
ABSTRACT
New clinical trials are now evaluating the efficacy of neoadjuvant immunotherapy in the context of primary tumor surgery. Using the orthotopic 4T1.2 mouse model of spontaneously metastatic mammary cancer, we have shown that neoadjuvant immunotherapy and surgery was superior in the generation of tumor-specific CD8+ T cells and eradication of lethal metastases compared to surgery followed by adjuvant immunotherapy. However, the importance of host Batf3 and type I interferon (IFN) for long-term survival of mice following neoadjuvant immunotherapy is unknown. Here we demonstrated that loss of Batf3+ DCs or type I IFN receptor blockade in 4T1.2 tumor-bearing mice treated with neoadjuvant anti-PD-1+anti-CD137 immunotherapy reduced long-term survival with a corresponding reduction in tumor-specific CD8+ T cells producing effector cytokines in the primary tumor and in the periphery. Interestingly, we found all high-risk stage III melanoma patients relapsing after adjuvant or neoadjuvant ipilimumab+nivolumab within the OpACIN trial (NCT02437279) displayed low expression of Batf3+ DC-associated genes in pre-treatment tumor biopsies. Further focus should now be placed on validating the requirement of an intratumoral Batf3+ DC gene signature for response to neoadjuvant immunotherapy.
Introduction
Immune checkpoint antibodies reactive with CTLA4 and PD-1/PD-L1 used alone or in combination have demonstrated efficacy across many advanced/metastatic cancer types and are approved as standard-of-care for a number of cancers including melanoma and non-small cell lung cancer (NSCLC)Citation1. Recently, the efficacy of adjuvant anti-CTLA4 or anti-PD-1 has also been demonstrated in melanomaCitation2-Citation4, although a significant proportion of these treated patients eventually relapse. Using two mouse model of spontaneously metastatic triple negative breast cancer (TNBC), we have demonstrated that neoadjuvant immunotherapy followed by surgery of the primary tumor compared to surgery and adjuvant immunotherapy was superior in the eradication of lethal metastasesCitation5. Neoadjuvant immunotherapy was always superior to adjuvant immunotherapy in its ability to improve long-term survival of mice regardless of the immunotherapies used, including anti-CD25 to deplete Tregs, anti-PD-1 alone or in combination with agonistic anti-CD137. More recently our randomized phase I clinical trial (OpACIN; NCT02437279) comparing neoadjuvant versus adjuvant anti-CTLA4 and anti-PD-1 in macroscopic stage III melanoma showed that neoadjuvant ipilimumab+nivolumab followed by surgery was feasible and superior in its immune activating capacity to adjuvant immunotherapy, although it induced high toxicity ratesCitation6. High response rates but significant toxicities were also seen in a randomized phase II clinical trial of high-risk stage III/IV melanoma patients treated with neoadjuvant ipilimumab+nivolumabCitation7. This study also demonstrated that neoadjuvant ipilimumab+nivolumab yielded better response compared to neoadjuvant nivolumab monotherapyCitation7 similar to what our pre-clinical studies predictedCitation5. Furthermore, in another small study of 20 patients with resectable early-stage NSCLC, 45% (9/20), those who received neoadjuvant nivolumab had an unexpectedly high pathological complete responseCitation8.
In pre-clinical mouse models, the efficacy of neoadjuvant anti-PD-1+anti-CD137 depended on CD8+ T cells and partially on CD4+ T cells and NK cellsCitation5. We also demonstrated an increase in tumor-specific CD8+ T cells producing IFNγ in the periphery and the primary tumor were critical for the efficacy of neoadjuvant immunotherapyCitation5. However, the impact of innate immunity on neoadjuvant immunotherapy remains poorly understood. We therefore investigated the requirement of innate immune cells and the type I IFN pathway in mediating the efficacy of neoadjuvant immunotherapy both pre-clinically and in melanoma patients.
Results
Efficacy of neoadjuvant anti-PD-1+anti-CD137 is dependent on Batf3+ DCs and type I IFN
It has been shown that innate immune sensing of tumors occurs through activation of the STING pathway, which leads to type I interferon (IFN) production, dendritic cell (DC) activation, cross-presentation of tumor-associated antigens to CD8+ T cells, and T cell recruitment into the tumor microenvironmentCitation9,Citation10. Recent studies have also revealed a major role for Batf3 lineage-derived DCs in anti-tumor immunityCitation11,Citation12 and responses to immunotherapyCitation13-Citation16. Therefore, we examined how loss of Batf3+ DCs impacted on the efficacy of neoadjuvant anti-PD-1+anti-CD137 therapy (). In 4T1.2 tumor-bearing Batf3−/- mice treated with neoadjuvant immunotherapy, we observed a complete loss of long-term survivors compared to similar groups of tumor-bearing WT mice (). Similarly in the adjuvant setting, treatment of tumor-bearing WT mice with adjuvant anti-PD-1+anti-CD137 significantly prolonged survival compared to similar groups of treated Batf3−/- tumor-bearing mice (p = 0.0228) (). Interestingly, even in the absence of host Batf3, neoadjuvant immunotherapy significantly prolonged survival compared to adjuvant immunotherapy (p = 0.0271) ().
Figure 1. Deficiency in Batf3 impacts on the efficacy of neoadjuvant anti-PD-1 and anti-CD137 immunotherapies. (A, B) Groups of BALB/c WT or Batf3−/- mice (n = 5/grp) were injected with 5 × 104 4T1.2 mammary carcinoma cells in the mammary fat pad. A, As indicated in the schematic, groups of mice were treated with either neoadjuvant or adjuvant anti-PD-1 and anti-CD137 mAb (100 μg/mouse of each mAb, i.p.) on days 11 and 13 (neoadjuvant groups) or days 19 and 21 (adjuvant groups) with all primary tumors resected on day 16 while control IgG (cIg) (200 μg/mouse, i.p.) was given on days 11, 13, 19 and 21. A, The Kaplan-Meier curves for overall survival of each group are shown. Data pooled from 2 experiments with significant differences between indicated groups determined by log-rank sum test with exact p values shown. B, From one experiment in (A), peripheral blood was collected from the indicated groups of mice longitudinally for flow cytometry (n = 5/grp). Gating on live CD45.2+ cells of lymphocyte morphology, the proportion of gp70 tetramer+ CD8+ TCRβ+ cells is shown. Data presented as mean + SEM. A naive mouse was also included in this experiment. Significant differences between neoadjuvant anti-PD-1+ anti-CD137 treated WT mice and Batf3−/- mice on day 16 following tumor inoculation were determined by unpaired Welch’s t-test with exact p value indicated. C-E, In a similar experimental setup as (A), groups of WT and Batf3−/- mice (n = 4–6/grp) were treated with neoadjuvant anti-PD-1 and anti-CD137 mAb (100 μg/mouse of each mAb, i.p.) or cIg (200 μg/mouse, i.p.) on days 11 and 13 and were sacrificed on day 16 and their tumors, draining lymph node (dLN) and non-draining lymph node (ndLN) collected and single cell suspensions generated for flow cytometry. Gating on live CD45.2+ cells of lymphocyte morphology, the absolute numbers of gp70 tetramer+ CD8+ TCRβ+ cells in the (C) tumor, (D) dLN and (E) ndLN are shown. Data presented as mean ± SEM. Data pooled from 2 experiments with significant differences determined by two-way ANOVA with Dunnett’s post-test analysis with exact p value shown.
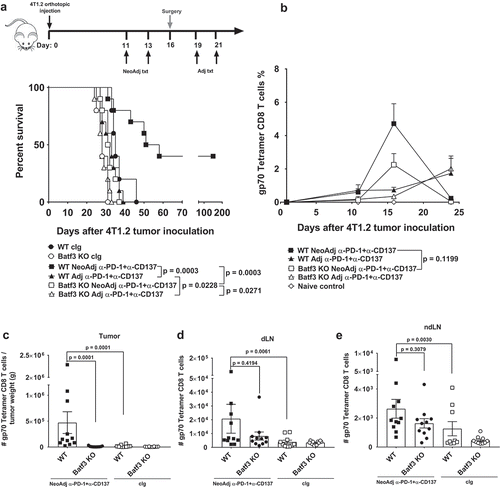
The murine leukemia virus (MuLV) which encodes for the envelope glycoprotein (gp70) is present in many mouse tumor cell lines. While gp70 is normally silent in normal mouse tissue, it can function as a tumor neoantigen in cell lines such as 4T1.2 that express MuLV where tumor-reactive gp70 tetramer-specific CD8+ T cells can be identifiedCitation5,Citation17,Citation18. The loss of efficacy in Batf3−/- mice correlated with a decrease in the proportion of peripheral blood gp70 tetramer-specific CD8+ T cells in tumor-bearing Batf3−/-, compared to WT mice, following neoadjuvant anti-PD1+anti-CD137 (). Despite this decrease, this data also demonstrated that non-Batf3+ APCs may play a role in the expansion of gp70 tetramer-specific CD8+ T cells in the blood following neoadjuvant immunotherapy, although this was not sufficient to generate any long-term survivors (). We also confirmed that a proportion gp70 tetramer-specific CD8+ T cells in the primary tumor, lung and spleen expressed PD-1 and/or CD137, whether they were treated with neoadjuvant immunotherapy or cIg (Supp. Fig. 1).
We previously demonstrated that early after neoadjuvant immunotherapy, but not adjuvant immunotherapy, gp70 tetramer-specific CD8+ T cells increased within the blood and this correlated with the efficacy of neoadjuvant immunotherapyCitation5. Similarly, when we assessed the tumor-infiltrating lymphocytes (TILs) in the resected primary tumor at day 16, we observed a significant decrease in the numbers and proportion of gp70 tetramer-specific CD8+ T cells after neoadjuvant treatment in Batf3−/- mice compared to WT mice ()(Supp. Fig. 2A). Interestingly, the numbers and proportion of gp70 tetramer-specific T cells in the tumour draining or non-draining lymph nodes were not significantly changed between these two groups ()(Supp. Fig. 2B, C). In addition, neoadjuvant immunotherapy significantly increased gp70 tetramer-specific T cell numbers in WT mice, compared to cIg treatment, but this increase was generally not observed between Batf3−/- mice treated with neoadjuvant immunotherapy or cIg (). In contrast, the total number of CD8+ T cells in the primary tumors of WT mice treated with neoadjuvant immunotherapy or cIg was similar, suggesting the increase in gp70 tetramer-specific T cells was specific (Supp. Fig. 3A).
Interestingly, while the total number of CD8+ T cells was lower in neoadjuvant anti-PD-1+anti-CD137 treated Batf3−/- compared to WT mice (Supp. Fig. 3A), this was not observed in the draining or non-draining lymph nodes (Supp. Fig. 3B, C), suggesting that the presence of Batf3 lineage-derived dendritic cells in the primary tumor may be critical for the efficacy of neoadjuvant immunotherapy. Finally, we also assessed changes in total CD4+ T cells in the primary tumor resected from neoadjuvant-treated Batf3−/- mice compared to WT mice and surprisingly observed no significant changes suggesting that the loss of Batf3 primarily appears to impact on CD8+ T cell numbers (Supp. Fig. 3D).
A caveat of using the Batf3−/- mouse is that they lack cross-presenting CD103+-CD8α+ DCs, which can impact on the initial priming of tumor-specific T cells. Given that these DCs are thought to be the major source of type I IFNsCitation19, we next examined whether the efficacy of neoadjuvant anti-PD-1+anti-CD137 therapy depended on type I IFN (). Strikingly, blocking type I IFN receptor in 4T1.2 tumor-bearing mice immediately prior to neoadjuvant immunotherapy resulted in the complete inability to generate long term-survivors compared to a cIg-treated group (). Similarly, blocking type I IFN receptor in neoadjuvant anti-PD-1 alone- or anti-PD-1+anti-CTLA4-treated mice significantly reduced their survival compared to similar neoadjuvant-treated groups that received cIg (). From the experiment in , a cohort of mice from the indicated groups were bled longitudinally and we observed a decrease in gp70 tetramer-specific T cells in mice treated with neoadjuvant immunotherapy and anti-IFNAR1 compared to the neoadjuvant immunotherapy and cIg-treated groups ().
Figure 2. Type I interferon signaling is required for the efficacy of neoadjuvant anti-PD-1 and anti-CD137 immunotherapy. A, Groups of BALB/c WT mice (n = 5–6/grp) were injected with 5 × 104 4T1.2 mammary carcinoma cells in the mammary fat pad. Groups of mice were treated with either neoadjuvant or adjuvant anti-PD-1 and anti-CD137 mAb (100 μg/mouse of each mAb, i.p.) or cIg (200 μg/mouse, i.p.) with all primary tumors resected on day 16 as indicated in the schematic. In some groups, mice were additionally treated with cIg or anti-IFNAR1 (500 μg/mouse of each mAb, i.p.) on day 10, 11, 13 (neoadjuvant groups) or days 18 and 19, 21 (adjuvant groups). The Kaplan-Meier curves for overall survival of each group are shown. Significant differences between indicated groups were determined by log-rank sum test with exact p values shown. Data pooled from 2 experiments. B, Groups of BALB/c WT mice (n = 5–10/grp) were injected with 2 × 104 4T1.2 mammary carcinoma cells in the mammary fat pad. Groups of mice were treated with either neoadjuvant anti-PD-1 alone or anti-PD-1 and anti-CTLA4 mAb (250 μg/mouse of each mAb, i.p.) or cIg (500 μg/mouse, i.p.) on day 12 and 14 with all primary tumors resected on day 16. In some groups, mice were additionally treated with cIg or anti-IFNAR1 (250 μg/mouse of each mAb, i.p.) on day 10, 12, 14 and 21. The Kaplan-Meier curves for overall survival of each group are shown. Significant differences between indicated groups were determined by log-rank sum test with exact p values shown. This experiment was performed once. C, From one experiment in (A), peripheral blood was collected from the indicated groups of mice longitudinally for flow cytometry (n = 5/grp) at the indicated time point for flow cytometry. Gating on live CD45.2+ cells of lymphocyte morphology, the proportion of gp70 tetramer+ CD8+ TCRβ+ cells is shown. Data presented as mean + SEM. A naive mouse was also included for this experiment. Significant differences between cIg or anti-IFNAR1 mAb treated mice on day 16 following tumor inoculation were determined by unpaired Welch’s t-test with exact p value indicated. Data representative of 2 independent experiments.
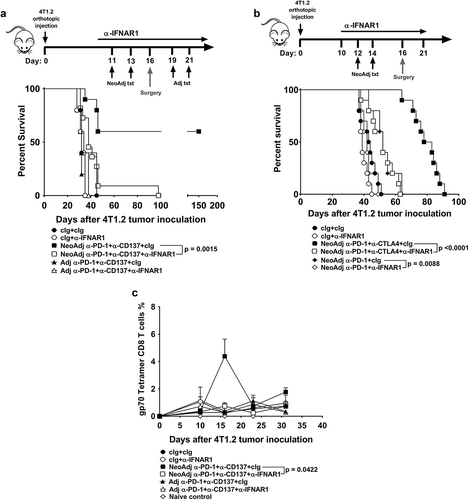
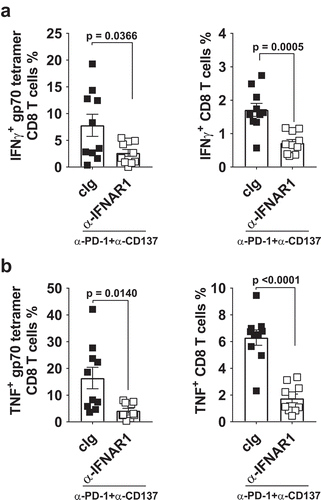
Although there was a trend for a decrease in the proportion and numbers of gp70 tetramer-specific CD8+ T cells in the primary tumors of mice treated with neoadjuvant immunotherapy and anti-IFNAR1 compared to cIg, this was not significantly different (Supp. Fig. 4A, B). Nevertheless, the proportion of activated gp70 tetramer-specific CD8+ T cells or total CD8+ T cells as defined by CD69 expression did not differ between these two groups (Supp. Fig. 4C, D). However, the decrease of blood gp70 tetramer-specific CD8+ T cells in neoadjuvant immunotherapy-treated mice that received anti-IFNAR1 translated into a reduced proportion of tumor-specific or total CD8+ T cells that produced IFNγ or TNF in the lungs, which may explain the loss of anti-tumor efficacy ().
Figure 3. Decreased proportion of IFNγ or TNF producing tumor-specific T cells in tumor-bearing mice treated with neoadjuvant immunotherapy and anti-IFNAR1. In a similar experimental setup as , groups of neoadjuvant anti-PD-1 and anti-CD137 treated mice (n = 5/grp) that were additionally treated with cIg or anti-IFNAR1 mAb were sacrificed on day 16 and their lungs collected and single cell suspensions generated for flow cytometry. Gating on live CD45.2+ cells of lymphocyte morphology, the proportions of gp70 tetramer+ CD8+ TCRβ+ T cells or total CD8+ TCRβ+ T cells in lungs that were IFNγ+ (A) or TNF+ (B) are shown. Data presented as mean ± SEM. Significant differences determined by unpaired Welch’s t-test with exact p value shown. Experiment pooled from 2 independent experiments.
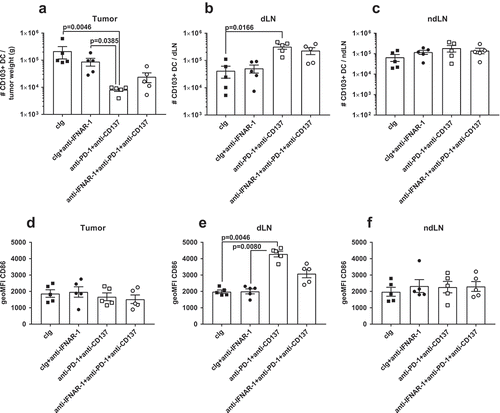
We next assessed for the presence of Batf3 lineage-derived CD103+ DCs in the primary tumor, draining lymph node (DLN) and non-draining lymph node (NDLN) of cIg- or neoadjuvant-treated mice that additionally received anti-IFNAR1 (). Using the gating strategy as previously describedCitation16, we observed a decrease in the number of CD103+ DCs in the tumor two days following neoadjuvant therapy and their concomitant increase in the DLN, but not the NDLN (). Furthermore, we also observed these CD103+ DCs in the DLN significantly up-regulated the co-stimulatory ligand CD86 and its expression was lower in neoadjuvant-treated mice that also received anti-IFNAR-1 (). In contrast, we did not observe any significant changes in CD86 expression on CD103+ DCs isolated from tumor or NDLN at this time point ().
Figure 4. CD103+ DCs increased in tumor draining lymph nodes and expressed higher levels of CD86 following neoadjuvant immunotherapy. Groups of BALB/c WT mice (n = 5/grp) were injected with 5 × 104 4T1.2 mammary carcinoma cells in the mammary fat pad before treatment with either anti-PD-1+ anti-CD137 or cIg (100 μg/mouse of each mAb, i.p.) or cIg (200 μg/mouse, i.p.) on day 10 and 12. Some groups of mice were additionally treated with anti-IFNAR1 (500 μg/mouse) on day 9. Tumors, draining lymph nodes (dLN) and non-draining lymph nodes (ndLN) were harvested two days following the final antibody treatment (on day 14) and single cell suspensions generated for flow cytometry. Gating on live CD45.2+ cells, MHC-II+CD11c+CD64lo/-Ly6C− cells, the absolute numbers of CD103+CD11b− DCs in the (A) tumor, (B) dLN, and (C) ndLN are shown. The geometric mean fluorescence intensity (MFI) of CD86 on CD103+ DCs in the (D) tumor, (E) dLN, and (F) ndLN are shown. Data presented as mean ± SEM. Significant differences determined by Kruskal-Wallis test with exact p values shown. Experiment was performed once.
Patients treated with adjuvant or neoadjuvant ipilimumab plus nivolumab in OpACIN trial that relapsed displayed low expression of Batf3+ DC associated gene signature
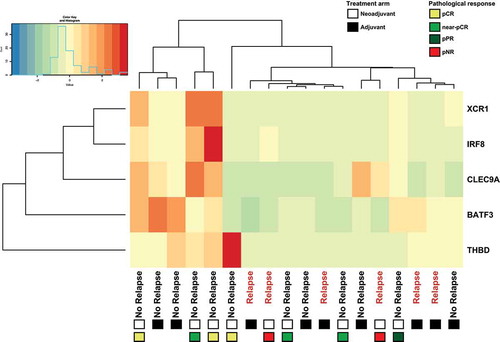
In our own randomized phase 1b clinical trial (OpACIN; NCT02437279)Citation6, we compared for the first time neoadjuvant versus adjuvant anti-CTLA4 (ipilimumab) plus anti-PD-1 (nivolumab) in patients with stage III melanoma with palpable lymph node metastases. In this trial, 20 patients were randomized to either 4 courses of adjuvant ipilimumab+nivolumab, or a split 2 courses neoadjuvant and 2 courses adjuvant. At median follow up of 25.6 months, the relapse free survival was 80% (8/10) and 60% (6/10) in the neoadjuvant and adjuvant arm respectivelyCitation6. The overall survival was 90% (9/10) compared to 70% (7/10) for neoadjuvant- and adjuvant-treated patients respectivelyCitation6. Furthermore neoadjuvant therapy expanded more tumor-resident T cell clones compared to adjuvant therapy. To determine if the expression of Batf3+ DC-associated genes (Batf3, Clec9a, Irf8, Thbd, Xcr1) as identified by Spranger et al.Citation13 correlated with responsiveness to therapy, RNA was extracted from pre-treatment tumor biopsies (). Interestingly, all patients who relapsed had a low expression of Batf3+ DC associated genes regardless of their treatment with adjuvant or neoadjuvant ipilimumab+nivolumab, further supporting our preclinical findings. Furthermore, we also found that all Batf3+ DC-associated genes with the exception of Thbd correlated significantly with a number of T-cell marker genes (Supp. Fig. 5).
Figure 5. Patients treated with adjuvant or neoadjuvant ipilimumab plus nivolumab in OpACIN trial that relapsed displayed low expression of Batf3+ DC-associated genes. Hierarchical clustering of Batf3+ DC-associated genes including Batf3 and the 4 genes (IRF8, THBD, CLEC9A, XRC1) that are included in the Batf3+ DC score as defined byCitation13. Gene-level expression values were computed as transcripts per million (tpm) and were normalized to z-scores before clustering. Positive values (red) indicate higher expression and negative values (green/blue) indicate lower expression. Every column represents one patient of which clinical outcome is depicted, the squares below the clinical outcome indicate the treatment arm: adjuvant (black) or neoadjuvant (white) and types of pathological response for neoadjuvant-treated patients. pCR (yellow); pathological complete response, near pCR (green), pPR (blue); pathological partial response, pNR (red); pathological non-response.
Discussion
In this study, we demonstrated that loss of Batf3+ lineage derived DCs and host non-responsiveness to type I IFN greatly reduced the efficacy of neoadjuvant anti-PD-1+anti-CD137 combination immunotherapy in an orthotopic 4T1.2 mouse model of spontaneously metastatic mammary cancer. This correlated with reduced levels of tumor-specific CD8+ T cells in the blood and primary tumors of neoadjuvant immunotherapy-treated Batf3-deficient mice or WT mice that received anti-IFNAR1 mAbs. Furthermore, the proportions of IFNγ/TNF-producing tumor-specific CD8+ T cells were reduced in neoadjuvant-treated tumor-bearing mice that could not respond to type I IFN. In our Phase Ib clinical trial, low expression of Batf3+ DC-associated genes appear to correlate with relapse regardless of whether patients received adjuvant or neoadjuvant immunotherapy.
We previously demonstrated an increase in tumor-specific CD8+ T cells producing IFNγ in the periphery and tumors were critical for the efficacy of neoadjuvant immunotherapyCitation5. However, the requirement for innate immune cells and pathways were not investigated. Recent studies suggests that innate immune sensing of tumors occurs through activation of the STING pathway, which leads to type I interferon (IFN) production, DCs activation, cross-presentation of tumor-associated antigens to CD8+ T cells, and T cell recruitment into the tumor microenvironmentCitation9. In our study, we demonstrated that the efficacy of neoadjuvant immunotherapy was abolished in Batf3−/− mice, which are deficient for cross-presenting cDCsCitation20 or in WT mice that were non-responsive to type I IFN. Our findings were consistent with a study from Sánchez-Paulete et al. where the curative potential of anti-PD-1+anti-CD137 therapy against established subcutaneous tumors was lost in tumor-bearing Batf3−/- mice. Specifically, they demonstrated that T cells primed by Batf3+ DC were a major population targeted by the immunotherapy Citation14. In another study of adoptive T cell therapy, tumor-residing Batf3+ DCs were demonstrated to be required for the migration of effector T cells into tumorsCitation13. Similarly, in our study we observed a complete loss of protection in neoadjuvant immunotherapy-treated Batf3−/- mice or neoadjuvant immunotherapy-treated WT mice that were unable to respond to type I IFN. In the Batf3−/- mice, this correlated with a significant decrease in the number of tumor-specific gp70-T cells and total CD8+ T cells in the primary tumor at the time of tumor resection, but not in the draining or non-draining lymph node. We also observed a trend towards a reduced expansion of gp70-T cells in the blood of neoadjuvant-treated Batf3−/- mice compared to neoadjuvant-treated WT mice. While other antigen presenting cells in Batf3−/- mice may have the capacity to prime tumor-specific CD8+ T cells, our data, similar to Sánchez-Paulete et al. and Spranger et al. supports the critical role that Batf3+ DCs play in the induction of anti-tumor immunity within tumors and immune cell recruitment into the tumor microenvironmentCitation13,Citation14. We also showed that neoadjuvant immunotherapy attenuated the migratory function of Batf3-lineage derived CD103+ DCs where they increased in the tumor draining lymph node and type I IFN was required for their up-regulation of the co-stimulatory molecule CD86, which likely is critical for the priming and/or expansion of tumor-specific T cells. As such, the expansion of peripheral blood gp70-T cells in neoadjuvant treated-mice that could not respond to type I IFN consequently had a reduced proportion of IFNγ/TNF producing T cells in their lungs resulting in a complete loss of long-term survival. Future experiments could utilize XCR1-DTR mice in which Batf3+ DCs can be conditionally depleted to confirm their requirement for the efficacy of neoadjuvant immunotherapyCitation21.
Although neoadjuvant anti-PD-1+anti-CD137 immunotherapy and surgery induced a significant proportion of long-term survivors, a proportion of treated mice relapse and die. One strategy to further improve survival is to administer radiotherapy followed by neoadjuvant anti-PD-1+anti-CD137. An elegant study by Rodrigueze-Ruiz et al., demonstrated that radiotherapy of 4T1 subcutaneous tumors followed by anti-PD-1+anti-CD137 was most effective in suppressing tumor growth and reducing spontaneous lung metastases compared to each treatment regime aloneCitation15. This was due to the ability of radiotherapy to upregulate CD137 and/or PD-1 expression on tumor-infiltrating T cells. In our study, we observed that a proportion of gp70 tetramer-specific T cells expressed CD137 and/or PD-1 which may be further upregulated following irradiation of the primary tumor.
Clinically, there is now a strong rationale to evaluate the use of neoadjuvant immunotherapies in patients with earlier stages of cancer that are at high risk of relapse given our pre-clinical data and recent clinical data in melanomaCitation6,Citation7 and early stage NSCLC where unexpectedly high response rate after neoadjuvant nivolumab monotherapy was observedCitation8. In particular, a key goal of neoadjuvant immunotherapy will be to identify immunological correlates of outcomes either from the primary tumor or in PBMCs in patients treated with neoadjuvant immunotherapy. Although our Phase 1b trial met its co-primary endpoints for safety/feasibility and tumor specific T cell expansion, almost all patients developed grade 3–4 immune-related adverse events (irAEs)(18/20) and therefore most patients received a maximum of 2 cycles of therapy similar to our pre-clinical scheduling. Therefore a subsequent phase 2 trial (OpACIN-neo; NTC02977052) assessed whether the efficacy of neoadjuvant checkpoint inhibition in combination could be preserved while reducing its toxicity. In this trial, patients were randomized between 3 different 6 week schedules of neoadjuvant ipilimumab+nivolumab. Preliminary results indicated that the schedule consisting of 2 cycles of ipilimumab (1 mg/kg) and nivolumab (3 mg/kg) was better tolerated with only 20% of patients developing one or more grade 3–4 irAEs while efficacy was preserved (pathological response rate: 77%)Citation22. In our current study, due to the small number of patients, our data is only suggestive that the Batf3+ DC-associated genes may enrich for responders. Pre-treatment biopsies obtained from the OPACIN-neo study which contain a larger cohort of patients (n = 90) may allow us to validate the requirement for an intratumoral Batf3+ DC-associated gene signature for response upon neoadjuvant immunotherapy and this may serve as an additional biomarker to the recently described IFNγ gene signatureCitation22 to identify poor responders.
Materials and methods
Mouse strains
BALB/c WT mice were bred or purchased from the Walter and Eliza Hall Institute for Medical Research. BALB/c Batf3-deficient mice were bred and maintained at QIMR Berghofer Medical Research Institute. Female mice greater than eight weeks old were used in all experiments, which were approved by the QIMR Berghofer Medical Research Institute Animal Committee.
Cell culture
BALB/c-derived 4T1.2 mammary carcinoma cells were cultured in RPMI 1640 containing 10% FCS, penicillin/streptomycin, and L-glutamine as previously describedCitation5. This cell line was routinely tested for mycoplasma but cell line authentication was not routinely performed.
Antibodies
Purified anti-mouse PD-1 mAb (RMP1-14), anti-mouse anti-CD137 (3H3), anti-mouse CTLA4 (UC10-4F10), control IgG (2A3) were obtained from BioXCell (West Lebanon) or Leinco Technologies (St. Louis). In all experiments 100 μg/mouse each of anti-PD-1 or anti-CD137 or 200 μg/mouse of cIg were administered i.p as indicated. In some experiments, 250 or 500 μg/mouse of anti-IFNAR1 (MAR1-5A3)(Leinco) was administered i.p at the indicated time points.
4T1.2 orthotopic model and treatment
4T1.2 tumor cells were inoculated into the mammary fat-pad of female BALB/c WT mice and resected as previously describedCitation5.
Flow cytometry analysis
Tumors, blood, lungs and lymph nodes were harvested from mice and processed for flow cytometry analysis as previously describedCitation5. For dendritic cell analysis, resected tumors were dissociated using a Gentle MACS Dissociator (Miltenyi Biotec). For surface staining, TILs or immune cell suspensions were stained with anti-CD45.2 FITC (104)(eBioscience), or anti-CD45.2 A780 (104)(eBioscience) or anti-CD45.2 Brilliant Violet 570 (clone 30-F11)(Biolegend), anti-TCRβ PerCP-Cy5.5 (H57-597)(eBioscience), anti-CD8α Brilliant Violet 711 (53–6.7)(Biolegend), anti-CD4 Brilliant Violet 605 (RM4-5)(Biolegend), anti-CD69 PE-Cy7 (H1.2F3)(eBioscience), H-2Ld tetramer to peptide SPSYVYHQF APC (MuLV env gp70 423–431)(NIH Tetramer Core facility) or anti-MHC-II Alexa780 (M5/114.15.2)(Biolegend), anti-CD11c APC (clone N418)(eBioscience), anti-CD64 PerCPCy5.5 (clone X54-5/7.1)(eBioscience), anti-Ly6C PECy7 (clone RB6-6C5)(eBioscience), anti-CD11b Brilliant Violet 650 (clone M1/70)(Biolegend), anti-CD103 eFlour 450 (clone 2E7)(Biolegend), and anti-CD86 PE (clone GL1)(eBioscience) and live/dead dye Zombie Aqua (Biolegend) or 7AAD (Biolegend) in the presence of anti-CD16/32 (2.4G2) to block FcR. To measure intracellular cytokine staining, single cell suspensions were incubated for 4 hrs in complete RPMI with monensin and brefeldin A (eBioscience). Samples were then surface stained before being fixed and permeabilized (BD CytoFix/CytoPerm Kit), and stained with anti-IFNγ AF488 (XMG1.2)(Biolegend) and anti-TNF PE (MP6-XT22)(Biolegend) or fluorescence minus one control for IFNγ staining and isotype control for TNF staining. To determine absolute counts in samples, liquid counting beads (BD Biosciences) were added directly before samples were run on the flow cytometer. All data were collected on a Fortessa 4 (Becton Dickinson) flow cytometer and analyzed with FlowJo v10 software (Tree Star, Inc.).
OpACIN trial
In the phase 1b OpACIN trial (NCT02437279), 20 patients with palpable stage III melanoma were randomized 1:1 to receive ipilimumab 3 mg/kg and nivolumab 1 mg/kg, either 4 courses after surgery (adjuvant arm), or 2 courses prior to surgery and 2 courses post-surgery (neoadjuvant arm). Co-primary endpoints were safety/feasibility and tumor specific T-cell expansion. All patients underwent a pre-treatment tumor biopsy out of which RNA was extracted for RNA-Seq analysis.
Study approval
The study was approved by the medical ethics committee of the Netherlands Cancer Institute. All subjects provided informed consent prior to their participation in the study.
RNA isolation and gene expression analysis
RNA was extracted from fresh frozen tumor material using the Allprep DNA/RNA kit (QIAGEN) for frozen material following manufacture’s protocol in a QIAcube (QIAGEN). Strand-specific libraries were generated using the TruSeq Stranded mRNA sample preparation kit according to the manufacturer’s instructions. Briefly, polyadenylated RNA from intact total RNA was purified using oligo-dT beads. Following purification the RNA was fragmented, random primed and reverse transcribed using SuperScript II Reverse Transcriptase with the addition of Actinomycin D. Second strand synthesis was performed using Polymerase I and RNaseH with replacement of dTTP for dUTP. The generated cDNA fragments were 3ʹ end adenylated and ligated to Illumina Paired-end sequencing adapters and subsequently amplified by 12 cycles of PCR. The libraries were analyzed on a 2100 Bioanalyzer using a 7500 chip (Agilent, Santa Clara, CA), diluted and pooled equimolar into a multiplex sequencing pool and stored at −20 °C. The libraries were sequenced with 65 bp single reads on a HiSeq2500 using V4 chemistry (Illumina Inc., San Diego). Raw reads were aligned to GRCh38 using STAR RNA-seq aligner after which gene expression levels were quantified by Salmon using default parameters for both applications.
Statistics
Statistical analysis was performed using GraphPad Prism software (San Diego, CA). Comparison of different groups was carried out using unpaired Welch’s t-test or two-way ANOVA with Dunnett’s post-test analysis or Kruskal-Wallis test as indicated. In some experiments, to determine what immunological effects were induced after respective neoadjuvant or adjuvant therapy, significance had to be performed on different days. Kaplan-Meier analyses with log-rank sum test were used for animal survival experiments. A p value of less than 0.05 was considered significant.
Author Contributions
JL designed and performed pre-clinical experiments, analyzed data, and edited the manuscript. JO’D and SA performed pre-clinical experiments and edited the manuscript. LF analyzed patient’s RNA-sequencing data. ER analyzed clinical data and correlated this to translational data, and edited the manuscript. MS designed and performed pre-clinical experiments, and edited the manuscript. CB designed and supervised the clinical study, analyzed data and edited the manuscript. MT designed and supervised the pre-clinical study, analyzed data, and wrote the manuscript.
Conflict of interest
M.J. Smyth declares scientific research agreements with Bristol-Myers Squibb (BMS), Tizona Therapeutics, and Aduro Biotech and advisory roles for Tizona Therapeutics and Elstar Therapeutics. C. Blank declares advisory roles for BMS, MSD, GSK, Novartis, Roche, Pfizer, Lilly and received research grants from Novartis and BMS. No potential conflicts of interest were disclosed by the other authors.
Supplemental Material
Download Zip (219.7 KB)Acknowledgments
The project was funded by a National Health and Medical Research Council of Australia (NH&MRC) Project Grant (1099546). M.J. Smyth is supported by a Senior Principal Research Fellowship (1078671) and Program Grant (1132519). The authors thank Liam Town and Kate Elder for mouse genotyping and maintenance during this study.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi:10.1126/science.aaa8172.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375:1845–1855. doi:10.1056/NEJMoa1611299.
- Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, Dalle S, Schenker M, Chiarion-Sileni V, Marquez-Rodas I, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377:1824–1835. doi:10.1056/NEJMoa1709030.
- Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A, Carlino MS, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med. 2018;378:1789-1801. doi:10.1056/NEJMoa1802357.
- Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, Young A, O'Donnell JS, Allen S, Smyth MJ, et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6:1382–1399. doi:10.1158/2159-8290.CD-16-0577.
- Blank CU, Rozeman EA, Fanchi LF, Sikorska K, van de Wiel B, Kvistborg P, Krijgsman O, van den Braber M, Philips D, Broeks A, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med. 2018;24:1655–1661. doi:10.1038/s41591-018-0198-0.
- Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, Cormier JN, Lewis C, Hwu WJ, Hanna E, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24:1649–1654.
- Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Zahurak M, Yang SC, Jones DR, Broderick S, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–1986. doi:10.1056/NEJMoa1716078.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi:10.1016/j.immuni.2013.07.012.
- Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, Chen X, Li XD, Deng L, Chen ZJ, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein alpha signaling. Immunity. 2017;47:363–373.e5. doi:10.1016/j.immuni.2017.07.016.
- Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27:96–108. doi:10.1038/cr.2016.149.
- Vanpouille-Box C, Galluzzi L. CD103(+) cells at the forefront of anticancer immunity. Oncoimmunology. 2017;6:e1356154. doi:10.1080/2162402X.2017.1356154.
- Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31:711–23 e4. doi:10.1016/j.ccell.2017.04.003.
- Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 2016;6:71–79. doi:10.1158/2159-8290.CD-15-0510.
- Rodriguez-Ruiz ME, Rodriguez I, Garasa S, Barbes B, Solorzano JL, Perez-Gracia JL, Labiano S, Sanmamed MF, Azpilikueta A, Bolanos E, et al. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mAbs and are dependent on CD8 T cells and crosspriming. Cancer Res. 2016;76:5994–6005. doi:10.1158/0008-5472.CAN-16-0549.
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44:924–938. doi:10.1016/j.immuni.2016.03.012.
- Scrimieri F, Askew D, Corn DJ, Eid S, Bobanga ID, Bjelac JA, Tsao ML, Allen F, Othman YS, Wang SC, et al. Murine leukemia virus envelope gp70 is a shared biomarker for the high-sensitivity quantification of murine tumor burden. Oncoimmunology. 2013;2:e26889. doi:10.4161/onci.26889.
- Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci U S A. 1996;93:9730–9735.
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi:10.1084/jem.20101159.
- Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, Kretzer NM, Durai V, Murphy KM. Transcriptional control of dendritic cell development. Annu Rev Immunol. 2016;34:93–119. doi:10.1146/annurev-immunol-032713-120204.
- Yamazaki C, Sugiyama M, Ohta T, Hemmi H, Hamada E, Sasaki I, Fukuda Y, Yano T, Nobuoka M, Hirashima T, et al. Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J Immunol. 2013;190:6071–6082. doi:10.4049/jimmunol.1202798.
- Rozeman EA, Menzies AM, van de Wiel BA, Adhikari C, Sikorska K, Krijgsman O, Eriksson H, Bierman C, Grijpink-Ongering LG, Gonzalez M, Broeks et al. OpACIN-neo: A Multicenter Phase 2 Study to identify the Optimal neo-Adjuvant Combination scheme of Ipilimumab (IPI) and Nivolumab (NIVO). Ann Oncol Suppl 2018:abstract LBA42.
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940. doi:10.1172/JCI91190.