
ABSTRACT
Invariant natural killer T (iNKT) cells are a small population of T lymphocytes that expresses an invariant T cell receptor with a unique specificity for glycolipid antigens. Their activation using the glycolipid α-galactosylceramide (α-GalCer) triggers innate and adaptive immune responses. The use of α-GalCer in preclinical models as a single antitumor treatment showed moderate effect, but its efficacy in cancer patients was less effective. In addition, this glycolipid induces long-term iNKT-cell anergy precluding the possibility of retreatment. Recently, the first murine iNKT-cell agonistic antibody, NKT14m, has been developed. Here, we analyzed, for the first time, the antitumor efficacy of NKT14m in a B-cell lymphoma model. In a therapeutic setting, a single dose of NKT14m had a moderate antitumor efficacy that was associated with an increase of IFN-γ producing iNKT cells even after a second dose of the NKT14m antibody. Importantly, the combination of a single dose of NKT14m with cyclophosphamide had a potent antitumor efficacy and long-lasting immunity in vivo. Our findings provide the first evidence of the in vivo antitumor efficacy of NKT14m antibody, showing that, either alone or in combination with chemotherapy, induces an effective antitumor response. These results open new opportunities for iNKT-cell mediated immunotherapy to treat B-cell lymphoma.
Introduction
Invariant natural killer T (iNKT) cells represent a small T-cell subset that has an invariant T-cell receptor (iTCR), which recognizes glycolipid antigens presented by the non-polymorphic MHC class I-like molecule CD1d.Citation1–Citation3 Preclinical studies have shown that iNKT cells can activate both innate and adaptive immune responses.Citation1,Citation2,Citation4,Citation5 In fact, activated iNKT cells can increase the activation and function of natural killer (NK) cells,Citation6,Citation7 CD4+ and CD8+ T cells,Citation6,Citation8–Citation10 dendritic cells (DC)Citation9,Citation10 and B cells,Citation11 coordinating a global immune response and promoting the killing of tumor cells. In addition, iNKT-cell activation also inhibits tumor-induced myeloid derived suppressor cells (MDSCs)Citation8 and tumor angiogenesisCitation12. Increased numbers of circulating and intra-tumor iNKT cells have been associated with an improved prognosis in different cancers and hematological malignancies,Citation13,Citation14 supporting the important role of iNKT cells in cancer immunosurveillance and its potential as a promising immunotherapeutic target.
The activation of these cells with the glycolipid α-galactosylceramide (α-GalCer) increases IFN-γ production and cytokine secretion (i.e., IL-12, IL-17, IL-21), which contributes to the enhancement of a T-cell mediated antitumor effect.Citation5,Citation15,Citation16 Different approaches using α-GalCer have been tested in several tumor models with success,Citation15,Citation17–Citation22 but their translation to the clinical setting proved to be less effective.Citation23–Citation29 Moreover, administration of α-GalCer induces long-term iNKT-cell anergy, causing unresponsiveness to sequential stimulation with this glycolipid,Citation18,Citation21,Citation30,Citation31 which can partially explain the lack of clinical effect in patients with cancer. To enhance antitumor responses mediated by iNKT-cell activation, DCs expressing CD1d were loaded ex vivo with this α-GalCer. This combination showed moderate antitumor efficacy in mice models, although better than α-GalCer alone.Citation15,Citation20,Citation32 In addition, its translation to the clinic demonstrated a sustained expansion of iNKT cells in patients with advanced cancer, as well as an increase in serum levels of IL-12 and IFN-γ, although no clinical responses were observed.Citation23–Citation26,Citation28,Citation29 Recently, the first iNKT-cell agonistic monoclonal antibody (mAb), the NKT14m that can activate murine iNKT cells, has been described.Citation30 This new murine IgG2a antibody activates iNKT cells in vivo by direct binding to the iTCR in fully immune-competent miceCitation30 and represents a surrogate antibody to the human specific iNKT cell activating antibody NKTT320.Citation33 Direct acting antibodies would represent an alternative to α-GalCer in cancer treatment with significant therapeutic potential. Injection of NKT14m into naive Balb/c mice triggers the IFN-γ production by iNKT cells comparable to α-GalCer but, in contrast to α-GalCer, the NKT14m did not induced iNKT-cell long-term anergy, allowing the readministration of this antibody.Citation30 While the activation of murine iNKT cells by agonistic NKT14m antibody has been described, the in vivo antitumor effect of NKT14m has not been previously reported. Here, we describe for the first time the antitumor efficacy of NKT14m-mediated direct iNKT cell activation against B-cell lymphoma, as single agent or in combination with chemotherapy.
Results
NKT14m has antitumor activity against B-cell lymphoma
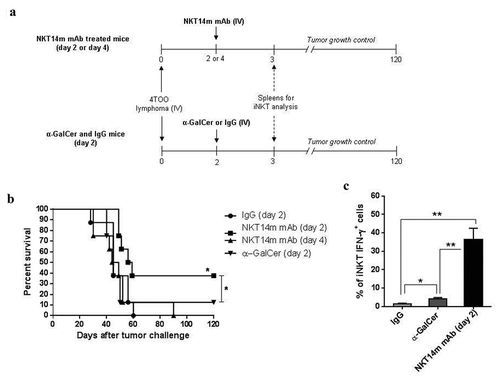
We first studied the in vivo antitumor efficacy of NKT14m antibody in a therapeutic setting (). The treatment with a single dose of NKT14m two days after injection of 4 × 105 4TOO tumor cells induced a moderate antitumor response in comparison with tumor-bearing mice injected with IgG (37% vs. 0% survival, respectively; p = 0.03) or mice treated with a single dose of α-GalCer (37% vs. 10% survival, respectively; p = 0.04) (). However, a delayed treatment (i.e., 4 days after tumor challenge) resulted in complete loss of efficacy. Importantly, we could detect a significant increase of IFN-γ-producing iNKT cells 24 hours after NKT14m treatment in comparison with mice treated with α-GalCer (36.28 ± 6.02 % vs. 4.15 ± 0.73%, respectively; p = 0.007) (). However, the treatment with NKT14m did not change de percentage of iNKT cells in spleen (1.54 ± 0.6% vs 1.35 ± 0.2%, p = 0.7) (Supplemental Figure S1). In addition, no significant increase in total numbers and IFN-γ production was observed for other immune cells such as NK, CD4+ and CD8+ T cells (Supplemental Figure S1). Collectively, this data shows that the NKT14m efficiently activates iNKT cells in vivo, inducing an improved antitumor response able to eliminate tumor cells compared to the one induced by the prototypic iNKT-cell agonist α-GalCer.
Figure 1. The agonistic NKT14m mAb induces an effective in vivo antitumor response against B-cell lymphoma.
(a) Diagram of treatment schedule with NKT14m mAb and spleens harvest for iNKT-cell analysis. Balb/c mice (n = 8/group) were injected with 4 × 105 4TOO tumor cells (iv) on day 0 and were treated 2 or 4 days later with a single dose of NKT14m mAb (100μg/mice, iv) or IgG (100μg/mice, iv). A group of mice received α-GalCer (2μg/mice, iv) two days after tumor challenge. Mice were monitored daily for survival. Spleens of control and treated mice were harvested 3 days after tumor challenge (24h after each treatment) to detect IFN-γ producing iNKT cells. (b) Survival analysis of mice treated as described in (a). Data represents survival from one of three independent experiments. *p < 0.05. (c) Splenocytes (n = 4) were analyzed by flow cytometry for IFN-γ producing iNKT cells 24 hours (day 3) after NKT14m mAb and α-GalCer injection. Data are represented as mean ± SEM. **p < 0.01.
In addition, mice treated with NKT14m antibody that eliminated the tumor were sacrificed at the end of the experiment and a pathology analysis was done. No abnormalities in spleen, liver, bone marrow, lymph nodes, and lungs were observed (data not shown). In contrast, mice that were not able to eliminate the tumor (either control or treated) showed lymphoma dissemination in the spleen, bone marrow, lymph nodes and liver and, in some cases, the tumor could also be detected in the ovary and intestine (data not shown). Tumor progression also induced mobility difficulties in some mice.
Retreatment with NKT14m increases the antitumor efficacy
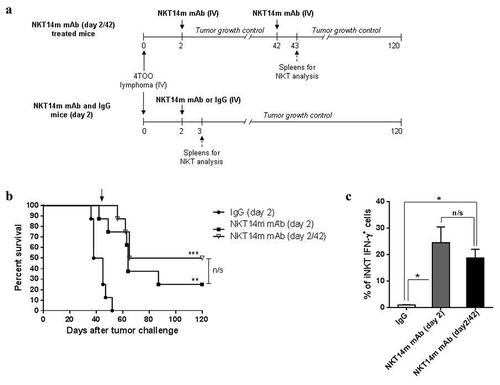
We next evaluated the possibility to administrate a second dose of NKT14m to improve the observed antitumor effect. Mice treated with a single dose of NKT14m two days after tumor challenge (4x105 4TOO tumor cells/mice) received a second dose of antibody 6 weeks after tumor injection (). Retreatment with NKT14m showed an improved antitumor effect (50% survival vs. 0% in control mice, p < 0.0001), which translate into better survival compared with those injected with a single dose of antibody (50% vs. 25%, respectively; p = 0.1) ( and Supplemental Figure S2). In addition, an increase of IFN-γ-producing iNKT cells was detected after a second dose of NKT14m (19 ± 0.95% vs. IgG control mice, p = 0.05) with no significant differences in the number of IFN-γ-producing iNKT cells after the first injection of the antibody (19 ± 0.95% after retreatment vs. 24.5 ± 2.2% after a single dose at day 2, p = 0.5) (), demonstrating that iNKT cells remained functional after antibody readministration.
Figure 2. Retreatment with NKT14m mAb enhances the antitumor efficacy.
(a) Timeline of retreatment with NKT14m mAb and spleens harvest for iNKT-cell analysis. Balb/c mice (n = 8/group) were injected with 4 × 105 4TOO tumor cells (iv) and, 2 days later, treated with a single dose of NKT14m mAb (100μg/mice, iv) or control IgG (100μg/mice, iv). One group of treated mice was injected again with a single dose of NKT14m mAb (100μg/mice, iv) 42 days after tumor inoculation (NKT14m mAb day2/42). Mice were daily followed for survival. Spleens of control and treated mice were harvested 24 hours after each treatment (day 3 for NKT14m mAb and IgG groups, and day 43 for mice receiving antibody retreatment) for iNKT-cell analysis. (b) Survival analysis of mice treated as described in (a). Arrow indicates the day of retreatment. Data represents survival from one of three independent experiments. **p < 0.01; ***p < 0.001; n/s: no significance. (c) Splenocytes (n = 4) from mice treated with NKT14m or IgG at day 2, and mice that received a second dose of the antibody 42 days after tumor challenge, were analyzed by flow cytometry for IFN-γ producing iNKT cells, 24 hours after treatment in all cases (day 3 and day 43, respectively). Data are represented as mean ± SEM. *p < 0.05; n/s: no significance.
The combination of NKT14m and cyclophosphamide induces a potent antitumor immune response in a B-cell lymphoma mice model
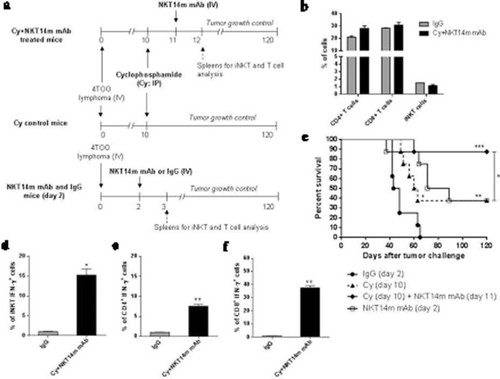
After showing that NKT14m induced an in vivo antitumor effect in a B-cell lymphoma mice model, we sought to improve the NKT14m efficacy using clinically relevant strategies. To this end, we evaluated the therapeutic efficacy of a combination of cyclophosphamide (Cy) with NKT14m. Mice were treated with Cy 10 days after tumor challenge followed by a single dose of NKT14m 24 hours after Cy administration (). In this setting, we did not observe any reduction in the proportion and total numbers of iNKT, CD4+ and CD8+ T cells in spleen of treated mice 24 hours after antibody administration (day 12), suggesting that Cy does not have any detrimental effect on these cell subsets ( and supplemental figure S3), as other studies have shown.Citation34 In addition, toxicity was not observed in mice receiving cyclophosphamide (i.e., no changes in body weight, motility, behaviour and organ morphology). NKT14m combined with Cy significantly enhanced the antitumor effect observed with Cy alone (87% vs. 37% survival, respectively; p = 0.03) (), and promoted a better tumor control than NKT14m alone. IFN-γ-producing iNKT cells were increased in mice treated with Cy and NKT14m, in both, spleen (15.3 ± 1.2% vs. 1,96 ± 0.4% control mice, p = 0.006; ) and liver (20,3 ± 1.7% vs. 1,05 ± 0.6% in control mice, p = 0,007) (Supplemental figure S4), where we also observed an iNKT-cell expansion after treatment (38.6 ± 2.0% vs. 18.9 ± 1,7% in control group, p = 0.006) (Supplemental Figure S4). In addition, a significant increase of IFN-γ-producing CD4+ and CD8+ T cells were found in mice treated with the combination of Cy and NKT14m (CD4+ T cells: 7.5 ± 0.05% vs. IgG treated mice, p = 0.006; CD8+ T cells: 37.5 ± 1.7% vs. IgG treated mice, p = 0.002) ().
Figure 3. Treatment with cyclophosphamide following NKT14m mAb administration induces a potent antitumor response against B-cell lymphoma.
(a) Timeline of mice treatment with cyclophosphamide (Cy) and NKT14m mAb, and spleens harvest for iNKT and T-cell analysis. Balb/c mice (n = 8/group) were treated with Cy (70mg/kg; ip) 10 days after tumor challenge. One group of Cy treated mice received a single dose of NKT14m mAb (100μg/mice, iv) 24 hours after Cy injection (Cy+ NKT14m mAb). Another group of mice received a single dose of NKT14m mAb (100μg/mice, iv) 2 days after tumor challenge. Mice were monitored daily for survival. Spleens of control IgG and Cy+ NKT14m mAb groups were harvested 24 hours after each treatment (day 3 and day 12, respectively) for iNKT and T-cell analysis. (b) Percent of iNKT, CD4+ and CD8+ T cells were analyzed from spleens (n = 3) of mice treated with Cy+ NKT14m mAb and control IgG at day 12 after tumor challenge. n/s: no significance. (c) Survival analysis of treated mice as described in (a). Data represents survival from one of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. Splenocytes (n = 3) from Cy+ NKT14m treated mice were analyzed by flow cytometry for IFN-γ producing (d) iNKT, (e) CD4+ and (f) CD8+ T cells, 24 hours (day 12) after Cy+ NKT14m treatment. Data are represented as mean ± SEM. *p < 0.05; **p < 0.01.
A long-lasting immunity is conferred by NKT14m in combination with cyclophosphamide
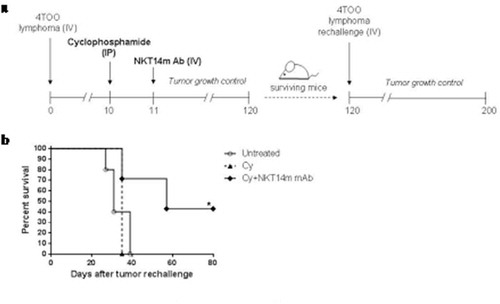
To test whether NKT14m with Cy could induce a long-term immunity against B-cell lymphoma, mice that survived after the first tumor injection were challenged again with 4 × 105 4TOO tumor cells (). 43% of immunized mice remained tumor-free after tumor rechallenge (p = 0.01), whereas Cy treated were not able to reject the tumor (), suggesting the establishment of a long-lasting immunity in surviving mice.
Figure 4. Long-term immunity is induced by the combination of cyclophosphamide and NKT14m mAb.
(a) Mice immunized with Cy alone or Cy+ NKT14m treatment that survived the first 4TOO tumor injection (n = 3 and n = 7, respectively) were rechallenged with a second dose of tumor cells (4x105 cells/mouse, iv) at day 120. Mice were followed daily for survival. (b) Survival analysis of mice rechallenged with 4TOO tumor cells as described in (a). A group of untreated age-matched mice received an injection of 4 × 105 4TOO tumor cells as control. *p < 0.05.
Discussion
iNKT cells can trigger a coordinated innate and adaptive immune response contributing to tumor immune-surveillance,Citation1,Citation2,Citation4,Citation5 making its activation an attractive approach for cancer immunotherapy. iNKT cells can eliminate tumor cells directly by recognition of CD1d-tumor lipid antigen complex in the case of CD1d+ tumors,Citation2,Citation35 due to its degranulation capacity and cytotoxic activity. In addition to direct lysis, iNKT cells could activate both innate and adaptive immune cells through rapid cytokine secretion upon activation (i.e. IFN-γ, IL-17, IL-4, TNF-α), after the recognition of self or foreign lipid antigens presented by CD1d+ cells (i.e., DCs, B cells and macrophages).Citation2,Citation4 One of the most important cytokine produced by iNKT cells is IFN-γ, which helps to stimulate DCs that can further activate other antitumor-promoting effector cells, such as NK cells and cytotoxic CD8+ T lymphocytes.Citation4,Citation36 IFN-γ secreted by iNKT cells also has direct implications in CD4 and CD8 T-cell activation and differentiation. Moreover, iNKT cells could promote M1 macrophage polarization and regulate the suppressive activities of neutrophils.Citation4,Citation36
Stimulation of iNKT cells with α-GalCer administration has been shown to induce effective antitumor responses in some murine tumor models,Citation15,Citation17,Citation22 and induction of strong iNKT-cell activation and a potent cytokine secretion.Citation15,Citation21,Citation22 Despite of that, previous studies have shown that continuous administration of α-GalCer induces iNKT-cell anergy.Citation21,Citation30,Citation31 In addition, a large number of clinical studies in cancer focused on the activation of iNKT cells using α-GalCer approaches did not observe potent antitumor responses.Citation23–Citation29 α-GalCer was tested in a clinical trial with solid cancer patients and only transient iNKT cell activation was detected in a minority of patients.Citation24,Citation27 In other cases of solid tumors and hematological malignancies, a sustained expansion of iNKT cells and an increase in serum levels of IFN-γ and IL-12 were observed, although no antitumor responses were noted.Citation23,Citation25,Citation26,Citation28
A novel agonistic antibody that activates iNKT cells in a CD1d-independent manner, the NKT14m, has been developedCitation30 and represents a promising tool to activate iNKT cells in vivo. We demonstrated, for the first time, the antitumor efficacy of NKT14m antibody in the setting of cancer. An effective therapeutic response against B-cell lymphoma was obtained using a single dose of the novel antibody (), while no significant antitumor effect was observed with a single dose α-GalCer administration, as shown by previous studies.Citation19,Citation32,Citation37,Citation38 In fact, the poor antitumor efficacy of α-GalCer alone in our tumor model mostly resembles the clinical scenario, where patients treated with this glycolipid did not experiment any response although they had a moderate iNKT-cell expansion and activation with increased IFN-γ production.Citation23-Citation26 NKT14m antibody induced a potent iNKT-cell activation with a higher increase of IFN-γ production than α-GalCer (), suggesting that this antibody could be a better option to improve responses in cancer patients. In this study, we did not observe an increase in neither total numbers nor IFN-γ+ NK cells (Supplemental Figure S1), suggesting that they are not activated by iNKT-cell stimulation throughout the antibody. In addition, CD4+ and CD8+ T cells did not seem to be affected by NKT14m administration (Supplemental Figure S1), since IFN-γ+ CD4+ and CD8+ T cells were not significantly increased after treatment. However, mice treated with the NKT14m antibody 4 days after tumor injection were not able to eliminate the tumor (). This lack of tumor control was mostly due to the aggressiveness of the tumor model itself, and not to a defect in IFN-γ production by iNKT cells, since we were able to detect IFN-γ in a number of time points after administration of NKT4m antibody (i.e., day 3, 12, and 43 after tumor challenge).
Since NKT14m did not induced long-term iNKT-cell anergy,Citation30 we tried to improve its efficacy with the administration of a second dose of antibody, six weeks after tumor injection, which is the time previously shown to assure proper reactivation of iNKT cells.Citation30 In this situation, we observed again an activation of iNKT cells in vivo (i.e., increase of IFN-γ production) with an improved therapeutic efficacy ( and Supplemental Figure S2). This is in line with previous dataCitation30 and shows for the first time that readministration of NKT14m can induce iNKT cell reactivation in vivo and increases survival in a therapeutic setting of cancer. The possibility of retreatment with NKT14m makes it a promising therapeutic strategy and represents a critical advantage with important clinical implications over α-GalCer, which induces a potent iNKT-cell anergy that prevents further retreatment.Citation18,Citation21,Citation30,Citation31 In this regard, the use of NKT14m would allow to developing strategies of maintenance treatment like other antibodies, which hopefully would contribute to relapse prevention and increase survival of B-cell lymphoma patients.
In our study, the use of cyclophosphamide in the combined therapeutic approach did not reduce the percentage or total numbers of iNKT and T cells in vivo ( and Supplemental Figure S3), suggesting that the chemotherapeutic dose used in our experiments was not toxic. These observations are in line with previous studies, which have demonstrated that cyclophosphamide does not induce any significant changes in the total numbers of these different cell subsets.Citation34 Although it was reported that cyclophosphamide has immunostimulatory activitiesCitation39,Citation40 and can induce the activation of CD4+ and CD8+ T cells, including iNKT cells,Citation34 its administration alone in our B-cell lymphoma model did not have any antitumor effect (), in line with previous data.Citation34 The combination treatment with NKT14m antibody and cyclophosphamide resulted in a greater antitumor effect compared to either treatment alone (). In contrast to previous studies using α-GalCer,Citation34 iNKT-cell activation with NKT14m antibody seems to be more potent and its combination with a chemotherapeutic agent, such as cyclophosphamide, has a synergistic activity increasing IFN-γ-producing iNKT and T cells (), and establishing a long-lasting memory (), suggesting that this combination could foster a coordinated antitumor response which translates into a better outcome.
There are many immune mechanisms that could underlie the potent antitumor efficacy of the combination treatment. Cyclophosphamide induces immunogenic cell death (ICD) of tumor cells, a process that enhances the uptake and presentation of tumor antigens by DCs.Citation8,Citation34,Citation41 Simultaneous release of necrotic cells and their associated tumor antigens could increase tumor-infiltrating DCs and induce its maturation and activation,Citation34,Citation42 enhancing iNKT and T-cell priming. Previous reports also shown that cyclophosphamide could induce a reduction of FoxP3+ TregsCitation40,Citation43 but, despite this regulatory T-cell control and the ICD activity, cyclophosphamide on its own is not sufficient to induce an effective long-lasting immunity.Citation34 However, tumor cells cultured with cyclophosphamide have shown to be more sensitive to iNKT-cell killing in vitro,Citation44,Citation45 which may explain the enhanced antitumor effect seen in our model with this combination. In addition, tumor-bearing mice treated with cyclophosphamide exhibited an increase of iNKT cells in the tumor mass.Citation34 The increased tumor-infiltrating iNKT cells with an early activation of DCs and T cells, together with a non-immunosuppressive tumor microenvironment, establish a favorable scenario to enhance iNKT-cell immunostimulatory functions.
Our data demonstrate that NKT14m antibody, either alone or in combination with cyclophosphamide, stimulates iNKT cells to produce mainly IFN-γ. It is known that Th1-immunity is directly involved in stimulating antitumor responses and promoting tumor recognition and elimination.Citation46,Citation47 The major product of Th1-mediated immunity, IFN-γ, could induce the upregulation of MHC-type I and II, increasing the activation and recruitment of CD4+ T cells and cytotoxic T lymphocytes (CTLs).Citation48–Citation50 Moreover, this cytokine is directly involved in differentiation of T cells and stabilizes Th1-cell phenotype. IFN-γ could activate M1 macrophages and NK cells and increases their cytotoxic activity. This cytokine also supports the activity of IL-12, a crucial cytokine to maintain the communication between DCs and iNKT cells, since IL-12 also induces IFN-γ production by iNKT cells, stablishing a positive feedback.Citation2,Citation4 In addition, IFN-γ has antitumor properties by itself inducing tumor cell apoptosis by different mechanisms, for example upregulating the expression of caspases (i.e., caspase 1, 3 and 8) or increasing the secretion of Fas and Fas ligand.Citation51 Importantly, IFN-γ also plays an important role in tumor angiogenesis suppression.Citation12,Citation52
Although the antitumor effect of our therapeutic treatments were not tested in IFN-γ-deficient mice, it may be speculated, in light of our data, that iNKT-cell activation by NKT14m antibody establishes an IFN-γ production that could induce direct and indirect immune responses against B-cell lymphoma. This iNKT-cell activation together with cyclophosphamide activity could increase DC maturation, CD4 and CD8 T-cell activation and tumor cytotoxicity, leading to a synergistic and coordinated tumor immune response.
The results obtained with the combination treatment may have relevant clinical consequences since cyclophosphamide is a chemotherapeutic agent largely used to treat B-cell lymphoma and this therapeutic strategy offers a promising treatment that can be rapidly translated to the clinical scenario. In addition, we did not observed any toxic effect of cyclophosphamide or NKT14m antibody during the study; mice presented normal body weight, motility, behaviour and organ morphology, demonstrating that the therapy with NKT14m antibody and its combination with cyclophosphamide is safe. Furthermore, our data showing absence of toxicity of NKT14m antibody is in line with previous data regarding the use of this antibody in tumor-free mice.Citation30
In summary, our findings demonstrate that the novel NKT14m antibody can induce an important iNKT-cell mediated antitumor response associated with potent iNKT-cell activation in a therapeutic setting of B-cell lymphoma. More importantly, NKT14m combined with a standard chemotherapy, such as cyclophosphamide, shows an enhanced antitumor efficacy. The availability of a humanized agonistic iNKT-cell antibody for clinical use, the NKTT320 mAb,Citation33 opens new possibilities for using iNKT-cell agonists for the treatment of patients with B-cell lymphoma in combination with current chemotherapy drugs, which warrants further testing in clinical trials.
Materials and methods
Mice
Female Balb/c mice (6–7 weeks of age; Charles River) were used for in vivo experiments. Animals were housed under specific pathogen-free conditions at the Laboratory Animal Facility at Hospital Sant Pau (Barcelona). All experiments and care of animals were conducted according to European Animal Care guidelines and approved by the Ethical Committee of Animal Experimentation at Hospital Sant Pau.
Tumor cell lines
4TOO is a Balb/c plasmacytoma cell line expressing MHC class I H-2d molecules provided by Dr. M. Khuel (NCI, Bethesda, MD). Tumor cells were maintained in complete medium consisting of RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 50 μM β-2-mercaptoethanol (Life Technologies). Cells were grown in suspension culture at 37ºC in 5% CO2. 4TOO tumor cells were thawed from a common frozen stock and grown in vitro in complete medium for 3 days before use. On the day of tumor injection, cells were washed with complete medium and diluted to the appropriate concentration in 0.1ml of phosphate buffered saline (PBS) per mouse.
In vivo vaccination experiments with NKT14m mAb
Mice (8 per group) were injected intravenously (iv) with 4TOO tumor cells (4x105 cells/mouse) and, two or four days later, were treated with the NKT14m mAb (100μg/mice, iv; NKT Therapeutics Inc.). Control mice received IgG (100μg/mice, iv) or α-GalCer (2μg/mice, iv), two days after tumor injection. In some cases, mice were retreated with 100μg of NKT14m antibody 42 days after tumor challenge.
For combination studies, mice received a treatment that combined a single dose of cyclophosphamide (Cy, 70mg/kg; Sigma-Aldrich, catalog C7397) intraperitoneally (ip) 10 days after tumor injection, and a single dose of NKT14m mAb (100μg/mice; iv) 24 hours after cyclophosphamide treatment. In some experiments, mice were rechallenged with a second dose of 4TOO tumor cells (4x105 cells/mouse, iv) 120 days after the first tumor injection. In rechallenge studies, an age-matched mice group were used as untreated control.
Animals were followed daily for survival and sacrificed when moribund with tumor burden.
Flow cytometry and intracellular IFN-γ detection
Spleens of treated and control mice were harvested 24 hours after treatment in all cases; a detailed timepoint of tissues harvest is shown in –. Spleens were disaggregated in 5mL of complete medium by mechanical procedures. Splenocytes were collected, filtered through a 70μm cell strainer (BC Falcon, Cultek S.L.U.) and centrifuged at 1500 rpm during 5 minutes. Erythocytes were lysed using an ammonium chloride solution (Pharmlyse Buffer, Pharmingen, BD Bioscience) during 3 minutes in agitation at room temperature. Finally, cells were centrifuged again during 5 minutes at 1500 rpm. Splenocytes were counted and maintained in 10mL of complete medium until use.
Unloaded and α-GalCer analogue (PBS-57)-loaded CD1d tetramers were kindly provided by the NIH Tetramer Core Facility (Atlanta, GA) and were used for iNKT cell detection (1.2μg for 106 cells in 100μl), together with anti-mouse TCRβ antibody (REA318; Miltenyi Biotec, catalog 130–104-861). CD4+ and CD8+ T cells were identified using the anti-CD4 (GK1.5; Miltenyi Biotec, catalog 130–102-779), anti-CD8 (53–6.7; Miltenyi Biotec, catalog 130–102-804) antibodies and anti-CD44 (IM7.8.1; Miltenyi Biotec, catalog 130–102-982) was used for memory T-cell detection. NK cells were defined as CD3− NKp46+ cells, using the anti-CD3 (Clone REA641, catalog 130–109-880) and anti-NKp46 (29A1.4.9, catalog 130–102-842) antibodies, both from Miltenyi Biotec.
Splenocytes were stained for 30 minutes at 4ºC in PBS containing 1% bovine serum albumin (BSA; Sigma-Aldrich) and 0.01% NaN3 (Sigma-Aldrich) (staining buffer). For intracellular staining of IFN-γ, splenocytes were fixed and permeabilized following the manufacturer´s instructions (BD Cytoperm/Cytofix kit, BD Bioscience). Cells were stained using the anti-mouse IFN-γ mAb (AN.18.17.24; Miltenyi Biotec, catalog 130–102-340), for 30 minutes at 4ºC. All data was acquired on a MACSQuant Analyzer 10 (Miltenyi Biotec). Flow cytometer was automatically set-up daily accordingly to Miltenyi Biotec instructions. The compensation was performed automatically by the flow cytometer and was verified by our experimental controls (unstained cells, isotype antibodies and cells from control mice). Data from flow cytometry was analysed using the FlowJo Version 10 software (TreeStar), following the gating strategy showed in supplemental figures S5 and S6.
Statistical analysis
Results are expressed as the mean ± SEM. Kaplan-Meier plots were used to analyze mice survival and the significant differences between survival curves were assessed by the log- rank test. For all other data, t test was performed to analyze the differences between groups. All statistical analysis and graphics were performed using GraphPad Prism 6 (Graph Pad Software Inc.).
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests.
Author Contributions
LEG: design, experimental, analytical performance, and writing of the paper. CAF: experimental performance. AC: experimental performance. RS: contributed vital reagents. JS: analytical performance. JB: study´s conception, design, analytical performance, and writing of the paper. All the authors edited and approved the manuscript for submission.
Supplemental Material
Download Zip (291.2 KB)Supplemental material
Supplementary material for this article can be accessed here.
Additional information
Funding
References
- Terabe M, Berzofsky JA. The role of NKT cells in tumor immunity. Adv Cancer Res. 2008;101:277–348. doi:10.1016/S0065-230X(08)00408-9.
- Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi:10.1038/nri3174.
- Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, Kawano T, Pelikan SB, Crowe NY, Godfrey DI. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661–668.
- Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol. 2013;13:101–117. doi:10.1038/nri3369.
- Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. 2009;9:28–38. doi:10.1038/nri2451.
- Mattarollo SR, West AC, Steegh K, Duret H, Paget C, Martin B, Matthews GM, Shortt J, Chesi M, Bergsagel PL, et al. NKT cell adjuvant-based tumor vaccine for treatment of myc oncogene-driven mouse B-cell lymphoma. Blood. 2012;120:3019–3029. doi:10.1182/blood-2012-04-426643.
- Eberl G, MacDonald HR. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol. 2000;30:985–992. doi:10.1002/(SICI)1521-4141(200004)30:4<985::AID-IMMU985>3.0.CO;2-E.
- Gebremeskel S, Clattenburg DR, Slauenwhite D, Lobert L, Johnston B. Natural killer T cell activation overcomes immunosuppression to enhance clearance of postsurgical breast cancer metastasis in mice. Oncoimmunology. 2015;4. doi:10.1080/2162402X.2015.1008371.
- Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–279. doi:10.1084/jem.20030324.
- Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171:5140–5147.
- Galli G, Pittoni P, Tonti E, Malzone C, Uematsu Y, Tortoli M, Maione D, Volpini G, Finco O, Nuti S, et al. Invariant NKT cells sustain specific B cell responses and memory. Proc Natl Acad Sci USA. 2007;104:3984–3989. doi:10.1073/pnas.0700191104.
- Hayakawa Y, Takeda K, Yagita H, Smyth MJ, Van Kaer L, Okumura K, Saiki I. IFN-gamma-mediated inhibition of tumor angiogenesis by natural killer T-cell ligand, alpha-galactosylceramide. Blood. 2002;100:1728–1733.
- Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, Imamura M. Increased intratumor Valpha24-positive natural killer T cells: a prognostic factor for primary colorectal carcinomas. Clin Cancer Res. 2005;11:7322–7327. doi:10.1158/1078-0432.CCR-05-0877.
- Gorini F, Azzimonti L, Delfanti G, Scarfo L, Scielzo C, Bertilaccio MT, Ranghetti P, Gulino A, Doglioni C, Di Napoli A, et al. Invariant NKT cells contribute to chronic lymphocytic leukemia surveillance and prognosis. Blood. 2017;129:3440–3451. doi:10.1182/blood-2016-11-751065.
- Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol. 2002;3:867–874. doi:10.1038/ni827.
- Spada FM, Koezuka Y, Porcelli SA. CD1d-restricted recognition of synthetic glycolipid antigens by human natural killer T cells. J Exp Med. 1998;188:1529–1534.
- Kobayashi E, Motoki K, Uchida T, Fukushima H, Koezuka Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol Res. 1995;7:529–534.
- Chang WS, Kim JY, Kim YJ, Kim YS, Lee JM, Azuma M, Yagita H, Kang C-Y. Cutting edge: programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol. 2008;181:6707–6710.
- Chung Y, Qin H, Kang CY, Kim S, Kwak LW, Dong C. An NKT-mediated autologous vaccine generates CD4 T-cell dependent potent antilymphoma immunity. Blood. 2007;110:2013–2019. doi:10.1182/blood-2006-12-061309.
- Matsuyoshi H, Hirata S, Yoshitake Y, Motomura Y, Fukuma D, Kurisaki A, Nakatsura T, Nishimura Y, Senju S. Therapeutic effect of alpha-galactosylceramide-loaded dendritic cells genetically engineered to express SLC/CCL21 along with tumor antigen against peritoneally disseminated tumor cells. Cancer Sci. 2005;96:889–896. doi:10.1111/j.1349-7006.2005.00123.x.
- Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, Joyce S, Van Kaer L. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. 2005;115:2572–2583. doi:10.1172/JCI24762.
- Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii S. Cross-presentation of glycolipid from tumor cells loaded with alpha-galactosylceramide leads to potent and long-lived T cell mediated immunity via dendritic cells. J Exp Med. 2007;204:2641–2653. doi:10.1084/jem.20070458.
- Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, Hutchinson A, Geller M, Liu N, Annable R, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. 2005;201:1503–1517. doi:10.1084/jem.20042592.
- Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, von Blomberg BME, Scheper RJ, van der Vliet HJJ, van Den Eertwegh AJM, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. 2002;8:3702–3709.
- Nieda M, Okai M, Tazbirkova A, Lin H, Yamaura A, Ide K, Abraham R, Juji T, Macfarlane DJ, Nicol AJ. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood. 2004;103:383–389. doi:10.1182/blood-2003-04-1155.
- Uchida T, Horiguchi S, Tanaka Y, Yamamoto H, Kunii N, Motohashi S, Taniguchi M, Nakayama T, Okamoto Y. Phase I study of alpha-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer. Cancer Immunol Immunother. 2008;57:337–345. doi:10.1007/s00262-007-0373-5.
- Schneiders FL, Scheper RJ, von Blomberg BM, Woltman AM, Janssen HLA, van Den Eertwegh AJM, Verheul HMW, de Gruijl TD, van der Vliet HJ. Clinical experience with α-galactosylceramide (KRN7000) in patients with advanced cancer and chronic hepatitis B/C infection. Clin Immunol. 2011;140:130–141. doi:10.1016/j.clim.2010.11.010.
- Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, Iizasa T, Nakayama T, Taniguchi M, Fujisawa T. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. 2005;11:1910–1917. doi:10.1158/1078-0432.CCR-04-1453.
- Richter J, Neparidze N, Zhang L, Nair S, Monesmith T, Sundaram R, Miesowicz F, Dhodapkar KM, Dhodapkar MV. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood. 2013;121:423–430. doi:10.1182/blood-2012-06-435503.
- Scheuplein F, Lamont DJ, Poynter ME, Boyson JE, Serreze D, Lundblad LK, Mashal R, Schaub R, Leite-de-Moraes M. Mouse invariant monoclonal antibody NKT14: A novel tool to manipulate iNKT cell function In Vivo. PLoS ONE. 2015;10:e0140729. doi:10.1371/journal.pone.0140729.
- Singh AK, Gaur P, Das SN. Natural killer T cell anergy, co-stimulatory molecules and immunotherapeutic interventions. Hum Immunol. 2014;75:250–260. doi:10.1016/j.humimm.2013.12.004.
- Escriba-Garcia L, Alvarez-Fernandez C, Tellez-Gabriel M, Sierra J, Briones J. Dendritic cells combined with tumor cells and alpha-galactosylceramide induce a potent, therapeutic and NK-cell dependent antitumor immunity in B cell lymphoma. J Transl Med. 2017;15:115. doi:10.1186/s12967-017-1219-3.
- Das R, Scheuplein F, Guan P, Schaub R, Nichols KE. Cancer immunotherapeutic potential of NKTT320, a novel human invariant natural killer T-cell activating monoclonal antibody. In: Res C, editor. Proceedings of the 106th annual meeting of the American association for cancer research, 2015, Philadelphia (PA). Cancer Res; 75 (15 Suppl): Abstract 4294.
- Gebremeskel S, Lobert L, Tanner K, Walker B, Oliphant T, Clarke LE, Dellaire G, Johnston B. Natural Killer T-cell immunotherapy in combination with chemotherapy-induced immunogenic cell death targets metastatic breast cancer. Cancer Immunol Res. 2017;5:1086–1097. doi:10.1158/2326-6066.CIR-17-0229.
- K TT H, Nakahara F, Matsumoto A, Kurokawa M, Ogawa S, Oda H, Hirai HCS. CD1d expression level in tumor cells is an important determinant for anti-tumor immunity by natural killer T cells. Leuk Lymphoma. 2006;47:2218–2223. doi:10.1080/10428190600682688.
- Nair S, Dhodapkar MV. Natural killer T cells in cancer immunotherapy. Front Immunol. 2017;8:1178. doi:10.3389/fimmu.2017.01178.
- Gibbins JD, Ancelet LR, Weinkove R, Compton BJ, Painter GF, Petersen TR, Hermans IF. An autologous leukemia cell vaccine prevents murine acute leukemia relapse after cytarabine treatment. Blood. 2014;124:2953–2963. doi:10.1182/blood-2014-04-568956.
- Liu K, Idoyaga J, Charalambous A, Fujii S, Bonito A, Mordoh J, Wainstok R, Bai X-F, Liu Y, Steinman RM. Innate NKT lymphocytes confer superior adaptive immunity via tumor-capturing dendritic cells. J Exp Med. 2005;202:1507–1516. doi:10.1084/jem.20050956.
- Weir GM, Hrytsenko O, Stanford MM, Berinstein NL, Karkada M, Liwski RS, Mansour M. Metronomic cyclophosphamide enhances HPV16E7 peptide vaccine induced antigen-specific and cytotoxic T-cell mediated antitumor immune response. Oncoimmunology. 2014;3:e953407. doi:10.4161/21624011.2014.953407.
- Salem ML, Kadima AN, El-Naggar SA, Rubinstein MP, Chen Y, Gillanders WE, Cole DJ. Defining the ability of cyclophosphamide preconditioning to enhance the antigen-specific CD8+ T-cell response to peptide vaccination: creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J Immunother. 2007;30:40–53. doi:10.1097/01.cji.0000211311.28739.e3.
- Vacchelli E, Aranda F, Eggermont A, Galon J, Sautes-Fridman C, Cremer I, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology. 2014;3:e27878. doi:10.4161/onci.27878.
- Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–434.
- Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res. 2012;72:3439–3444. doi:10.1158/0008-5472.CAN-11-3912.
- Mattarollo SR, Kenna T, Nieda M, Nicol AJ. Chemotherapy pretreatment sensitizes solid tumor-derived cell lines to V alpha 24+ NKT cell-mediated cytotoxicity. Int J Cancer. 2006;119:1630–1637. doi:10.1002/ijc.22019.
- Fallarini S, Paoletti T, Orsi Battaglini N, Lombardi G. Invariant NKT cells increase drug-induced osteosarcoma cell death. Br J Pharmacol. 2012;167:1533–1549. doi:10.1111/j.1476-5381.2012.02108.x.
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi:10.1038/35074122.
- Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998;95:7556–7561.
- Curtsinger JM, Agarwal P, Lins DC, Mescher MF. Autocrine IFN-gamma promotes naive CD8 T cell differentiation and synergizes with IFN-alpha to stimulate strong function. J Immunol. 2012;189:659–668. doi:10.4049/jimmunol.1102727.
- Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, Corthay A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. 2011;2:240. doi:10.1038/ncomms1239.
- Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, Haraldsen G, Bogen B. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005;22:371–383. doi:10.1016/j.immuni.2005.02.003.
- Fulda S, Debatin KM. IFNgamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene. 2002;21:2295–2308. doi:10.1038/sj.onc.1205255.
- Beatty G, Paterson Y. IFN-gamma-dependent inhibition of tumor angiogenesis by tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-gamma. J Immunol. 2001;166:2276–2282.