CD3+ CD4+ NK1.1+ Foxp3−) (Fig. S5E). In the LN of either normal or tumor-bearing MHC-II KO mice, Tregs represented 60% of the remaining CD4+ T-cells vs of the 35% Teffs and approximately 3–5% of the NKT-cells (Fig. S5E and F).
A larger proportion of lymphocytes was observed in the tumors of MHC-II KO mice ( and ), with a strong increase in both the number and frequency of CD8+ T-cells and dramatically reduced numbers of Teffs and Tregs. However, although the absolute number of Tregs was drastically reduced in MHC-II KO tumors, their proportions within total CD4+ T-cells was slightly higher than those in the tumors of WT mice ( and and Fig. S5G).
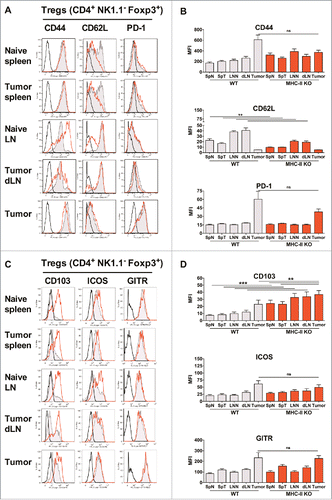
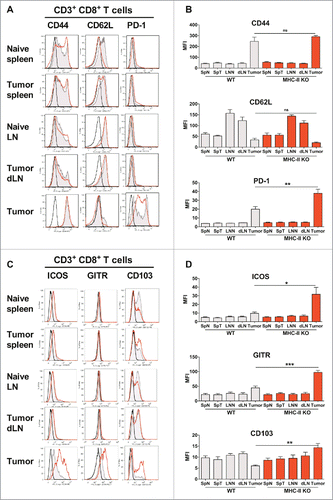
We then analyzed the phenotype of the T-cells in MHC-II KO mice and found an increased level of CD44 on the few remaining Teffs of naive or tumor-bearing MHC-II KO mice, both in spleen and LN, in association with a decreased level of CD62L in these lymphoid organs, suggesting that they were highly activated, as confirmed by their upregulation of ICOS, CD103, CD39 and CD73 (Fig. S7). A significant decrease in CD62L was also observed for tumor Tregs (), but in contrast to WT mice, CD44 was not significantly increased on the Tregs infiltrating the tumor. The expression of PD-1, ICOS and GITR was similar in both mouse strains ().
Figure 5. Similar activation status of Tregs in WT and MHC-II KO mice. WT C57BL/6J and MHC-II KO mice were injected on day 0 with 6 × 105 TC-1 cells, and cell suspensions were prepared after 25 days from spleens, dLNs and tumors and analyzed by flow cytometry. The spleens and lymph nodes from naive mice were used as controls. The surface expression of CD44, CD62 L, PD-1, CD103, ICOS and GITR on Tregs from the indicated organs is shown in A and C. Filled grey lines represent WT mice, open red lines represent MHC-II KO mice, while labeling with isotype controls is represented by black lines. Data from one representative experiment are shown. The MFI ± SEM shown in B and D represents cumulative results from 4 independent experiments with n = 7–11 mice per group (WT: grey histograms and MHC-II KO mice: red histograms). ** p < 0.01 and *** p < 0.001 as determined by the Mann-Whitney test.
MHC-II KO regressor mice develop an efficient E7-specific CD8+ T-cell response
In contrast to the dramatically reduced numbers of CD4+ T-cells, higher numbers of CD8+ T-cells infiltrated the tumors of MHC-II KO mice ( and ), which could explain the controlled tumor growth in these mice. The CD8+ T-cells infiltrating the tumors from both WT and MHC-II KO mice had significantly increased expression of CD44 and decreased expression of CD62L (). By contrast, compared to WT mice, the CD8+ T-cells infiltrating the tumor from MHC-II KO expressed a significantly higher level of PD-1 as well as ICOS, GITR and CD103, suggesting a higher level of activation of these cells in these mice, which could explain their increased capacity to control the TC-1 tumor growth.
Figure 6. CD8+ T lymphocytes are activated in MHC-II KO mice and recruited to the tumor, despite the absence of CD4+ T-cells. WT C57BL/6J and MHC-II KO mice were injected on day 0 with 6 × 105 TC-1 cells, and cell suspensions were prepared after 25 days from spleens, dLN and tumors and analyzed by flow cytometry. The spleens and lymph nodes from naive mice were used as controls. The surface expression of CD44, CD62L, PD-1, ICOS, GITR and CD103 on CD8+ T-cells purified from the indicated organs is shown in A and C. Filled grey lines represent WT mice, open red lines represent MHC-II KO mice, while labeling with isotype controls is represented by black lines. Data from one representative experiment are shown. The MFI ± SEM shown in B and D (WT: grey histograms and MHC-II KO: red histograms) represents cumulative results from 4 independent experiments (n = 7–11 mice per group). * p < 0.05, ** p < 0.01 and *** p < 0.001 as determined by the Mann-Whitney test.
To further evaluate the cytotoxic functions of these CD8+ T-cells, we next quantified the CD8+ T-cells specific for the HPV-16 E7 oncoprotein, which developed in TC-1 tumor-bearing mice. First, we compared the percentage of E7-specific CD8+ T-cells that developed in naïve WT and MHC-II KO mice after immunization with the CyaA-E7 vaccine. Despite the absence of CD4+ Teffs, MHC-II KO mice developed an efficient E7-specific CTL response, which was even significantly higher than the response of WT mice ().
Figure 7. MHC-II KO mice develop strong specific anti-tumor CD8+ T-cell responses after vaccination or during tumor growth, independently of CD4+ T-cell help. (A) Naive WT and MHC-II KO mice were vaccinated on day 0 with either CyaA-E7 (50 µg/mouse) and CpG-B-DOTAP (30 and 60 µg/mouse, respectively) or PBS. On day 7, the mice were killed, cell suspensions were prepared from their spleens and the percentages of tetramer-E7+ CD8+ T-cells were analyzed by flow cytometry. Cumulative results from 3 independent experiments are shown (n = 9 mice per group). (B-D) WT and MHC-II KO mice were injected on day 0 with 6 × 105 TC-1 cells, cell suspensions were prepared from tumors after 20 days, and the percentages of tetramer-E7+ CD8+ T-cells were analyzed by flow cytometry. Mice vaccinated on day 13 were used as a positive control. The results represent cumulative data from 2 independent experiments. The percentages of tetramer E7-positive CD8+ T-cells in spleen (B), LN (C) (n = 6–8 mice per group) and tumor (D) (n = 5–8 mice per group) are shown. (E-F) MHC-II KO mice were injected on day 0 with 6 × 105 TC-1 cells. In E, the red curves represent tumor growth up to day 20 in mice that started to reject their tumor (regressor mice), while the black curves represent tumor growth in mice with growing tumors (progressor mice). (F) Mice were killed at day 20, and the percentages of tetramer E7-positive CD8+ T-cells were analyzed in the tumors of progressor and regressor MHC-II KO mice (n = 4 mice per group). * p< 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001 as determined by the Mann-Whitney test.
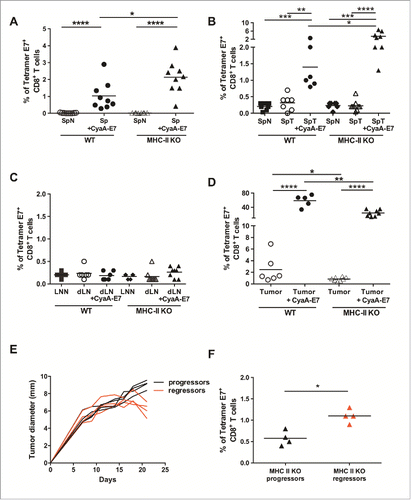
In tumor-bearing WT and MHC-II KO mice, the E7-specific CD8+ T-cell responses which developed in the spleen and LN in the absence of immunization were very low and comparable to the responses of naïve mice without tumors ( and ). However, tumor-bearing MHC-II KO mice that were immunized with the CyaA-E7 vaccine developed significantly higher E7-specific CTL responses in the spleen than the WT mice. In the absence of vaccination, E7-specific CD8+ T-cells were detectable in the tumors of both WT and MHC-II KO mice and dramatically increased in mice immunized with the CyaA-E7 vaccine ().
As previously observed in , in some MHC-II KO mice grafted with TC-1 cells, the tumor regressed spontaneously (regressor mice), in contrast to mice in which tumor growth was not controlled (progressor mice) (). Comparison of the E7-specific CD8+ T-cell responses infiltrating the tumors of regressor and progressor MHC-II KO mice demonstrated that rejection of the tumor was associated with significantly higher percentages of E7-specific CD8+ T-cells ().
Overall, these data clearly established that in CD4+ Teff-deficient MHC-II KO mice, increased numbers of CD8+ T-cells infiltrated the tumor, with a more activated phenotype, suggesting that the increased CTL responses that developed in these mice are responsible for the enhanced control of tumor growth.
Finally, we evaluated whether Tregs CD4+ Foxp3+ T cells which are found in spleen, LN and tumor of MHC class II deficient mice could affect anti-tumor responses of these mice. Wild-type C57BL/6or MHC-II KO were injected with TC-1 cells and treated either by anti-CD25 (clone PC61-5.3) or by control isotype as previously described.Citation33 This strategy has been shown to successfully attenuate in vivo Treg functions, without total depletion of this cell subset, in large numbers of various experimental models, including infections with virus, bacteria and parasites, as well as in anti-tumor immune responses.Citation34 As shown in Fig. S8, the growth of the TC-1 tumor was clearly delayed in MHC-II KO mice treated by anti-CD25, as compared to mice treated with anti-isotype antibodies, as observed in wild-type C57BL/6.
Discussion
Radiotherapy and chemotherapeutic agents are widely used, alone or in combination, for the treatment of cancer patients. Chemotherapeutic agents have an established beneficial effect against tumor cells, but they may also affect the immune system. Their impact can be beneficial: indeed, cyclophosphamide induces IFN type I responses against tumorsCitation35–37 and Treg depletion,Citation38 while anthracyclines favor tumor-specific CD8+ T-cell responses. Conversely, they can also compromise the anti-tumor immune response by inducing myelo- or lympho-ablative side effects. Immunotherapy is now used in combination with other anti-cancer agentsCitation39–44 based on the demonstration of the importance of the immune infiltrate at tumor site.Citation37,41,45–49 Indeed, several strategies aim to deplete or inhibit MDSCsCitation50 or Tregs, which massively accumulate at the tumor site and/or in the blood of cancer patients and inhibit the anti-tumor immune response.Citation51–53 Several strategies have been developed for depleting Tregs or suppressing their functions, such as anti-CD25 antibody or anti-CTLA-4, or -GITR antibodies. Although these strategies have provided promising results, none of them resulted in complete tumor regression. Thus, they need to be improved and used in combination with other immunotherapeutic agents. To achieve such an objective, a better understanding of Treg recruitment and function is needed.
Therefore, the aim of our study was to fully characterize the phenotype and function of tumor-infiltrating Tregs and to decipher the mechanism(s) responsible for their recruitment to the tumor site in order to develop strategies to inhibit their accumulation or their suppressive activity.
In the present study, using the TC-1 mouse model of HPV-related carcinoma, we demonstrated that tumor-infiltrating Tregs were highly activated, with increased expression of immunosuppressive molecules. Both intratumoral Teffs and Tregs expressed high levels of PD-1, but anti-PD-1 antibody treatment did not impact the growth of the TC-1 tumor nor restore the therapeutic effect of the CyaA-E7 vaccine. To analyze the mechanisms by which Tregs are recruited to the tumor site, we used MHC-II KO mice with drastically reduced numbers of CD4+ effector T-cells. We demonstrated that these mice still had significant numbers of Tregs in the lymphoid organs and that these Tregs were recruited to the tumor. In MHC-II KO mice, the growth of the TC-1 tumor was delayed, which correlated with a strong increase in the intratumoral recruitment of CD8+ T-cells. In addition, in mice that spontaneously rejected their tumor, the infiltration of E7-specific CD8+ T-cells was significantly higher than that in MHC-II KO mice with growing tumors. These results demonstrate that tumor-specific CD8+ T-cells can be efficiently activated and recruited in the absence of MHC class II molecules and of CD4+ T-cell help.
Using Foxp3-GFP mice, to clearly distinguish Tregs from Teffs, we demonstrated that tumor-infiltrating Tregs were highly activated with significantly increased expression of suppressive molecules, including CD39, CD103 and PD-1. Although tumor-infiltrating Tregs as well as Teffs had significantly increased expression of PD-1, the treatment of tumor-bearing mice with an anti-PD-1 mAb did not induce tumor regression or enhance the therapeutic effect of the CyaA-E7 vaccine. These results were surprising given that a beneficial effect of anti-PD-1/PD-L1 treatment was demonstrated in the same mouse model of HPV-related cervical carcinoma. Indeed, it was reported in two previous studies using the TC-1 model that the combination of PD-1 or PD-L1 blockade with anti-tumor vaccine improved the therapeutic effect of the vaccine.Citation54,55 This discrepancy with our results could be explained by the different experimental conditions used. Indeed, in these studies, mice received repeated injections of the vaccine few days after tumor grafting (days 9 to 12 or 8, 15 and 22), whereas in our study, TC-1 tumor-bearing mice received a single injection of the CyaA-E7 vaccine at day 14 or 24 when tumors were larger (> 7 mm). In addition, we started the injection of the anti-PD-1 mAb on day 13 or 23, in contrast to day 8 in the study by Mkrtichyan et al.Citation54 We have previously established that the antitumor efficacy of the CyaA-E7 vaccine gradually decreased when the time between TC-1 tumor graft and vaccination increased and the vaccine had no effect if the tumor diameter was greater than 8 mm,Citation18 demonstrating the difficulty to overcome escape mechanisms at late stages of tumor growth.
Another hypothesis to explain the lack of efficacy of the treatment by the anti-PD-1 mAb could be that TC-1 tumor cells express high levels of PD-L1Citation56 which could compete with the anti-PD-1 mAb for binding to PD-1 at the surface of Teffs. This competition could be more marked in large tumors. The absence of beneficial effect of PD-1 blockade could be also due to the compensatory upregulation of other immune checkpoints such as CTLA-4, TIM-3 or LAG-3 on treated T-cells.Citation57,58 Dual or a triple blockade might thus be required to induce an improved anti-tumor immune response.Citation58
CD39 expression was also significantly increased on tumor Tregs, but since CD73 is significantly reduced, these results suggest that tumor Tregs do not exert their immunosuppressive activity through the up-regulation of intracellular AMP via the CD39-CD73 pathway. Recent studies have shown that highly suppressive Tregs that accumulate in tumors are characterized by increased expression of ICOS.Citation24,25 Since we also observed increased expression of ICOS on tumor and dLN Tregs, this finding suggests that inhibition of the ICOS-ICOS-L pathway could be a promising strategy for enhancing the anti-tumor immune response. Similar results were obtained for the CD103 molecule, which was significantly increased on tumor and dLN Tregs. CD103 was shown to be a hallmark of tumor-infiltrating Tregs.Citation23 However, this integrin, which is required for T-cell homing to the intestinal mucosa, is also expressed by T lymphocytes in epithelial tissuesCitation59 and by tissue-resident innate lymphoid cells and innate-like T cells.Citation60 Importantly, resident memory cells (Trm), defined by the expression of CD103 and CD69, were recently shown to play a key role in the efficacy of cancer vaccine to inhibit tumor growth.Citation61 In agreement with these findings, the expression of CD103 was also significantly increased on tumor-infiltrating Teffs.
Furthermore, we demonstrated that tumor Tregs had significantly increased expression of T-bet. According to recent data showing that Tregs increase the expression of transcription factors involved in the differentiation of the effector cells they are suppressing,Citation62–64 we hypothesized that tumor Tregs had increased suppressive activity towards Teffs, which were mainly Th1 (T-bethigh) in the tumor.
Unexpectedly, however, our data showed that in a polyclonal assay, Tregs purified from tumors had a similar suppressive ability as Tregs isolated from the spleen of naive mice. However, we have previously demonstrated that the Tregs that infiltrated the TC-1 tumor displayed CDR3 spectratyping profiles characteristic of biased and strongly perturbed repertoires, typical of clonal expansion.Citation19 Thus, it could be suggested that Tregs may have increased suppressive activity only toward certain antigenic specificities, which cannot be detected in a polyclonal assay.
This previous study has also established that the TCR repertoire of tumor-Tregs shares public sequences with Tregs from dLN but not with those from spleen or from naive mice.Citation19 Indeed, in six independent experiments, we demonstrated public sequences that are shared by all TC-1 tumor-infiltrating Treg samples. These results were confirmed with the B16-OVA melanoma tumor. The public sequences were, however, completely different between the two tumor models, corroborating the observation that tumor-infiltrating Tregs are specific for tumor antigens for each tumor type. Furthermore, these sequences were not shared by tumor-infiltrating Teff cells, suggesting that the conversion process does not actively contribute to Treg enrichment of the tumor. Our present results showing that the tumor-infiltrating Tregs were nTregs confirm this hypothesis.
The demonstration that the TCR repertoire of tumor Tregs is skewed towards public sequences present in the dLN suggests that Tregs accumulate in the tumor tissue through an antigen-driven mechanism that is specific to each tumor model and different from that of Teff cells. To test this hypothesis, we used a strain of MHC-II KO mice that lacks all classical MHC class II genesCitation32 and shows nearly complete absence of CD4+ T-cells in the spleen and LN.Citation32 The few remaining CD4+ T-cells had an activated phenotype characterized by high levels of CD44 and low levels of CD62L. In these KO mice, CD8+ T-cells differentiated normally.Citation32
In the present study, we confirmed that CD4+ T-cells were detectable in the spleen and LN of MHC-II KO mice in agreement with other studies.Citation20 In addition, we demonstrated that the few remaining CD4+ T-cells observed in the spleen of naive or tumor-bearing MHC-II KO mice consisted of conventional Teffs (40%, CD4+ NK1.1− Foxp3−), Tregs (20%, CD4+ NK1.1− Foxp3+) and NKT-cells (40%, CD3+ CD4+ NK1.1+ Foxp3−). In the LN of either normal or tumor-bearing MHC-II KO mice, Tregs represented 60% of the remaining CD4+ T-cells. These results are in agreement with previous studies that demonstrated the presence of CD4+ Foxp3+ T-cells in the spleen and LN, but not in the thymus, of MHC-II KO mice,Citation21,22 and also established that these Tregs are functional. These results demonstrate that Tregs can develop and express CD4 in the absence of conventional MHC class II molecules. In addition, we also established that in MHC-II KO mice, Tregs infiltrated the tumor, although to a lesser extent than in WT Tregs and, thus, independently of antigen-presentation by MHC-II molecules on APCs or on the TC-1 tumor cells. The activation of Tregs and their recruitment to the tumor site in MHC-II KO mice could be due to homeostatic expansion. However, our previous demonstration that the TCR repertoire of tumor Tregs is skewed towards public sequences also present in the dLN suggest that Tregs accumulate into the tumor tissue through an antigen-driven mechanismCitation19 and, therefore, support the view that Tregs use restriction element(s) that differ from MHC class II molecules and remain to be identified.
In MHC-II KO mice, compared with wild-type mice, spleen and LN Tregs and Teffs were highly activated, as shown by the significantly increased expression of CD44 and the reduced expression of CD62L.
In contrast to the dramatically reduced numbers of CD4+ T-cells, higher numbers of CD8+ T-cells were present in the spleen and LN of MHC-II KO mice and infiltrated the tumor. This finding is in agreement with previous studies and is probably due to a compensatory effect.Citation20,32 These tumor-infiltrating CD8+ T-cells were more activated than their WT counterpart and also showed significantly higher expression of PD-1, ICOS, GITR, CD39 and CD103. These observations suggest that the increased recruitment and activation of CD8+ T-cells in MHC-II KO, despite the quasi-absence of CD4+ T-cells, are responsible for the more efficient control of the TC-1 tumor growth. Indeed, the CD8+ T-cells expressing higher levels of GITR, ICOS or CD103 have a more efficient cytotoxic response.Citation65,66 This result also suggests that Tregs did not affect the recruitment of CD8+ T-cells in tumor-bearing mice. However, the treatment of MHC-II KO mice by anti-CD25 antibodies clearly delayed the growth of the TC-1 tumor, as observed in wild-type C57BL/6 mice, supporting the hypothesis that Tregs negatively regulated the response of CD8+ T-cells in these mice. The enhanced expression of immunosuppressive molecules (CD39) and of exhaustion markers (PD-1) by these CD8+ T-cells could also explain why only 13% of the MHC-II KO mice completely rejected their tumor.
After immunization with the CyaA-E7 vaccine, both normal and tumor-bearing MHC-II KO developed greater numbers and frequencies of E7-specific CD8+ T-cells in the spleen, in agreement with our previous demonstration that the induction of CTL responses by the CyaA vector did not require T-cell help.Citation67 Importantly the MHC-II KO mice that spontaneously rejected their tumors had a significantly higher percentage of E7-specific CD8+ T-cells at the tumor site, which could explain the improved control of tumor progression in these mice. In addition, the delayed tumor growth in MHC-II KO mice can also be explained by their increased frequency of CD4+ NKT-cells, especially in the spleen.
In conclusion, this study represents an extensive analysis of the phenotype, gene expression profile, function and recruitment of tumor-infiltrating Tregs, and clearly evidences that tumor-Tregs are highly activated nTregs that are recruited to the tumor independently of MHC-II molecules. Our results also show that molecules overexpressed by intratumoral Tregs, such as ICOS, deserve further exploration for the development of strategies aiming to deplete or suppress the activity of Tregs.
Materials and methods
Mice and tumors
Specific pathogen-free six- to eight-week-old female C57BL/6J mice were purchased from Charles River. Foxp3-GFP knockinCitation68 and B6.129 S2-H2dlAb1-Ea/J (MHC-II KO)Citation32 mice on a C57BL/6J background were kindly given by B. Malissen (Centre d'Immunologie de Marseille-Luminy, Marseille, France) and M. Albert (Institut Pasteur, Paris, France), respectively. All animals were kept in the Pasteur Institute animal facilities under pathogen-free conditions with water and food ad libitum. The animal experiments were conducted in compliance with French and European regulations for the protection of animals used for scientific purposes (Project authorization number: 00668.02).
TC-1 tumor cells expressing HPV-16 E6 and E7 proteinsCitation69 and derived from primary mouse lung epithelial cells were obtained from the ATCC LGC (Promochem). TC-1 cells were injected subcutaneously (s.c.) into the shaved left flank of C57BL/6J mice (6 × 105 TC-1 cells/mouse for C57BL/6J and MHC-II KO mice and 1 × 106 cells/mouse for Foxp3-GFP mice). The tumor size represents the average of two perpendicular diameters (millimeters) and was measured with a digital caliper (Mitutoyo).
Reagents
The detoxified CyaA of Bordetella pertussis carrying a truncated form of E7 protein from HPV-16 (CyaA-E7) was prepared as previously described.Citation70 CpG-B 1826 (5-TCCATGACGTTCCTGACGTT-3) was synthesized by Proligo and mixed with 60 μg N-[1-(2,3-dioleoyloxyl)propyl]-NNNtrimethylammoniummethyl sulfate (DOTAP; Roche) in 100 μL Opti-MEM.
Rat anti-mouse PD-1 (clone RMP1-14) and control IgG2 a (clone 2A3) monoclonal antibodies (mAbs) were purchased from BioXCell (West-Lebanon, USA) and injected intra-peritoneally (i.p.; 250 µg/mouse). Anti-PD-1 or its control mAb were injected one day before vaccination with CyaA-E7/CpG-B-DOTAP and every 3 days after vaccination (total of 4 injections/mouse: days 13, 17, 20 and 23 or days 23, 27, 30 and 33).
Purified anti-CD3 (clone 145-2C11) mAb was purchased from BD Pharmingen and used to coat the culture plates (96-wells, round bottom) at a concentration of 5 µg/mL.
Cell isolation
Peripheral lymph nodes from naive mice (LNN; maxillary, axillary and inguinal) or inguinal tumor-draining LN (dLN) were harvested, mechanically disrupted and filtered to obtain single-cell suspensions. Spleens from both naive (SpN) and tumor-bearing mice (SpT) were harvested, treated for 20 minutes with 400 U/mL collagenase D and 50 µg/mL DNase I (Boehringer Mannheim), mechanically disrupted and filtered to obtain single-cell suspensions. The tumors were harvested and dissociated using the gentleMACS dissociator (Miltenyi Biotec, Paris, France, program mimpTumor-01-01). The dissociated tumors were then incubated for 45 minutes with 400 U/mL collagenase D and 50 µg/mL DNase I and filtered to obtain single-cell suspensions.
Flow cytometry analysis
The following mAbs were used for FACS staining: APC- or APC eF780-conjugated anti-CD3e (clone 145-2C11), pacific-blue-conjugated anti-CD4 (clone RM4-5 or GK1.5) and PerCP-Cy5.5-conjugated anti-CD8a (clone 53–6.7), together with APC-conjugated anti-CD44 (clone IM7), anti-CD45RB (clone C363.16A), streptavidin, APCeF780-conjugated CD45.2 (clone 104), PE-Cy7-conjugated anti-CD19 (clone 6D5), anti-CD62L (clone MEL-14), anti-CD69 (clone HI.2F), anti-CD28 (clone 37.51), PE-conjugated anti-CD39 (clone 24DMS1), anti-CD73 (clone TY/11.8), anti-CD103 (clone 2E7), anti-GITR (clone DTA-1) anti-PD-1 (clone J43), anti-ICOS (clone 7E.17G9), anti-Neuropilin-1 (clone 761705) biotin-conjugated PD-L1 (clone M1H5) and relevant isotype antibodies were purchased from BD Pharmingen and eBioscience. The pacific orange Live/dead® fixable aqua dead cell stain kit (Invitrogen, molecular probes®, France) was used to stain dead cells, according to the manufacturer's protocol before fixing them for intracellular staining. PE-conjugated anti-CTLA-4 (clone UC10-4F10-11) and anti-T-Bet (clone 4B10) and isotype antibodies were purchased from BD Pharmingen and eBioscience and used for intracellular staining. Foxp3+ cells were detected by intracellular staining with FITC-conjugated anti-Foxp3 mAb (clone FJK-16), according to the manufacturer's protocol (eBioscience). Streptavidin-PE-conjugated H-2Db/E749-57 tetramers were purchased from MBL International Corporation. The cells were acquired on a CyAn Coultronics (Beckman Coulter) flow cytometer and analyzed with FlowJo software (Tree Star).
Ex vivo tetramer staining
The mice were i.v. immunized with CyaA-E7 (50 μg/mouse) together with CpG-B and DOTAP (30 μg and 60 μg/mouse, respectively). Seven days after immunization, spleens were harvested, mechanically disrupted and filtered to obtain single-cell suspensions. Cells were then stained with anti-CD3, anti-CD8a and the H-2Db/E749-57 tetramers (MBL International Corporation) or the control according to the manufacturer's recommendations.
For the tumor-bearing mouse experiment, tumors, dLN and spleens were harvested 20 days after TC-1 inoculation and mechanically disrupted as described above. LN and spleen from naive mice were used as negative controls, and dLN, spleen and tumor from TC-1-bearing mice vaccinated on day 13 were used as positive controls. The obtained cell suspensions were then stained with anti-CD45, anti-CD3, anti-CD8 a and the H-2Db/E749-57 tetramers.
Purification of Teffs and Tregs
LN, spleen, and tumor-cell suspensions from Foxp3-GFP knock-in mice were incubated with anti-CD4 or anti-CD90.2–coated magnetic beads (Miltenyi Biotec) according to the manufacturer's instructions. After incubation, the cells were washed, and CD4+ or CD90.2+ cells were selected on an automated magnetic cell sorter (AutoMACS; Miltenyi Biotec) using the possel_s program. Positive fractions composed of 75% to 90% positive cells were stained with anti-CD4, anti-CD8a, anti-CD3 and the 7-aminoactinomycin D (7AAD) viability dye. The cells were then washed and FACS-sorted into live (7AAD−) CD3+CD8−CD4+Foxp3-GFP− effector T-cells (Teffs) or CD3+CD8−CD4+Foxp3-GFP+ regulatory T-cells (Tregs) on an Aria III sorter (BD). The purity of each cell type was higher than 93%.
T-cell suppression and proliferation assays
Highly purified Tregs and Teffs were co-cultured in 96-well plates (TPP) in triplicate and polyclonally activated with plate-bound anti-CD3 antibody (5 µg/mL; clone 145-2C11; BD Pharmingen) for 72 hours at a 1:1 Tregs/Teffs ratio. The Tregs were then diluted in 2-fold steps to obtain Tregs/Teffs ratios from 1:2 to 1:128.
Proliferative responses were measured by pulsing the cultures with 1 µCi/well [methyl-3H] thymidine (Perkin Elmer, France) for the last 18 hours of culture. The results are expressed as the mean ± SEM of duplicate or triplicate measurements. IFNγ production was also analyzed in the culture supernatants by ELISA.
Quantitative gene expression analysis
Total RNA was extracted from highly purified FACS sorted Teff and Treg cell samples using the RNeasy Plus MicroKit (Qiagen). First-strand cDNA synthesis was conducted using oligo-dT and RNase H-reverse transcriptase SuperScript II (Invitrogen). Quantitative analysis of cDNA samples was performed using real-time PCR. cDNA samples were mixed with the RT2-SYBR® Green qPCR Master Mix (Qiagen) and distributed in ready-to-use PCR array plates (apoptosis, Th17 for autoimmunity and inflammation and mTOR signaling PCR Arrays; Qiagen). Quantitative real-time PCR was followed by the CFX96™ detection system from Bio-Rad.
Statistical analysis
Prism software (GraphPad Software, Inc.) was used to calculate the statistical significance for differences in particular measurements between the groups. The two-tailed Mann-Whitney's test, Mantel–Cox log-rank or ANOVA tests were used. P values less than 0.05 were considered statistically significant.
Disclosure of potential conflict-of-interests
The authors declare no competing financial interests.
Author contributions
N.C. contributed to the design of the project, developed the methodology, performed the research, analyzed and interpreted the data and wrote the paper. A.T., B.D., M.O., C.F and A.SP. performed research and analyzed data. D.L. provided the CyaA-E7 vaccine. C.L. designed, analyzed and interpreted the data and supervised the study and wrote the paper. We thank M. Davi for expert technical assistance in preparing the CyaA-E7 protein.
Financial support
This work was supported by grants from the Ligue Nationale Contre le Cancer (Equipe Labellisée 2017). N.C. was supported by Conseil Régional IdF / Cancéropôle IdF and Fondation de France.
2017ONCOIMM0420R-s01.pdf
Download PDF (45.8 MB)References
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi:10.1126/science.1203486. PMID:21436444
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71. doi:10.1146/annurev-immunol-031210-101324. PMID:21219185
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi:10.1146/annurev.immunol.25.022106.141623. PMID:22224781
- Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. 2012;3:51. doi:10.3389/fimmu.2012.00051. PMID:22566933
- Wing JB, Sakaguchi S. Multiple treg suppressive modules and their adaptability. Front Immunol. 2012;3:178. doi:10.3389/fimmu.2012.00178. PMID:22754556
- Wilke CM, Wu K, Zhao E, Wang G, Zou W. Prognostic significance of regulatory T cells in tumor. Int J Cancer. 2010;127:748–58. PMID:20473951
- Menetrier-Caux C, Gobert M, Caux C. Differences in tumor regulatory T-cell localization and activation status impact patient outcome. Cancer Res. 2009;69:7895–8. doi:10.1158/0008-5472.CAN-09-1642. PMID:19808962
- Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009;27:186–92. doi:10.1200/JCO.2008.18.7229. PMID:19064967
- Tzankov A, Meier C, Hirschmann P, Went P, Pileri SA, Dirnhofer S. Correlation of high numbers of intratumoral FOXP3+ regulatory T cells with improved survival in germinal center-like diffuse large B-cell lymphoma, follicular lymphoma and classical Hodgkin's lymphoma. Haematologica. 2008;93:193–200. doi:10.3324/haematol.11702. PMID:18223287
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi:10.1038/nm1093. PMID:15322536
- Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–93. doi:10.1200/JCO.2006.09.4565. PMID:17577038
- Kobayashi N, Hiraoka N, Yamagami W, Ojima H, Kanai Y, Kosuge T, Nakajima A, Hirohashi S. FOXP3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clin Cancer Res. 2007;13:902–11. doi:10.1158/1078-0432.CCR-06-2363. PMID:17289884
- Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. Localisation pattern of Foxp3+ regulatory T cells is associated with clinical behaviour in gastric cancer. Br J Cancer. 2008;98:148–53. doi:10.1038/sj.bjc.6604149. PMID:18087278
- Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi MB, Harpole DH, Jr, Patz EF, Jr. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006;107:2866–72. doi:10.1002/cncr.22282. PMID:17099880
- van der Burg SH, Piersma SJ, de Jong A, van der Hulst JM, Kwappenberg KM, van den Hende M, Welters MJ, Van Rood JJ, Fleuren GJ, Melief CJ, et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A. 2007;104:12087–92. doi:10.1073/pnas.0704672104. PMID:17615234
- Munger K. The role of human papillomaviruses in human cancers. Front Biosci. 2002;7:d641–9. doi:10.2741/A800. PMID:11861215
- Chaoul N, Fayolle C, Desrues B, Oberkampf M, Tang A, Ladant D, Leclerc C. Rapamycin impairs antitumor CD8+ T-cell responses and vaccine-induced tumor eradication. Cancer Res. 2015;75:3279–91. doi:10.1158/0008-5472.CAN-15-0454. PMID:26122844
- Berraondo P, Nouze C, Preville X, Ladant D, Leclerc C. Eradication of large tumors in mice by a tritherapy targeting the innate, adaptive, and regulatory components of the immune system. Cancer Res. 2007;67:8847–55. doi:10.1158/0008-5472.CAN-07-0321. PMID:17875726
- Sainz-Perez A, Lim A, Lemercier B, Leclerc C. The T-cell receptor repertoire of tumor-infiltrating regulatory T lymphocytes is skewed toward public sequences. Cancer Res. 2012;72:3557–69. doi:10.1158/0008-5472.CAN-12-0277. PMID:22573714
- Grusby MJ, Johnson RS, Papaioannou VE, Glimcher LH. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science. 1991;253:1417–20. doi:10.1126/science.1910207. PMID:1910207
- Kish DD, Gorbachev AV, Fairchild RL. Regulatory function of CD4+CD25+ T cells from Class II MHC-deficient mice in contact hypersensitivity responses. J Leukocyte Biol. 2007;82:85–92. doi:10.1189/jlb.0207089. PMID:17412917
- Bochtler P, Wahl C, Schirmbeck R, Reimann J. Functional adaptive CD4 Foxp3 T cells develop in MHC class II-deficient mice. J Immunol. 2006;177:8307–14. doi:10.4049/jimmunol.177.12.8307. PMID:17142726
- Anz D, Mueller W, Golic M, Kunz WG, Rapp M, Koelzer VH, Ellermeier J, Ellwart JW, Schnurr M, Bourquin C, et al. CD103 is a hallmark of tumor-infiltrating regulatory T cells. Int J Cancer. 2011;129:2417–26. doi:10.1002/ijc.25902. PMID:21207371
- Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med. 2004;199:1479–89. doi:10.1084/jem.20040179. PMID:15184501
- Vocanson M, Rozieres A, Hennino A, Poyet G, Gaillard V, Renaudineau S, et al. Inducible costimulator (ICOS) is a marker for highly suppressive antigen-specific T cells sharing features of TH17/TH1 and regulatory T cells. J Allergy Clin Immunol. 2010;126:280–9, 9 e1-7. doi:10.1016/j.jaci.2010.05.022. PMID:20624644
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi:10.1146/annurev.immunol.26.021607.090331. PMID:18173375
- Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol. 2013;6:74. doi:10.1186/1756-8722-6-74. PMID:24283718
- Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209:1723–42, S1. doi:10.1084/jem.20120914. PMID:22966001
- Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209:1713–22, S1-19. doi:10.1084/jem.20120822. PMID:22966003
- Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, Rubio MT, MacIntyre E, Lepelletier Y, Hermine O, et al. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol. 2009;39:1466–71. doi:10.1002/eji.200839040. PMID:19499532
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Eng J Med. 2012;366:2443–54. doi:10.1056/NEJMoa1200690.
- Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, Mathis D, Fugger L. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A. 1999;96:10338–43. doi:10.1073/pnas.96.18.10338. PMID:10468609
- Jaron B, Maranghi E, Leclerc C, Majlessi L. Effect of attenuation of Treg during BCG immunization on anti-mycobacterial Th1 responses and protection against Mycobacterium tuberculosis. PLoS One. 2008;3:e2833. doi:10.1371/journal.pone.0002833. PMID:18665224
- Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, Cui X, Cummings TJ, Bigner DD, Gilboa E, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res. 2006;12:4294–305. doi:10.1158/1078-0432.CCR-06-0053. PMID:16857805
- Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, Natalino F, Belardelli F, Foà R, Proietti E. Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients' blood cells: implications for cancer chemoimmunotherapy. Clin Cancer Res. 2013;19:4249–61. doi:10.1158/1078-0432.CCR-12-3666. PMID:23759676
- Schiavoni G, Mattei F, Di Pucchio T, Santini SM, Bracci L, Belardelli F, Proietti E. Cyclophosphamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood. 2000;95:2024–30. PMID:10706870
- Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D'Urso MT, Belardelli F, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011;71:768–78. doi:10.1158/0008-5472.CAN-10-2788. PMID:21156650
- Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–8. doi:10.1007/s00262-006-0225-8. PMID:16960692
- Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64. doi:10.1038/nm.2999. PMID:23202296
- Lake RA, Robinson BW. Immunotherapy and chemotherapy–a practical partnership. Nat Rev Cancer. 2005;5:397–405. doi:10.1038/nrc1613. PMID:15864281
- Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–41. doi:10.1016/j.immuni.2013.03.003. PMID:23562161
- Ma Y, Adjemian S, Yang H, Catani JP, Hannani D, Martins I, Michaud M, Kepp O, Sukkurwala AQ, Vacchelli E, et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology. 2013;2:e24568. doi:10.4161/onci.24568. PMID:23894718
- Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Anticancer radioimmunotherapy. Oncoimmunology. 2013;2:e25595. doi:10.4161/onci.25595. PMID:24319634
- van der Most RG, Robinson BW, Lake RA. Combining immunotherapy with chemotherapy to treat cancer. Discov Med. 2005;5:265–70. PMID:20704886
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–95. doi:10.1016/j.immuni.2013.10.003. PMID:24138885
- Hiraoka K, Miyamoto M, Cho Y, Suzuoki M, Oshikiri T, Nakakubo Y, Itoh T, Ohbuchi T, Kondo S, Katoh H. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br J Cancer. 2006;94:275–80. doi:10.1038/sj.bjc.6602934. PMID:16421594
- Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:2654–66. doi:10.1056/NEJMoa051424. PMID:16371631
- Pages F, Galon J, Fridman WH. The essential role of the in situ immune reaction in human colorectal cancer. J Leukocyte Biol. 2008;84:981–7. doi:10.1189/jlb.1107773. PMID:18559950
- Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pagès F, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71:1263–71. doi:10.1158/0008-5472.CAN-10-2907. PMID:21303976
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68. doi:10.1038/nri3175. PMID:22437938
- Chaput N, Darrasse-Jeze G, Bergot AS, Cordier C, Ngo-Abdalla S, Klatzmann D, Azogui O. Regulatory T cells prevent CD8 T cell maturation by inhibiting CD4 Th cells at tumor sites. J Immunol. 2007;179:4969–78. doi:10.4049/jimmunol.179.8.4969. PMID:17911581
- Daniel D, Meyer-Morse N, Bergsland EK, Dehne K, Coussens LM, Hanahan D. Immune enhancement of skin carcinogenesis by CD4+ T cells. J Exp Med. 2003;197:1017–28. doi:10.1084/jem.20021047. PMID:12695493
- Zabala M, Lasarte JJ, Perret C, Sola J, Berraondo P, Alfaro M, Larrea E, Prieto J, Kramer MG. Induction of immunosuppressive molecules and regulatory T cells counteracts the antitumor effect of interleukin-12-based gene therapy in a transgenic mouse model of liver cancer. J Hepatol. 2007;47:807–15. doi:10.1016/j.jhep.2007.07.025. PMID:17935823
- Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, Cook L, Khleif SN. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol. 2011;41:2977–86. doi:10.1002/eji.201141639. PMID:21710477
- Liu Z, Zhou H, Wang W, Fu YX, Zhu M. A novel dendritic cell targeting HPV16 E7 synthetic vaccine in combination with PD-L1 blockade elicits therapeutic antitumor immunity in mice. Oncoimmunology. 2016;5:e1147641. doi:10.1080/2162402X.2016.1147641. PMID:27471615
- Tang A, Dadaglio G, Oberkampf M, Di Carlo S, Peduto L, Laubreton D, Desrues B, Sun CM, Montagutelli X, Leclerc C. B cells promote tumor progression in a mouse model of HPV-mediated cervical cancer. Int J Cancer. 2016;139:1358–71. doi:10.1002/ijc.30169. PMID:27130719
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi:10.1038/ncomms10501. PMID:26883990
- Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. 2017;6:e1249561. doi:10.1080/2162402X.2016.1249561. PMID:28197366
- Agace WW, Higgins JM, Sadasivan B, Brenner MB, Parker CM. T-lymphocyte-epithelial-cell interactions: integrin alpha(E)(CD103)beta(7), LEEP-CAM and chemokines. Curr Opin Cell Biol. 2000;12:563–8. doi:10.1016/S0955-0674(00)00132-0. PMID:10978890
- Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell. 2016;164:365–77. doi:10.1016/j.cell.2016.01.002. PMID:26806130
- Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun. 2017;8:15221. doi:10.1038/ncomms15221. PMID:28537262
- Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. 2012;37:501–10. doi:10.1016/j.immuni.2012.05.031. PMID:22960221
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi:10.1038/ni.1731. PMID:19412181
- Yu F, Sharma S, Edwards J, Feigenbaum L, Zhu J. Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat Immunol. 2015;16:197–206. doi:10.1038/ni.3053. PMID:25501630
- Clouthier DL, Zhou AC, Watts TH. Anti-GITR agonist therapy intrinsically enhances CD8 T cell responses to chronic lymphocytic choriomeningitis virus (LCMV), thereby circumventing LCMV-induced downregulation of costimulatory GITR ligand on APC. J Immunol. 2014;193:5033–43. doi:10.4049/jimmunol.1401002. PMID:25281716
- Wallin JJ, Liang L, Bakardjiev A, Sha WC. Enhancement of CD8+ T cell responses by ICOS/B7 h costimulation. J Immunol. 2001;167:132–9. doi:10.4049/jimmunol.167.1.132. PMID:11418641
- Guermonprez P, Fayolle C, Rojas MJ, Rescigno M, Ladant D, Leclerc C. In vivo receptor-mediated delivery of a recombinant invasive bacterial toxoid to CD11 c + CD8 alpha -CD11bhigh dendritic cells. Eur J Immunol. 2002;32:3071–81. doi:10.1002/1521-4141(200211)32:11%3c3071::AID-IMMU3071%3e3.0.CO;2-A. PMID:12385027
- Wang Y, Kissenpfennig A, Mingueneau M, Richelme S, Perrin P, Chevrier S, Genton C, Lucas B, DiSanto JP, Acha-Orbea H, et al. Th2 lymphoproliferative disorder of LatY136 F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J Immunol. 2008;180:1565–75. doi:10.4049/jimmunol.180.3.1565. PMID:18209052
- Lin KY, Guarnieri FG, Staveley-O'Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–6. PMID:8548765
- Preville X, Ladant D, Timmerman B, Leclerc C. Eradication of established tumors by vaccination with recombinant Bordetella pertussis adenylate cyclase carrying the human papillomavirus 16 E7 oncoprotein. Cancer Res. 2005;65:641–9. PMID:15695409