
ABSTRACT
Most chronic viruses evade T-cell and natural killer (NK) immunity through downregulation of immune surface markers. Previously we showed that Pomalidomide (Pom) increases surface expression of major histocompatibility complex class I (MHC-I) in Kaposi sarcoma-associated herpesvirus-infected latent and lytic cells and restores ICAM-1 and B7-2 in latent cells. We explored the ability of Pom to increase immune surface marker expression in cells infected by other chronic viruses, including human T-cell leukemia virus type-1 (HTLV-1), Epstein-Barr virus (EBV), human papilloma virus (HPV), Merkel cell polyoma virus (MCV), and human immunodeficiency virus type-1 (HIV-1). Pom increased MHC-1, ICAM-1, and B7-2/CD86 in immortalized T-cell lines productively infected with HTLV-1 and also significantly increased their susceptibility to NK cell-mediated cytotoxicity. Pom enhancement of MHC-I and ICAM-1 in primary cells infected with HTLV-1 was abrogated by knockout of HTLV-1 orf-1. Pom increased expression of ICAM-1, B7-2 and MHC class I polypeptide related sequence A (MICA) surface expression in the EBV-infected Daudi cells and increased their T-cell activation and susceptibility to NK cells. Moreover, Pom increased expression of certain of these surface markers on Akata, Raji, and EBV lymphoblastic cell lines. The increased expression of immune surface markers in these virus-infected lines was generally associated with a decrease in IRF4 expression. By contrast, Pom treatment of HPV, MCV and HIV-1 infected cells did not increase these immune surface markers. Pom and related drugs may be clinically beneficial for the treatment of HTLV-1 and EBV-induced tumors by rendering infected cells more susceptible to both innate and adaptive host immune responses.
Introduction
Thalidomide (Thal), lenolidomide (Len) and pomalidomide (Pom) are structurally related immunomodulatory drugs that are effective in the treatment of multiple myeloma and certain other diseasesCitation1. Our group previously demonstrated that Thal and to a greater extent Pom have clinical activity in patients with Kaposi sarcoma (KS),Citation2,Citation3 and Len has also been found to have activity in KS.Citation4-Citation6 KS is caused by Kaposi sarcoma-associated herpesvirus (KSHV), also called human herpesvirus-8 (HHV-8), a gammaherpesvirus that is also the cause of several other diseases including primary effusion lymphoma (PEL), and a form of multicentric Castleman disease (MCD). Pom and Len are approved for the treatment of multiple myeloma, and Len is also approved for treating mantle cell lymphoma and 5q myelodysplastic syndrome.Citation6,Citation7 The principal target of these drugs is cereblon, an E3 ubiquitin ligase that provides substrate specificity for certain cullen-4 (CUL4) ubiquitin ligases.Citation8,Citation9 Many of the anti-tumor and immunomodulatory effects of these drugs can be attributed to increased degradation of Ikaros family zinc finger protein 1 and 3 transcription factors (IKZF-1 and IKZF-3), also called Ikaros and Aiolos, respectively, and to downstream effects related in part to the downregulation of interferon regulatory factor-4 (IRF4) and c-MycCitation10,Citation11 However, the activity of Len against 5q myelodysplastic syndrome as well as the activity of Pom against PEL can be at least partially attributed to cereblon-mediated degradation of casein kinase 1α.Citation1 The anti-tumor effects of these drugs can be attributed to their direct cytotoxic effects on tumor cells, and to enhanced overall T-cell and natural killer (NK) cell-mediated immune function.Citation1,Citation11,Citation12
While the initial interest in testing these drugs in KS was based on in vitro reports of the anti-angiogenic activity of Thal,Citation13 the mechanism(s) for the activity of these drugs against KS is still unclear. In investigating potential mechanisms, we found that they prevented the KSHV-mediated downregulation of surface immune recognition molecules on KSHV-infected PEL lines,Citation14 specifically downregulation of major histocompatibility class-1 (MHC-I) during lytic infection, and downregulatioin of intracellular adhesion molecule-1 (ICAM-1) and B7-2 (also known as CD86) during latent infection. MHC-I is primarily involved in antigen presentation to and activation of CD8-positive cytotoxic T-cells, while ICAM-1 and B7-2 are involved in the activation of both T-cells and natural killer (NK) cells. ICAM-1 is primarily a cell-adhesion molecule and helps increase T and NK cell activity either by increasing cell-cell adhesion or through downstream signaling pathway resulting from its binding to its receptor lymphocyte function-associated antigen-1 (LFA-1).Citation15-Citation17 B7-2, one of the essential co-stimulatory molecules, binds to its receptor, CD28, and enhances the TCR/CD3-mediated activation of T-cells.Citation18 B7-2 also increases NK activity through CD28-dependent as well as independent signalingCitation19-Citation21 Essentially all human viruses that establish chronic infections have evolved mechanisms to counteract both innate and adaptive host responses, in part by decreasing the expression of MHC-I and other cell surface molecules involved in immune recognition (for reviews seeCitation22,Citation23). In the case of KSHV, escape from immune recognition is mediated in part by K3 and K5, two viral lytic proteins. K3 and K5 are ubiquitin ligases that destroy surface MHC-I, ICAM-1, B7-2 and a number of other surface markers including ICAM-1 and B7-2 through ubiquitination and degradation.Citation24 K5 is also expressed at low levels during latent infectionCitation25,Citation26 making PEL cells resistant to NK and T cell-mediated cytotoxicity.Citation26 By blocking the downregulation of MHC-I, ICAM-1, and B7-2, Pom and Len could potentially thwart the ability of KSHV to render the cells invisible to these immunologic control mechanisms. A detailed analysis of the effects of Pom and Len on surface immune markers revealed that Pom blocked downregulation of MHC-I that was induced by transfected K3, but not K5. Further studies identified several potential contributing mechanisms for these effects in cells, including a modest increase in HLA mRNA expression and decreased upregulation of K3 in cells induced to lytic replication.Citation14
To determine whether these effects were specific for KSHV or could also be seen with other chronic viruses, we investigated the effects of Pom on expression of these surface markers in cells infected by human T-cell leukemia virus type 1 (HTLV-1), Epstein Barr virus (EBV), human papillomavirus (HPV), Merkel cell polyomavirus (MCV), and human immunodeficiency virus (HIV-1). These viruses utilize a variety of mechanisms to downregulate surface markers. Decreased expression of MHC-I by HTLV-1 is mediated by open reading frame-I (orf-I)-encoded p12 protein, which binds to MHC-I in the endoplasmic reticulum and redirects it for degradation.Citation27 In addition, the p12 cleavage product p8, which downregulates the T-cell receptorCitation28, works in concert with p12 to render infected cells invisible to immune recognition.Citation29 HTLV-1-encoded orf-I proteins also downregulate ICAM-1 and ICAM-2 as well as ligands for NK cell activating receptors, NCR and NKG2DCitation30 and thus decrease the susceptibility of HTLV-1 infected cells to NK cell-mediated cytotoxicity.
EBV has also evolved multiple mechanisms to avoid immune surveillance. The EBV-encoded lytic proteins BILF1 and BDLF3 increase degradation of MHC-I.Citation31,Citation32 Also, the latently-expressed EBV membrane protein 2A (LMP2A) can induce downregulation of MHC-I through the sonic hedgehog pathway,Citation33 and EBV downregulates several surface markers in primary infected B-cells including B7-2.Citation34 Other viruses use different strategies. For example, HPV E5 protein binds to MHC-I in the endoplasmic reticulum and prevents its trafficking to the plasma membrane,Citation35 and it has been reported that HPV E7 can inhibit MHC-I transcription.Citation23 There is evidence that MCV downregulates MHC-I expression through multiple mechanisms involving the small and large T-antigens.Citation36 For HIV-1,the viral encoded Nef protein downregulates MHC-I and other cellular proteins by routing them to the endosomal degradation compartmentCitation37 and there is evidence that HIV-1 Vpu can modulate MHC-II/CD74 expression by interacting with its cytoplasmic tail.
With this background, we studied the changes induced by Pom on MHC-I, ICAM-1 and B7-2 expression in cells infected with the viruses listed above. In addition, we studied the functional consequences of this Pom-induced ICAM-1 and/or B7-2 upregulation on NK cell-mediated cytotoxicity as well as T-cell activation. We focused on Pom because it had the strongest activity of the three drugs in vitro on KSHV-induced surface immune expressionCitation14 and has shown substantial clinical activity in KS.Citation3
Results
Pomalidomide increases expression of immune surface markers in certain HTLV-1-infected cell lines
We previously demonstrated that Pom prevented virus-induced downregulation of MHC-I and restored expression of the NK ligands and T-cell activation markers, B7-2 and ICAM-1, in KSHV-infected PEL cells.Citation14 Extending this work, we sought to explore whether Pom has similar effects on cells infected with other chronic viruses. We first treated the HTLV-1-transformed CD4+ T-cell line, MT-2 which constitutively produces HTLV-1,Citation38 with 0.5, 1.0, and 10 µM Pom for 3 and 10 days. Cell viability (live cell number/total cell number) remained above 90% during this period (Fig. S1A and S1B), although the number of live cells in Pom-treated cultures was less than control cells, especially at higher doses (maximum decrease approximately 62% with 10 µM Pom after 10 days), indicating that Pom has some inhibition of MT-2 cell growth (Fig. S1C and S1D). After 3 days of treatment with 1 µM or 10 µM Pom, MT-2 cells on average exhibited a 1.6 and 1.8 fold increase, respectively, in MHC-I surface expression as assessed by flow cytometry ( and ). Pom was also found to increase both ICAM-1 and B7-2 expression in MT-2 cells in a dose-dependent manner ( and ). ICAM-1 expression increased 1.6-fold with 1 µM Pom (p < 0.05) and 1.8 fold with 10 µm Pom (p = 0.056), respectively () while B7-2 increased 1.5 fold (p < 0.01) and 1.7 fold (p < 0.05) with 1 and 10 µM Pom (). MHC-I expression in MT-2 cells was also examined after 10 days of treatment with Pom and even greater increases in MHC-I were observed, with increases of 1.7 and 3-fold (p < 0.05) for 1 and 10 µM Pom, respectively (Fig. S2A-S2B).
Figure 1. Pom increases MHC-I, ICAM-1 and B7-2 surface expression in HTLV-1 infected MT-2 cells but not in HTLV-1 infected TLOM1 cells. Indicated MT-2 cells (A,B) or TLOM1 cells (C,D) were treated for 3 days and then analyzed for surface expression markers. (A,C) Shown are representative histograms of DMSO control (solid black line) and 10 µM Pom treated (dashed line) MT-2 (A) or TLOM1 (C) cells for MHC-I, ICAM-1, and B7-2. Isotype controls are shown shaded in grey. (B,D). Fold change in MHC-I, ICAM-1, and B7-2 for MT-2 cells (B) or TLOM1 cells (D) treated for three days with 0, 1 and 10 µM Pom. Shown are the averages ± the standard deviations of 4 separate experiments for MT-2 cells and 3 separate experiments for TLOM1 cells. Asterisks indicate p values as follows compared to DMSO control: * p < 0.05, **p < 0.01.
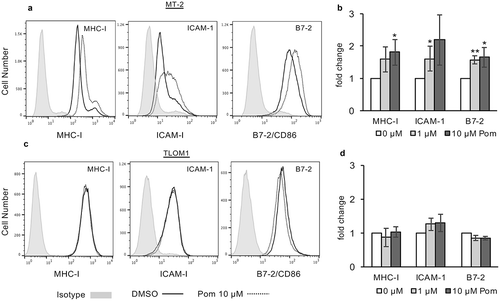
Anadditional HTLV-1-producing cell line, C91PL,Citation39 was also examined. While Pom had little effect on MHC-I expression at day 3, the MHC-I surface expression increased 1.63-fold over control (DMSO-treated) cells by day 10 (Fig. S3A and S3B). We next examined TLOM1 cells, which are infected with HTLV-1 but do not express Tax protein or produce transmittable virus.Citation40 Among the three HTLV-1 infected lines, TLOM1 cells routinely had the highest baseline median fluorescence intensity (MFI) expression of MHC-I and ICAM-1 () and by contrast to MT-2 cells, exposure of TLOM1 cells to Pom did not result in significant changes in MHC-I, ICAM-1, or B7-2 expression ( and ). Also, MHC-I expression in TLOM1 cells remained unchanged by Pom even after ten days of Pom treatment (Fig. S4A-B).
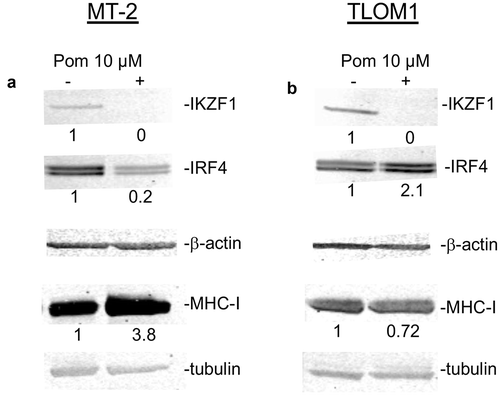
Downstream of its interaction with cereblon, Pom can downregulate the transcriptional repressors IKZF1 and IKZF3 as well as IRF4 in certain cells.Citation1,Citation10 Treatment of MT-2 cells with Pom decreased IKZF1 to undetectable levels and substantially decreased IRF4 expression (20% of control levels) (). The decrease in IKZF1 and IRF4 coincided with an increase in cellular MHC-I expression (3.8-fold) as assessed by western blot (). Interestingly, while Pom treatment also decreased IKZF1 in TLOM1 cells to undetectable levels, it increased IRF4 levels in those cells ). Also, by contrast with MT-2 cells, Pom caused a small decrease in cellular MHC-I expression in TLOM1 cells (72% of control levels) (). Pom also decreased IKZF1 expression in C91PL cells to undetectable levels, but interestingly caused no change in IRF4 expression (Fig. S3C). Pom also increased cellular MHC-I by 1.6-fold in C91PL cells treated for 10 days as assessed by western blot (Fig. S3D). Overall, these data suggest that Pom can increase MHC-I surface expression in cells actively producing HTLV-1 virus (MT-2 and C91PL) but not in at least one cell line (TLOM1) that does not express Tax or actively produce HTLV-1.
Figure 2. Effect of Pom on cellular IKZF1, IRF4 and MHC-I in MT-2 cells and TLOM1 cells. Immunoblots for IKZF1, IRF4 and beta actin in MT-2 cells (A) or TLOM1 cells (B) treated for 10 days with 10 µM Pom or DMSO control. Relative protein levels for IRF4 and MHC-I were determined based on the loading controls (beta-actin or tubulin) using the Licor system and the relative values are shown below the images.
Pomalidomide increases NK cell-mediated cytotoxicity of MT-2 cells but not TLOM1 cells
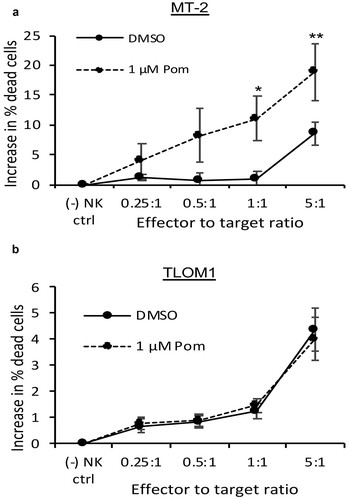
Because B7-2 and ICAM-1 regulate NK cell activity, we next sought to determine if the Pom-induced increases in B7-2 and ICAM-1 would result in increased NK cell-mediated cytotoxicity. MT-2 cells and TLOM1 cells were cultured with Pom or DMSO control for 5 days and then NK cell-mediated cytotoxicity was assessed using YTS NK effector cells,Citation41 which are sensitive to B7-2 expression but, because they do not express killer cell immunoglobulin like receptors (KIR)s, are not inhibited by MHC-I.Citation19,Citation26 Essentially no NK cell-mediated cytotoxicity was observed against DMSO-exposed control MT-2 cells at effector-to-target (E:T) cell ratios of up to 1:1 and only about 7% cytotoxicity was observed at an E:T ratio of 5:1 (). By contrast, cells exposed to 1 µM Pom showed increased NK-cell mediated toxicity up to a maximum of 19% as the effector-to-target ratio was increased, and this increase was statistically significant compared to DMSO treated cells at 1:1 and 5:1 E:T ratios (). By contrast, Pom treatment of TLOM1 cells did not lead to increased NK cell-mediated cytotoxicity at any of the effector to target ratios used (). These data provide evidence that Pom-induced increases in NK surface markers result in increased susceptibility of the cells to NK cell-mediated cytotoxicity.
Figure 3. Pom increases NK cell-mediated cytotoxicity against MT-2 HTLV-1-producing cells but not of TLOM1 HTLV-1 nonproducing cells. MT-2 cells (A) or TLOM1 cells (B) were treated with DMSO control or 1 µM Pom for 5 days and then assayed for NK cell-mediated cytotoxicity using YTS effector cells with effector-to-target ratios ranging from 0.25:1 to 5:1. The data represent the average of 3 independent experiments ± the standard deviation. Asterisks indicate p values as follows compared with the DMSO control: * p < 0.05, ** p < 0.01. In these experiments, expression of B7-2 increased from 2.9–3.6 fold in MT-2 cells treated for 5 days with 1 µM Pom but was not increased in TLOM-1 cells.
Pomalidomide increases MHC-I and ICAM-1 in HTLV-1-infected primary T-cells
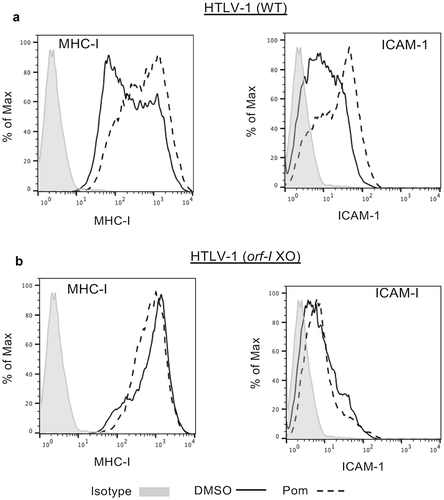
We also sought to determine if Pom affected surface marker expression in primary T-cells infected de novo with HTLV-1. Previous studies have shown that the orf-I p12 protein of HTLV-1 is in large part responsible for down-regulating MHC-I and ICAM-1 expression in primary T-cellsCitation30 and renders HTLV-1 infected cells less susceptible to cytotoxic T-cells and NK cell-mediated cytotoxicity.Citation29,Citation42 CD4+ primary T-cells were infected with wild type (WT) HTLV-1 or an HTLV-1 with orf-I/p12 knocked out. Infection was verified by determining virus levels in the supernatant of established cells after infection as previously described.Citation43 Cells infected with WT HTLV-1 had 4-fold lower MHC-I surface expression as compared to cells infected with the orf-I knockout virus (MFI of 206 for WT as compared to 870 for orf-I knockout) (compare solid line tracing in to solid line in ). Treatment of WT HTLV-1 infected T-cells with 1 µM Pom led to a 2.67-fold increase in MHC-I expression (, dashed line). However, Pom did not increase MHC-I expression in cells infected with HTLV-1 lacking orf-I (MFI of 722 with Pom and 870 without Pom) (, dashed line). We also examined the effect of Pom on ICAM-1 expression in these infected primary CD4+ cells. As with MHC-I, Pom increased ICAM-1 expression in HTLV-1 productively infected primary CD4 + T-cells by 2.7-fold (MFI increased from 9.04 to 24.4) () but did not increase ICAM-1 in the HTLV-1 orf-I knockout-infected cells (MFI 6.67 in orf-I KO vs 6.19 in WT cells) (). These data suggest that orf-I/p12 contributes to MHC-I and ICAM-1 downregulation in primary T-cells infected with HTLV-1 and that Pom interferes with virus-induced downregulation of both these surface markers.
Figure 4. Pom increases MHC-I and ICAM-1 expression in WT HTLV-1 but not orf-I/P12 knockout HTLV-1-infected CD4+ primary T-cells. CD4+ primary T-cells infected with WT HTLV-1 (A) or orf-I knockout HTLV-1 (B) were treated with DMSO control (solid black line) or 1 µM pomalidomide (dashed line) for four days. Cells were then analyzed by flow cytometry for MHC-I (left panel) and ICAM-1 (right panel) expression. The isotype control is shown shaded in grey. Shown are representative results from two separate experiments for WT infected cells but only one experiment for orf-I knockout-cells due to limitation of available cells.
Pom upregulates surface markers in EBV-infected cell lines
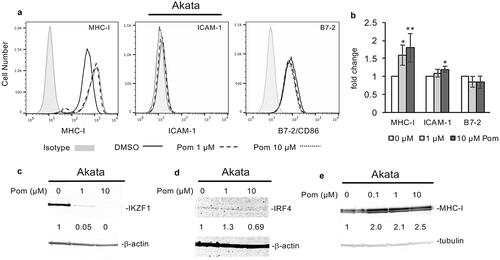
We previously demonstrated that Pom could prevent MHC-I downregulation during KSHV lytic replication and increase MHC-I expression in JSC-1, a PEL cell line that is dually infected with EBV and KSHV.Citation14 To explore these effects in EBV-infected cells that lack KSHV co-infection, we utilized the EBV-infected Burkitt lymphoma lines Akata, Daudi, and Raji. Treatment of unstimulated Akata cells with Pom at 1 µM or 10 µM had little effect on cell viability or live cell numbers (Fig. S5A and B). ( and ). However, four independent experiments revealed that Pom at 1 µM and 10 µM significantly increased MHC-I expression in Akata cells a mean of 1.6 and 1.8 fold, respectively, while having little effect (less than a 25% change) on the other two markers ( and ). Pom treatment of Akata cells also led to a dramatic reduction in the level of IKZF1 protein () and variable effects on IRF4 levels () although the basal level of IRF4 in Akata cells was extremely low in these cells, consistent with previous observations.Citation44,Citation45 Pom also increased cellular MHC-I expression up to 2.5-fold in a dose dependent fashion as determined by western blot ().
Figure 5. Pom increases MHC-I surface expression in EBV-infected akata cells but does not increase ICAM-1 or B7-2 expression. Akata cells were treated for 2 days with DMSO control, 1 µM Pom or 10 µM Pom. (A) Representative histograms of each surface marker for DMSO control (solid line), 1 µM Pom (dashed line) or 10 µM Pom (dotted line) treated cells. The isotype controls are shown shaded in grey. (B) The fold change in MHC-I, ICAM-1, and B7-2 in Akata cells treated for three days with 0, 1 and 10 µM Pom. Shown are the averages ± standard deviations of 4 separate experiments for MHC-I, (only 3 experiments at 10 µM) and 3 separate experiments for ICAM-1 and B7-2. Asterisks indicate p values as follows compared to DMSO control: * p < 0.05, ** p < 0.01. (C,D) Nuclear extracts were prepared 2 days after treatment and analyzed for IKZF1 (C) and IRF4 (D) by immunoblot. β-actin was measured as a loading control. (E) Immunoblot for MHC-I with tubulin as a loading control from cytoplasmic extracts from Akata cells treated for 2 days with 0, 0.1, 1.0 and 10 µM Pom. In C, D and E, the relative levels of IKZF1, IRF4, and MHC-I are indicated under the blots and are relative to the DMSO treated controls using the Licor system.
EBV is known to further downregulate MHC-I expression during lytic activationCitation31,Citation32 and so we assessed the effects of Pom on MHC-I expression in Akata cells induced to lytic replication with anti-human IgG (anti-IgG).Citation46,Citation47 We did not detect a significant decrease of MHC-I surface expression as assessed by flow cytometry analysis in anti-human IgG treated cells (less than a 5% decrease in MHC-I) (Fig. S5C). However, Pom treatment of anti-human IgG-treated cells at 10 µM increased MHC-I surface expression 1.5-fold over that for anti-human IgG alone (Fig. S5C). Anti-human IgG-treatment of Akata cells also decreased MHC-I cellular protein approximately 25% by western blot (Fig. S5D, compare lane 1 to lane 4), and this decrease was prevented with Pom treatment, which resulted in substantial increases in MHC-I (Fig. S5D, compare lane 4, 5 and 6). Cellular MHC-I levels increased 6.3-fold in the presence of 10 µM Pom as compared to uninduced cells and increased 8.4-fold as compared to the anti-IgG-induced control cells based on western blot (Fig. S5D). Induction of lytic replication by anti-human IgG in these experiments was verified by assessing the lytic EBV BMRF1 gene product by western blot (Fig. S5E, lanes 4–6). Although Pom has been reported to induce EBV lytic replication in certain other EBV-infected cell lines such as B95.8 and Daudi,Citation48 we did not detect induction of lytic replication (assessed by BMRF1 induction) in Akata cells by Pom in these experiments (Fig. S5E, lanes 1–3), nor did it have a substantial effect on the levels of BMRF1 lytic protein induced by anti-human IgG (Fig S6E, lanes 4–6).
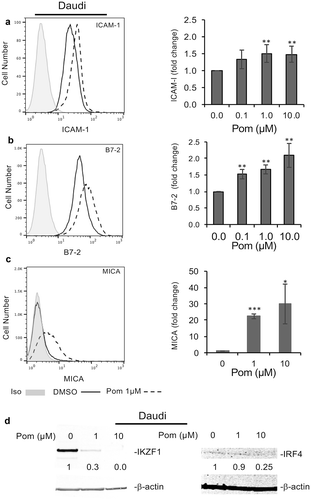
We also investigated the effect of Pom on the Daudi cell line. Daudi cells do not express beta 2-microglobulin and thus do not express surface MHC-I.Citation49,Citation50 They do, however, express B7-2 and ICAM-1, and we explored the effect of Pom on these surface proteins as well as expression of MICA, which can increase recognition by both NK cells and CD8 + T-cells and is normally decreased in EBV infected cells.Citation34 Treatment of Daudi cells for 48 hours with 0.1, 1, and 10 µM Pom led to an increase in ICAM-1 expression of up to 1.5-fold and this was statistically significant at 1 and 10 µM Pom (p < 0.01) (). Also, B7-2 expression increased as much as 2.1-fold and this was statistically significant at 0.1, 1 and 10 µM Pom (p < 0.01) (). MICA surface expression in Daudi cells was close to isotype controls but significantly increased more than 20 fold with 1 and 10 µM Pom (P < 0.005 and < 0.05 respectively) (). Cell viability was minimally decreased by Pom during the 2-day culture period, although live cell number was almost 40% lower as compared to DMSO- treated control cells (Fig. S6). We also assessed the effect of Pom on IKZF1 and IRF-4 expression in Daudi cells. Pom at 1 and 10 µM dramatically decreased IKZF1 (). Consistent with previous reports,Citation51 IRF4 levels were very low in these cells, and Pom appeared to decrease these levels further ().
Figure 6. Pom increases ICAM-1, B7-2, and MICA expression in EBV-infected Daudi cells. Daudi cells were treated for two days with DMSO control or Pom (0, 0.1 1, or 10 µM). Shown are representative histograms of each surface marker for (A) ICAM-1, (B) B7-2, and (C) MICA for cells treated with DMSO (solid line) or 1 µM Pom (dashed line). The isotype control is shown in grey. The average fold changes for these markers are shown in the bar graphs to the right. Shown on the graphs are the averages ± standard deviation from five independent experiments for ICAM-1 and B7-2, and three independent experiments for MICA. Asterisks indicate p values as follows compared to DMSO control: *p < 0.05, **p < 0.01, and ***p < 0.005. (D) Levels of IKZF1 and IRF4 in the nuclear extracts of cells treated for 2 days. β-Actin was measured as a loading control. The levels of IKZF1 and IRF4 relative to DMSO control are indicated under the blots and are relative to the DMSO treated control.
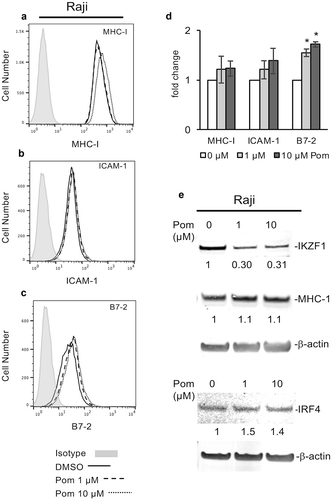
The effects of Pom on EBV-infected Raji cells was also studied. Raji cells treated with Pom at 1 and 10 µM for two days showed a non-significant trend for slightly increased MHC-I, while B7-2 increased 1.6-fold and 1.7-fold for 1 and 10 µM Pom, respectively (p < 0.05) ( to D). Similar to Akata and Daudi cells, Pom reduced IKZF1 expression in Raji cells (). However, unlike the other lines, Pom at 10 µM did not cause a complete loss of IKZF1 in Raji cells (69% reduction at 10 µM) (), and it had essentially no effect on cellular MHC-I (). As reported previously,Citation51 IRF4 levels were very low in these cells and, unlike most other lines tested, Pom treatment did not decrease IRF4 levels ().
Figure 7. Effect of Pom on MHC-I, ICAM-1, and B7-2 surface expression in raji cells. (A-C) Raji cells were treated for two days with DMSO control or Pom (0, 1, or 10 µM). A representative surface expression histogram for (A) MHC-I, (B) ICAM-1, and (C) B7-2 is shown for DMSO (solid line), 1 µM Pom (dashed line), and 10 µM Pom (dotted line). The isotype control is shown in grey. (D) The average fold change in MHC-I, ICAM-1 and B7-2 expression. The data represents the averages ± standard deviations from three independent experiments. The asterisk indicates p < 0.05. (E) Nuclear and cytoplasmic extracts were prepared 2 days after treatment and analyzed for MHC-I (cytoplasmic) and IKZF1 and IRF4 (nuclear) expression. β-Actin was measured as a loading control. The levels of MHC-I, IKZF1 and IRF4 relative to the DMSO-treated control are indicated under the blots. Note the viability of Raji cells remained > 95% for all treatments.
We next studied the effect of Pom on MHC-I, B7-2, MICA/B, and ULBP4 surface expression in lymphoblastoid cell lines (LCLs) established by infecting primary B-cells with EBV; these immune markers are known to be decreased by EBV infection.Citation34 LCLs from four different donors were treated with 0.3, 1, or 3 µM Pom for three days and then analyzed for surface marker expression. Although there was a tendency for increased MHC-I expression with Pom in two independent experiments, the MHC-I expression was already relatively high in all four LCL clones (Fig. S7A). However, Pom led to 1.25–1.5 fold increase in B7-2 and MICA/B in all four different clones in two independent experiments (Fig. S7B and S7C). By contrast, changes in ULBP4 were not consistent among the different clones and overall there was a tendency for decreased expression in the presence of Pom (Fig. S7D).
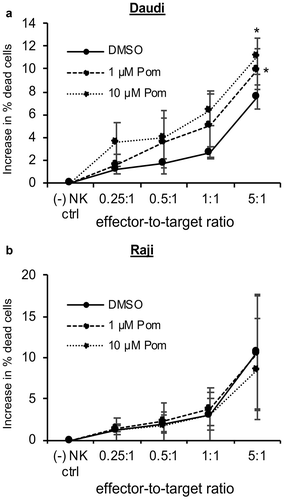
Pomalidomide increases NK cell-mediated cytotoxicity of Daudi cells but not Raji cells
We next assessed Pom’s effect on the cell-mediated cytotoxicity of YTS NK cells against Daudi and Raji cells. Daudi and Raji cells were treated with DMSO control or Pom at 1 or 10 µM for two days and then the ability of YTS NK cells to kill these cells was assayed. Treatment of Daudi cells with 1 µM Pom led to an increase in YTS cell-mediated cytotoxicity, and this further increased with 10 µM Pom (). The increase in cell death was significant (p < 0.05) for 1 and 10 µM Pom at the 5:1 effector to target ratio. However, Pom at 1 or 10 µM did not lead to increased NK cell-mediated cytotoxicity of Raji cells ().
Figure 8. Pom increases NK cell-mediated cytotoxicity against Daudi cells but not raji cells. Daudi cells (A) or Raji cells (B) were treated with DMSO control, 1 µM Pom or 10 µM Pom for 2 days and then assayed for NK cell-mediated cytotoxicity with YTS effector cells using effector-to-target ratios of 0.25:1 to 5:1. Shown are the average results ± standard deviation from 3 independent experiments for each cell line. The asterisk indicates p < 0.05 compared with DMSO control.
Pomalidomide increases T-cell activation by EBV-infected cells
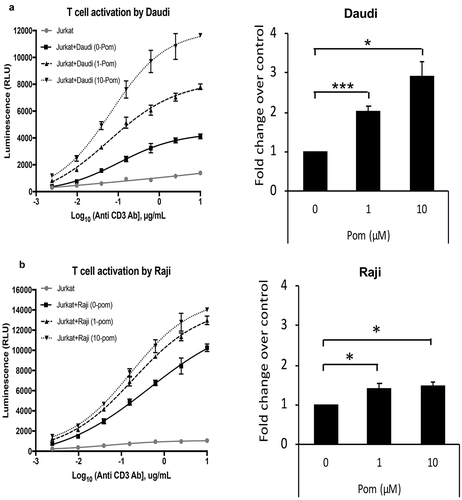
In addition to activating NK cells, ICAM-1 and B7-2 also bind to their receptors LFA-1 and CD28, respectively on T-cells and can provide important co-stimulatory signals for T-cell activation. Therefore, we wanted to test whether increases in surface expression of these markers by Pom resulted in enhanced T-cell activation. To perform T-cell activation assays, we utilized a Jurkat T-cell line that expresses luciferase under the control of an IL-2 promoter. These cells can be stimulated independent of MHC-mediated antigen-presentation using anti-CD3 antibody, and this stimulation is enhanced in the presence of additional CD28 co-stimulation by B7-1 or B7-2 cellular ligands. Daudi and Raji cells were treated with Pom for two days and then co-incubated with the anti-CD3 antibody-stimulated Jurkat T-cells for 6 hours. Both Daudi and Raji control cells activated T-cells more than just anti-CD3 antibody alone ( and ). Additionally, Daudi cells treated with 1 µM Pom increased T-cell activation by more than 2 fold over DMSO-treated control cells and this increased to almost 3 fold in the presence of 10 µM Pom (). Pom treatment also significantly increased T-cell activation by Raji cells, although the extent of activation was less than that observed for Daudi cells (). The increased activation by Daudi cells as compared to Raji cells correlates well with the extent to which Pom upregulates B7-2 in these two cell lines. This data suggests that the Pom-induced increases in B7-2 may lead to increased T-cells activation by increasing the CD28 costimulatory signaling pathway.
Figure 9. Pom increases T-cell activation by Daudi and raji cells. Daudi (A) or Raji (B) cells were treated with 0, 1, or 10 µM Pom for 2 days and co-incubated with Jurkat IL-2 reporter T-cells in the presence of various concentrations of anti-CD3 antibody. Luminescence, a measure of T cell activation, was measured after 6 hours. This experiment was performed three times. Data from one representative experiment for each cell line is shown in the left panel. The activation is expressed as relative luminescence unit (RLU) and plotted as a 4PL regression graph and the error bars represent standard deviations from technical replicates. Right panel shows average fold changes in T-cell activation by 1 and 10 µM Pom-treated Daudi cells (A) or Raji cells (B) relative to DMSO control in the presence of 0.16µM anti-CD3 antibody. Error bars represent standard deviations from three independent experiments. *p < 0.05, ***p < 0.005.
Pomalidomide does not increase MHC-I, ICAM-1, or B7-2 in HPV, HIV-1, and MCV-infected cell
Human papillomaviruses downregulate MHC-I surface expression largely through the effect of E5 and E6 proteins.Citation52,Citation53 We tested the effect of 1 and 10 µM Pom in the HPV-infected cervical carcinoma cell line CaSki and observed that they did not increase the expression of MHC-I (Fig. S8A) or ICAM-1(Fig. S8B). B7-2 was not expressed in these cells over isotype controls and Pom did not induce expression of B7-2 (Fig. S8C). The effect of Pom on the expression of MHC-I, IKZF-1, and IRF4 was also evaluated by western blot. MHC-I cellular protein levels were essentially unchanged (Fig. S8D) and neither IKZF-1 nor IRF4 protein were detected with or without Pom in nuclear or cytoplasmic extracts (Fig. S8E). Also, Pom only marginally decreased cell viability and live cell number in CaSki cells over this time period (Fig. S8F and S8G).
MHC-I surface expression is decreased in HIV-1-infected cells, and there is evidence that this is mediated by HIV-1 Nef and Vpu.Citation54,Citation55 The effect of Pom on MHC-I surface expression was examined in MOLT4 cells infected with HIV-1IIIB (MOLT4/IIIB). These cells had moderate MHC-I expression, which was not increased by treatment of these cells with Pom at 1 or 10 µM over a ten-day period (Fig. S9A). ICAM-1 and B7-2 surface expression was essentially undetectable in DMSO treated MOLT4/IIIB cells and these markers were also essentially unchanged by treatment with Pom (Fig. S9A). Recently it was shown that MCV-infected Merkel cell carcinoma (MCC) lines have downregulated MHC-I expression.Citation36 We tested the effect of 1 and 10 µM Pom treatment for 3 days on MHC-I, ICAM-1, and B7-2 expression in MCV-infected WaGa and MCC13 cells. However, Pom treatment did not lead to significant effects on these surface markers as assessed by flow cytometry analysis (Fig. S9B and S9C).
Discussion
While investigating possible mechanisms for the activity of Thal, Len, and Pom in patients with KS,Citation2-Citation4,Citation56,Citation57 we found that these drugs increase the surface expression of MHC-I in PEL cell lines latently and lytically infected with KSHV and also restore the NK cell and T-cell co-stimulatory ligands, ICAM-1 and B7-2, in cells latently infected with KSHV.Citation14 Of the three drugs, Pom was most active. Here we extend these studies and show that Pom can also increases MHC-I, ICAM-1, and/or B7-2 surface expression in HTLV-1 and EBV infected cell lines (). We further report that Pom increases immune surface marker expression in HTLV-1-infected primary CD4+ T-cells and EBV infected LCLs. In addition, we demonstrate that the Pom-induced increases in NK cell and T-cell-activating ligands are associated with increased NK cell-mediated cytotoxicity of HTLV-1-infected and EBV-infected cells as well as increased T-cell activation by EBV-infected cells. By contrast, Pom had little or no effect on these immune surface markers in the HPV, MCV, or HIV-1-infected cell lines that we tested ().
Table 1. Effect of Pomalidomide on immune surface markers in virus infected cells*.
In these experiments, Pom pretreatment of EBV-infected Daudi cells resulted in increased ICAM-1, B7-2 and MICA expression as well as increased T-cell activation and increased cell-mediated cytotoxicity to YTS NK cells. By contrast, Pom pretreatment of EBV infected Raji cells led to minimal increases in ICAM-1 and B7-2, and lesser increases in T-cell activation and no increase in NK cell-mediated cytotoxicity. The difference in YTS NK toxicity between the two lines can not simply be attributed to the lack of expression of MHC-I on Daudi cells because YTS NK cells lack KIRs needed to recognize MHC-I and downregulate NK activityCitation41. Additional experiments will be needed to understand the variability between different cell lines.
MHC-I plays an important role in T cell cytotoxicity, and ICAM-1 and B7-2 provide activation signals to NK cells as well as provide important co-stimulatory signals for T-cell-activation. Viruses have evolved mechanisms to simultaneously suppress MHC-I, ICAM-1, and B7-2, thus rendering infected cells relatively invisible to both cytotoxic T-cells and NK cells. By simultaneously increasing all three of these markers, Pom can potentially render virus-infected tumor cells more susceptible to attack by the immune system. MHC-I expression it is required for the T-cell-mediated cytotoxicity of cells expressing foreign antigens, yet MHC-I is generally inhibitory to NK cell-mediated cytotoxicity. This said, NK cell killing of cells can occur even in the presence of normal MHC-I expression, especially when activating receptors in the NK cells are engaged by their activating ligands.Citation58,Citation59 Also, there is evidence that NK killing of cells infected by another herpesvirus, cytomegalovirus, is not inhibited by MHC-1 expression on the target cells.Citation59 Moreover, tumor cells with upregulated MHC-I can still respond to IRF-1-induced NK cell-dependent elimination.Citation60 The importance of NK cells in controlling EBV tumorigenesis has also been demonstrated by others who have shown that depletion of human NK cells promotes EBV-associated tumorigenesis.Citation61
Our previous studies suggested that the main effects of these drugs on immune surface markers does not result from their interacting with KSHV viral proteins but more likely is an indirect effect of their alteration on host cell pathways. This conclusion is bolstered by the finding in the present study that Pom also increases MHC-I, ICAM-1, and B7-2 in EBV- and HTLV-1-infected cells. Although the current study was not specifically undertaken to explore in detail the mechanism(s) by which Pom affects surface marker expression, our results do provide some potential clues. The finding that cellular levels of MHC-I as assessed by western blot were enhanced by Pom, in addition to its surface expression, suggests that Pom does not simply enhance transport of existing protein to the surface but rather may involve enhanced production and/or interference with a common degradation pathway. This said, it is unclear why we did not see increased expression in cells infected with HIV, HPV, or MCV. It is noteworthy that upregulation of surface markers by Pom was most obvious in cells with low levels of basal expression of these markers. It is possible that changes in the balance between production and degradation only results in increased surface expression in those cases where viral infection has reduced surface expression by shifting the balance towards degradation.
With regard to the results seen with HTLV-1, Tax enhances MHC-I productionCitation30 while the orf-I gene products p8/p12 can override this effect by interacting with MHC-I and ICAM-1 in the endoplasmic reticulum and redirecting them for degradation.Citation30,Citation62,Citation63 Therefore, Pom could enhance basal production of these markers, potentiate the enhancing effect of Tax on ICAM-1 and MHC-I expression, or inhibit their degradation by p8/p12. Our data do not permit differentiating between these possible mechanisms. On the one hand, Pom had little or no effect in TLOM1 cells, which do not produce HTLV-1 and also do not express Tax. On the other hand, we observed that Pom could upregulate MHC-I and ICAM-1 in CD4+ primary T-cells infected with a WT HTLV-1 virus but not the HTLV-I orf-I knockout virus. While it is possible that Pom somehow interferes with the activity of p12, it is more likely that Pom’s effects are only revealed when the surface markers are relatively suppressed by virus.
One of the principal results of the binding of Pom to cereblon is increased destruction of the transcription factors IKZF3 and IKZF1 and decreases in IRF4 expression.Citation10,Citation11 A recent study suggests that the effects of Pom on IRF4 can occur by a novel mechnaism unrelated to its effects on IKZF3, IKZF1, or CK1α.Citation64 Cell lines in which Pom induced increases in MHC-I often had Pom-induced decreases in IRF4 expression (). However, we also saw increases in surface markers in cells such as Akata and Daudi that produce little to no IRF4, so the role of IRF4, if any, remains unclear. We also considered a more direct mechanism involving IKZF1 downregulation; however, our data revealed that in some cases loss of IKZF1 expression occurred in the absence of upregulation of immune surface markers. For example, TLOM1 cells treated with Pom had complete loss of IKZF1 protein expression, yet there were no increases in any of the surface markers. Interferon gamma (IFN-γ) can upregulate MHC-I, and in certain cells, Pom can lead to increased IFN-γ.Citation65 However, B cells generally do not produce IFN-γ, and we, and others,Citation66 also previously failed to detect IFN-γ in PEL lines exposed to Pom.Citation14 The related immunomodulatory drug, CC-122, has been reported to increase transcription of certain IFN-stimulated genes by an unclear mechanism independent of increases in IFN-α, IFN-β, or IFN-γ, and it is possible that a similar effect is occurring here.Citation67 Additional studies will be required to sort out the precise mechanisms most responsible for the changes observed and to explain the variation among cell lines and viruses.
The results reported in this study may have clinical utility. The increase in ICAM-1 and B7-2 expression by virus-infected tumor cells can potentially enhance the anti-tumor effects mediated by the broad Pom-induced upregulation of NK cell function. Also, the effects of Pom on MHC-I expression may be useful in combination with checkpoint inhibitor therapy. In recent years, checkpoint inhibition using antibodies directed at programmed cell death protein-1 (PD-1), PD-1 ligand (PD-L1), or cytotoxic T lymphocyte-associated antigen (CTLA-4) have been found to be active against a number of tumors by removing constraints in cytotoxic T-cells.Citation68-Citation71 Checkpoint inhibition therapy requires epitopes in the target cells that can be recognized by T-cells and have been shown to be especially effective against tumors with multiple mutations.Citation72 This therapy can also be useful in virus-associated tumors as the viruses provide foreign antigens and antibodies directed at anti-PD-1 or anti-PD-L1 have been shown to be effective against Merkel cell carcinomaCitation73 and Kaposi sarcoma.Citation74 However, potent viral-mediated mechanisms that downregulate expression of MHC-I can potentially reduce the effectiveness of these approaches for viral-associated tumors. By preventing virus-induced downregulation of surface markers, Pom can potentially render tumors caused by KSHV, HTLV-1, and EBV more visible to cytotoxic T-cells and thus susceptible to immunotherapy, especially when combined with the overall immune activation induced by Pom and related drugs. A potential advantage of Pom-mediated upregulation of surface proteins is that it is occurring in the virus-infected tumor cells and thus may be less likely to lead to some of the autoimmune toxicities observed with checkpoint inhibitors. Thus, the results here suggest that Pom may have at least three mechanisms of action in certain tumors: a direct cytostatic effect, general immunostimulation, and an upregulation of immune surface markers. The results here thus provide a rationale for the study of Pom or related drugs in tumors caused by these viruses. In addition to KS, there are preliminary results showing that Len has activity in acute T-cell leukemia/lymphoma caused by HTLV-1.Citation75 Finally, the results suggest that Pom could be useful in virus-induced tumors in combination with checkpoint inhibitors, which can enhance the T-cell response to tumor cells expressing foreign antigens.
Materials and methods
Cells and cell culture
HTLV-1 infected cell lines MT-2,Citation38 C91PL, and TLOM1 were obtained as previously described.Citation40,Citation42,Citation76 EBV cell lines Raji (#CCL-86) and Akata, and the HPV infected line CaSki were obtained from the American Type Culture Collection (ATCC, Manassas, VA). HIV-1-infected cells MOLT4/HIV-1IIIB were generated as described previouslyCitation77. The Merkle cell carcinoma lines WaGaCitation78 and MCC13Citation79 were provided by Isaac Brownell (National Institutes of Health, Bethesda, MD). Suspension cell lines and CaSki cells were grown in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 15 % fetal-bovine serum (FBS) (Thermo Scientific, Rockford, IL), 1 % penicillin/streptomycin glutamine (Sigma, St. Louis, MO) at 37 ºC with 5 % CO2. YTS cells were a kind gift from Jordan Orange MD, Baylor College of Medicine. YTS is s subclone of the NK tumor line that was established from a patient with acute lymphoblastic lymphomaCitation80. The adherent cell line MCC13 was cultured in DMEM (Invitrogen, Carlsbad, CA) with 10% FBS and 1 % penicillin/streptomycin glutamine.
Reagents
Stocks (20–100 mM) of pomalidomide (Celgene) were stored frozen in cell culture grade dimethyl sulfoxide (DMSO) (Sigma) and DMSO was used as a vehicle control. The effect of drug treatment on cell viability (live cell/total cell number) and growth (live cell number) was investigated over three passages. Cell viability was assessed by trypan blue staining. Cells grown for periods longer than 3 days were passed at 3 × 105 cells/ml in fresh media.
Flow cytometry analysis and antibodies
Analysis of cells for surface marker expression was carried out as described previously.Citation14 Briefly, control and drug-treated cells were exposed to FITC-labeled isotype antibody or antibodies toward HLA class I (A, B and C), ICAM-1, and B7-2 for 1 hr. Cells were washed three times with 10% FBS in phosphate buffered saline (PBS), suspended in 10% FBS/PBS and then analyzed with a flow cytometrycalibur™ Flow Cytometry system (BD Biosciences, San Jose, CA). Cells were examined using FlowJo flow cytometry analysis software (flowjo.com).
Western blotting
Whole cell lysates were prepared for control (DMSO) or drug-treated cells with M-PER (Pierce, Rockland, IL) in the presence of Halt Protease Inhibitors Cocktail (Pierce, Rockland, IL). Where indicated, nuclear and/or cytoplasmic extracts were prepared using the NE-PER Nuclear Extraction Reagent kit (Pierce) with Halt Protease Inhibitors and 1 mM ethylenediaminetetraacetic acid (EDTA). Protein concentrations were determined using the BCA assay (Pierce). Samples of equal protein content were subjected to LDS-PAGE (4 to 12 % NuPAGE Tris-Bis) (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes using iBlot (Life Technologies Grand Island, NY). The membranes were blocked with Odyssey blocking buffer (Licor, Lincoln, NE) for use in the Licor system. Blots were incubated with indicated antibodies: mouse anti-β-actin, mouse anti-β-tubulin, mouse anti-IKZF1 (Sigma), mouse anti-IRF4, (Cell Signaling, Beverly, MA), mouse anti-MHC-I (Santa Cruz Biotechnology, Dallas, TX), and mouse anti-BMRF1(Millipore Billerica, MA), and subjected to the appropriate secondary antibodies conjugated to green or red fluorescent dyes (Licor). Membranes were scanned, and images were processed. Quantitation of protein was determined using Image Studio software (Licor).
Generation and characterization of HTLV-1-infected primary human cells
Stable HTLV-1-producing 729.6 human lymphoblastoid B-cells were generated as described previously.Citation43 The 729–6 B-cell line infected with the pAB wild-type (WT) HTLV-1 was maintained in 10% FBS/RPMI 1640. Using negative selection beads (StemCell) CD4+ T-cells were isolated from uninfected peripheral blood mononuclear cells. Stable HTLV-1 producing CD4+ T-cell lines were established by co-cultivation of donor uninfected primary HLA.A2+/CD4+ T-cells with lethally γ-irradiated 729.6-HTLV-1 producing lines. T-cells were cultured in 20% FBS/RPMI 1640 and 100 U of interleukin-2 for several months. The production of HTLV-1 in the supernatant of the infected cell cultures was assessed by measuring the amount of MA (p19 Gag) protein by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (Zeptometrix, Buffalo, NY). Viral genomic sequences were verified by sequencing of the ClaI-SalI fragment as described previously.Citation29
Establishment of lymphoblastoid cell lines (LCLs) and LCL flow cytometry analysis
Infectious EBV stocks were prepared as previously described.Citation81 EBV viral stocks were titrated on Raji cells as described previouslyCitation82 and used at a multiplicity of infection (MOI) of 0.1. Human primary B cells were prepared from PBMCs by Ficoll-Hypaque gradient centrifugation with Ficoll-Paque PLUS (GE healthcare) and cultivated with virus stock for 18 h. After replacement with fresh medium, the infected cells were seeded at an initial density of 5 × 105 cells per ml. Resulting LCLs were incubated and expanded in 10% FBS/RPMI 1640 and used for experiments between 1 ~ 3 months after infection. To assess the effect of Pom on immune surface markers, LCLs (2 × 105 cells per ml) from four different LCL clones were treated with DMSO control or Pom at 0.3, 1, or 3 uM for three days. After immunostaining with fluorophore-conjugated antibodies, single-cell suspensions were measured with flow cytometryCanto II (BD) flow cytometers and the flow cytometryDiva software (BD). Dead cells were excluded with LIVE/DEAD Cell Viability Assays (Thermo Fisher). Acquired data were analyzed with FlowJo software Ver. 9.9 (FlowJo). The following fluoro- phore-conjugated antibodies reactive to human antigens were used: anti HLA-ABC-APC (W6/32), B7-2/CD86-PE (IT2.2), anti MICA/B-PE (6D4), anti Isotype-APC (MOPC-173) and Isotype-PE (MPC-11) (BioLegend, San Diego, CA). The anti ULBP4-PE (6E6) antibody was from Santa Cruz (Dallas,TX).
NK cell-mediated cytotoxicity assay
NK cytotoxicity mediated by the YTS natural killer cell line (effector cells) was assessed using a two-color fluorescence assay adapted from Hoppner et. al.Citation83 A green fluorescent membrane dye DiOC18(DiO) (Sigma) was used as an effector cell marker while a red fluorescent nuclear counterstain propidium iodide (PI) (Sigma) was used as a dead cell marker. Cells were plated with Pom or DMSO control at indicated concentrations for the indicated number of days. On the day of the assay, the effector cells were washed once with PBS, suspended at 106 cells per mL in PBS, and stained with DiO at a 20 µM final concentration by incubating at 37ºC for 30 to 60 minutes. The DiO-stained effector cells were washed with PBS three times and suspended at 2 × 106 cells per mL with complete media. The target cells were suspended at 2 × 106 cells per mL with complete media and mixed with the DiO-stained effector cells at various effector to target ratios by keeping the number of effector cells constant at 105 cells per well. This mixture was incubated at 37ºC for approximately 3.5 hours and then PI was added at a 75 µM final concentration. Target cells without effector cells were incubated in parallel to obtain a background level of target cell death (no NK control). The cells were then analyzed by flow cytometry using a flow cytometrycalibur™ Flow Cytometry system (BD Biosciences, San Jose, CA). The scatter gate was set to include all cellular events and analyzed in the FL1 and FL3 channels for DiO and PI respectively. The Dio-negative cells (total target cells) and the PI-positive cells in the Dio-negative fraction (dead target cells) were used to calculate the % of dead target cells. NK-induced cytotoxicity was then calculated as an increase in % dead cells by subtracting the % of dead target cells obtained in the absence of effector cells (no NK control) from that obtained in the presence of effector cells.
T-cell activation assays
An assay for T-cell activation was performed using the T-cell Activation Bioassay kit (Promega, cat# J1651), which uses IL-2 reporter Jurkat T-cells expressing a luciferase reporter gene under IL-2 promoter as effector T-cells. Cells were treated with the indicated concentrations of Pom for 2 days, after which T-cell activation was assessed using IL-2 reporter Jurkat T-cells according to the manufacturer’s recommended protocol. Briefly, 105 Jurkat T-cells per well of a 96-well plate were stimulated using various concentrations of anti-CD3 monoclonal antibody (OKT3 from ThermoFisher Scientific, Cat# 16–0037-81). Control or Pom-treated Daudi and Raji cells were co-incubated with the stimulated Jurkat cells at a 5:1 ratio (Jurkat to target) in a 37°C incubator. After 6 hours, bio-glo reagent was added and luminescence was measured using Victor X3 multilabel plate reader (PerkinElmer). Background luminescence from wells without cells was subtracted from all the wells containing cells. Luminescence data was plotted as a 4PL regression graph using GraphPad Prism software.
Statistics
Where indicated, the mean and standard deviation were calculated for experiments repeated 3 or more times. Statistical comparisons were performed using the Student’s two tailed paired T-test.
Disclosure of Potential Conflicts of Interest
Drs. Yarchoan and Davis are co-inventors on US Patent 10,001,483 entitled “Methods for the treatment of Kaposi’s sarcoma or KSHV-induced lymphoma using immunomodulatory compounds, and uses of biomarkers”. In is our understanding that foreign patents have also been filed for this invention. This invention was made as full-time employees of the US government under 45 Code of Federal Regulations Part 7. All rights, title, and interest to these patents have been or should by law be assigned to the U.S. Department of Health and Human Services. The government conveys a portion of the royalties it receives to its employee inventors under the Federal Technology Transfer Act of 1986 (P.L. 99-502). This research was supported in part by a CRADA between the NCI and Celgene Corporation.
Supplemental Material
Download PDF (6 MB)Acknowledgments
We thank Drs. Jerry Zeldis and Ken Arakawa of Celgene Corp. for their helpful discussions.
Supplemental material
Supplementary material for this article can be accessed here.
Additional information
Funding
References
- Lindner S, Kronke J. The molecular mechanism of thalidomide analogs in hematologic malignancies. J Mol Med (Berl). 2016;94:1327–1334. doi:10.1007/s00109-016-1450-z.
- Little RF, Wyvill KM, Pluda JM, Welles L, Marshall V, Figg WD, Newcomb FM, Tosato G, Feigal E, Steinberg SM, et al. Activity of thalidomide in AIDS-related kaposi’s sarcoma. J Clin Oncol. 2000;18:2593–2602. doi:10.1200/JCO.2000.18.13.2593.
- Polizzotto MN, Uldrick TS, Wyvill KM, Aleman K, Peer CJ, Bevans M, Sereti I, Maldarelli F, Whitby D, Marshall V, et al. Pomalidomide for symptomatic kaposi’s sarcoma in people with and without HIV infection: a phase I/II study. J Clin Oncol. 2016;34:4125–4131. doi:10.1200/JCO.2016.69.3812.
- Pourcher V, Desnoyer A, Assoumou L, Lebbe C, Curjol A, Marcelin AG, Cardon F, Gibowski S, Salmon D, Chennebault J-M, et al. Phase II trial of lenalidomide in HIV-infected patients with previously treated kaposi’s sarcoma: results of the ANRS 154 lenakap trial. AIDS Res Hum Retroviruses. 2017;33:1–10. doi:10.1089/AID.2016.0069.
- Shimabukuro K, Moore PC, Bui J, D’ittmer D, Ambinder R, Martinez-Maza O, et al. Lenalidomide is safe and effective in aids-associated kaposi sarcoma. 16th International Conference on Malignancies in HIV/AIDS; 2017; Bethesda, MD: National Cancer Institute.
- Liu LR, Qian SX. [Action mechanism of lenalidomide in hematological malignancies - review]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2012;20:1039–1041.
- Lacy MQ, Tefferi A. Pomalidomide therapy for multiple myeloma and myelofibrosis: an update. Leuk Lymphoma. 2011;52:560–566. doi:10.3109/10428194.2011.552139.
- Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, Karasawa S, Carmel G, Jackson P, Abbasian M, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–2335. doi:10.1038/leu.2012.119.
- Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, Chang X-B, Bjorklund CC, Fonseca R, Bergsagel PL, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118:4771–4779. doi:10.1182/blood-2011-05-356063.
- Bjorklund CC, Lu L, Kang J, Hagner PR, Havens CG, Amatangelo M, Wang M, Ren Y, Couto S, Breider M, et al. Rate of CRL4(CRBN) substrate ikaros and aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015;5:e354. doi:10.1038/bcj.2015.66.
- Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, Ito T, Ando H, Waldman MF, Thakurta A, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors ikaros and aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol. 2014;164:811–821. doi:10.1111/bjh.12708.
- Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, Todryk S, Chen R, Muller G, Stirling D, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother. 2009;58:1033–1045. doi:10.1007/s00262-008-0620-4.
- D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91:4082–4085.
- Davis DA, Mishra S, Anagho HA, Aisabor AI, Shrestha P, Wang V, Takamatsu Y, Maeda K, Mitsuya H, Zeldis JB, et al. Restoration of immune surface molecules in Kaposi sarcoma-associated herpes virus infected cells by lenalidomide and pomalidomide. Oncotarget. 2017;8:50342–50358. doi:10.18632/oncotarget.17960.
- Abraham C, Miller J. Molecular mechanisms of IL-2 gene regulation following costimulation through LFA-1. J Immunol. 2001;167:5193–5201.
- Bachmann MF, McKall-Faienza K, Schmits R, Bouchard D, Beach J, Speiser DE, Mak TW, Ohashi PS. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesion versus costimulation. Immunity. 1997;7:549–557.
- Van Seventer GA, Shimizu Y, Horgan KJ, Shaw S. The LFA-1 ligand ICAM-1 provides an important costimulatory signal for T cell receptor-mediated activation of resting T cells. J Immunol. 1990;144:4579–4586.
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi:10.1146/annurev.immunol.14.1.233.
- Azuma M, Cayabyab M, Buck D, Phillips JH, Lanier LL. Involvement of CD28 in MHC-unrestricted cytotoxicity mediated by a human natural killer leukemia cell line. J Immunol. 1992;149:1115–1123.
- Wilson JL, Charo J, Martin-Fontecha A, Dellabona P, Casorati G, Chambers BJ, Kiessling R, Bejarano MT, Ljunggren HG. NK cell triggering by the human costimulatory molecules CD80 and CD86. J Immunol. 1999;163:4207–4212.
- Luque I, Reyburn H, Strominger JL. Expression of the CD80 and CD86 molecules enhances cytotoxicity by human natural killer cells. Hum Immunol. 2000;61:721–728.
- Hewitt EW. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology. 2003;110:163–169.
- Westrich JA, Warren CJ, Pyeon D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017;231:21–33. doi:10.1016/j.virusres.2016.11.023.
- Lee HR, Brulois K, Wong L, Jung JU. Modulation of immune system by kaposi’s sarcoma-associated herpesvirus: lessons from viral evasion strategies. Front Microbiol. 2012;3:44. doi:10.3389/fmicb.2012.00044.
- Taylor JL, Bennett HN, Snyder BA, Moore PS, Chang Y. Transcriptional analysis of latent and inducible kaposi’s sarcoma-associated herpesvirus transcripts in the K4 to K7 region. J Virol. 2005;79:15099–15106. doi:10.1128/JVI.79.24.15099-15106.2005.
- Ishido S, Choi JK, Lee BS, Wang C, DeMaria M, Johnson RP, Cohen GB, Jung JU. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity. 2000;13:365–374.
- Johnson JM, Nicot C, Fullen J, Ciminale V, Casareto L, Mulloy JC, Jacobson S, Franchini G. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J Virol. 2001;75:6086–6094. doi:10.1128/JVI.75.13.6086-6094.2001.
- Fukumoto R, Andresen V, Bialuk I, Cecchinato V, Walser JC, Valeri VW, Nauroth JM, Gessain A, Nicot C, Franchini G. In vivo genetic mutations define predominant functions of the human T-cell leukemia/lymphoma virus p12I protein. Blood. 2009;113:3726–3734. doi:10.1182/blood-2008-04-146928.
- Pise-Masison CA, de Castro-Amarante MF, Enose-Akahata Y, Buchmann RC, Fenizia C, Washington Parks R, Edwards D, Fiocchi M, Alcantara LC, Bialuk I, et al. Co-dependence of HTLV-1 p12 and p8 functions in virus persistence. PLoS Pathog. 2014;10:e1004454. doi:10.1371/journal.ppat.1004454.
- Banerjee P, Feuer G, Barker E. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and −2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J Virol. 2007;81:9707–9717. doi:10.1128/JVI.00887-07.
- Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME, Wiertz EJHJ, Rowe M. The epstein-barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009;5:e1000255. doi:10.1371/journal.ppat.1000706.
- Quinn LL, Williams LR, White C, Forrest C, Zuo J, Rowe M. The missing link in epstein-barr virus immune evasion: the BDLF3 gene induces ubiquitination and downregulation of major histocompatibility complex class I (MHC-I) and MHC-II. J Virol. 2015;90:356–367. doi:10.1128/JVI.02183-15.
- Deb Pal A, Banerjee S. Epstein-barr virus latent membrane protein 2A mediated activation of sonic hedgehog pathway induces HLA class Ia downregulation in gastric cancer cells. Virology. 2015;484:22–32. doi:10.1016/j.virol.2015.05.007.
- Rancan C, Schirrmann L, Huls C, Zeidler R, Moosmann A. latent membrane protein lmp2a impairs recognition of ebv-infected cells by CD8+ T Cells. PLoS Pathog. 2015;11:e1004906. doi:10.1371/journal.ppat.1004906.
- Ashrafi GH, Haghshenas M, Marchetti B, Campo MS. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int J Cancer. 2006;119:2105–2112. doi:10.1002/ijc.22089.
- Paulson KG, Tegeder A, Willmes C, Iyer JG, Afanasiev OK, Schrama D, Koba S, Thibodeau R, Nagase K, Simonson WT, et al. Downregulation of MHC-I expression is prevalent but reversible in merkel cell carcinoma. Cancer Immunol Res. 2014;2:1071–1079. doi:10.1158/2326-6066.CIR-14-0005.
- Greenberg ME, Iafrate AJ, Skowronski J. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. Embo J. 1998;17:2777–2789. doi:10.1093/emboj/17.10.2777.
- Harada S, Koyanagi Y, Yamamoto N. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science (New York, NY. 1985;229:563–566. doi:10.1126/science.2992081.
- Popovic M, Lange-Wantzin G, Sarin PS, Mann D, Gallo RC. Transformation of human umbilical cord blood T cells by human T-cell leukemia/lymphoma virus. Proc Natl Acad Sci USA. 1983;80:5402–5406.
- Sugamura K, Fujii M, Kannagi M, Sakitani M, Takeuchi M, Hinuma Y. Cell surface phenotypes and expression of viral antigens of various human cell lines carrying human T-cell leukemia virus. Int J Cancer. 1984;34:221–228.
- Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, Capanni M, Ruggeri L, Benson DM, Blaser BW, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood. 2009;114:2667–2677. doi:10.1182/blood-2009-02-206532.
- Edwards D, Fenizia C, Gold H, de Castro-Amarante MF, Buchmann C, Pise-Masison CA, Franchini G. Orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses. 2011;3:861–885. doi:10.3390/v3060861.
- Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, Chung HK, Fukumoto R, Parks RW, Ferrari MG, et al. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood. 2010;116:3809–3817. doi:10.1182/blood-2010-05-284141.
- Wang L, Yao ZQ, Moorman JP, Xu Y, Ning S. Gene expression profiling identifies IRF4-associated molecular signatures in hematological malignancies. PLoS ONE. 2014;9:e106788. doi:10.1371/journal.pone.0106788.
- Lv DW, Zhang K, Li R. Interferon regulatory factor 8 regulates caspase-1 expression to facilitate epstein-barr virus reactivation in response to B cell receptor stimulation and chemical induction. PLoS Pathog. 2018;14:e1006868. doi:10.1371/journal.ppat.1006797.
- Takada K, Ono Y. Synchronous and sequential activation of latently infected epstein-barr virus genomes. J Virol. 1989;63:445–449.
- Daibata M, Humphreys RE, Takada K, Sairenji T. Activation of latent EBV via anti-IgG-triggered, second messenger pathways in the burkitt’s lymphoma cell line akata. J Immunol. 1990;144:4788–4793.
- Jones RJ, Iempridee T, Wang X, Lee HC, Mertz JE, Kenney SC, Lin HC, Baladandayuthapani V, Dawson CW, Shah JJ, et al. Lenalidomide, thalidomide, and pomalidomide reactivate the epstein-barr virus lytic cycle through phosphoinositide 3-kinase signaling and ikaros expression. Clin Cancer Res. 2016;22:4901–4912. doi:10.1158/1078-0432.CCR-15-2242.
- de Preval C, Mach B. The absence of beta 2-microglobulin in Daudi cells: active gene but inactive messenger RNA. Immunogenetics. 1983;17:133–140.
- Seong RH, Clayberger CA, Krensky AM, Parnes JR. Rescue of Daudi cell HLA expression by transfection of the mouse beta 2-microglobulin gene. J Exp Med. 1988;167:288–299.
- Teng Y, Takahashi Y, Yamada M, Kurosu T, Koyama T, Miura O, Miki T. IRF4 negatively regulates proliferation of germinal center B cell-derived burkitt’s lymphoma cell lines and induces differentiation toward plasma cells. Eur J Cell Biol. 2007;86:581–589. doi:10.1016/j.ejcb.2007.05.006.
- Campo MS, Graham SV, Cortese MS, Ashrafi GH, Araibi EH, Dornan ES, Miners K, Nunes C, Man S. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology. 2010;407:137–142. doi:10.1016/j.virol.2010.07.044.
- Kim DH, Kim EM, Lee EH, Ji KY, Yi J, Park M, Kim KD, Cho -Y-Y, Kang H-S. Human papillomavirus 16E6 suppresses major histocompatibility complex class I by upregulating lymphotoxin expression in human cervical cancer cells. Biochem Biophys Res Commun. 2011;409:792–798. doi:10.1016/j.bbrc.2011.05.090.
- Kerkau T, Bacik I, Bennink JR, Yewdell JW, Hunig T, Schimpl A, Schubert U. The human immunodeficiency virus type 1 (HIV-1) Vpu protein interferes with an early step in the biosynthesis of major histocompatibility complex (MHC) class I molecules. J Exp Med. 1997;185:1295–1305.
- Kasper MR, Collins KL. Nef-mediated disruption of HLA-A2 transport to the cell surface in T cells. J Virol. 2003;77:3041–3049.
- Martinez V, Tateo M, Castilla MA, Melica G, Kirstetter M, Boue F. Lenalidomide in treating AIDS-related kaposi’s sarcoma. AIDS (London, England) . 2011;25:878–880. doi:10.1097/QAD.0b013e328344c145.
- Steff M, Joly V, Di Lucca J, Feldman J, Burg S, Sarda-Mantel L, Peytavin G, Marinho E, Crickx B, Raymond E, et al. Clinical activity of lenalidomide in visceral human immunodeficiency virus-related kaposi sarcoma. JAMA Dermatol. 2013;149:1319–1322. doi:10.1001/jamadermatol.2013.5751.
- Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–171. doi:10.1038/35093109.
- Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev. 2001;1:41–49. doi:10.1038/35095564.
- Ksienzyk A, Neumann B, Nandakumar R, Finsterbusch K, Grashoff M, Zawatzky R, Bernhardt G, Hauser H, Kröger A. IRF-1 expression is essential for natural killer cells to suppress metastasis. Cancer Res. 2011;71:6410–6418. doi:10.1158/0008-5472.CAN-11-1565.
- Chijioke O, Muller A, Feederle R, Barros MH, Krieg C, Emmel V, Marcenaro E, Leung CS, Antsiferova O, Landtwing V, et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic epstein-barr virus infection. Cell Rep. 2013;5:1489–1498. doi:10.1016/j.celrep.2013.11.041.
- Johnson JM, Mulloy JC, Ciminale V, Fullen J, Nicot C, Franchini G. The MHC class I heavy chain is a common target of the small proteins encoded by the 3ʹ end of HTLV type 1 and HTLV type 2. AIDS Res Hum Retroviruses. 2000;16:1777–1781. doi:10.1089/08892220050193308.
- Bai XT, Nicot C. Overview on HTLV-1 p12, p8, p30, p13: accomplices in persistent infection and viral pathogenesis. Front Microbiol. 2012;3:400. doi:10.3389/fmicb.2012.00400.
- Patil A, Manzano M, Gottwein E. CK1alpha and IRF4 are essential and independent effectors of immunomodulatory drugs in primary effusion lymphoma. Blood. 2018;132:577–586. doi:10.1182/blood-2018-01-828418.
- Payvandi F, Wu L, Naziruddin SD, Haley M, Parton A, Schafer PH, Chen RS, Muller GW, Hughes CCW, Stirling DI. Immunomodulatory drugs (IMiDs) increase the production of IL-2 from stimulated T cells by increasing PKC-theta activation and enhancing the DNA-binding activity of AP-1 but not NF-kappaB, OCT-1, or NF-AT. J Interferon Cytokine Res. 2005;25:604–616. doi:10.1089/jir.2005.25.604.
- Gopalakrishnan R, Matta H, Tolani B, Triche T Jr., Chaudhary PM. Immunomodulatory drugs target IKZF1-IRF4-MYC axis in primary effusion lymphoma in a cereblon-dependent manner and display synergistic cytotoxicity with BRD4 inhibitors. Oncogene. 2016;35:1797–1810. doi:10.1038/onc.2015.245.
- Hagner PR, Man HW, Fontanillo C, Wang M, Couto S, Breider M, Bjorklund C, Havens CG, Lu G, Rychak E, et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood. 2015;126:779–789. doi:10.1182/blood-2015-02-628669.
- Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11:8. doi:10.1186/s13045-017-0552-6.
- Park J, Kwon M, Shin EC. Immune checkpoint inhibitors for cancer treatment. Arch Pharm Res. 2016;39:1577–1587. doi:10.1007/s12272-016-0850-5.
- Azoury SC, Straughan DM, Shukla V. Immune checkpoint inhibitors for cancer therapy: clinical efficacy and safety. Curr Cancer Drug Targets. 2015;15:452–462.
- Tsai KK, Zarzoso I, Daud AI. PD-1 and PD-L1 antibodies for melanoma. Hum Vaccin Immunother. 2014;10:3111–3116. doi:10.4161/21645515.2014.983409.
- Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–2501. doi:10.1056/NEJMc1713444.
- Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, Berry S, Chartash EK, Daud A, Fling SP, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. 2016;374:2542–2552. doi:10.1056/NEJMoa1603702.
- Galanina N, Goodman AM, Cohen PR, Frampton GM, Kurzrock R. Successful treatment of hiv-associated kaposi sarcoma with immune checkpoint blockade. Cancer Immunol Res. 2018;6:1129–1135. doi:10.1158/2326-6066.CIR-18-0121.
- Ishida T, Fujiwara H, Nosaka K, Taira N, Abe Y, Imaizumi Y, Moriuchi Y, Jo T, Ishizawa K, Tobinai K, et al. Multicenter phase ii study of lenalidomide in relapsed or recurrent adult T-Cell leukemia/lymphoma: ATLL-002. J Clin Oncol. 2016;34:4086–4093. doi:10.1200/JCO.2016.67.7732.
- Popovic M, Sarin PS, Robert-Gurroff M, Kalyanaraman VS, Mann D, Minowada J, Gallo R. Isolation and transmission of human retrovirus (human t-cell leukemia virus). Science (New York, NY. 1983;219:856–859. doi:10.1126/science.6600519.
- Hamamoto Y, Takamatsu K, Kobayashi S, Yamaguchi K, Yamamoto N, Kobayashi N. Characterization of human T-cell lines harboring defective human immunodeficiency virus type 1. Virus Genes. 1989;3:141–152.
- Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, Moore PS, Becker JC. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84:7064–7072. doi:10.1128/JVI.02400-09.
- Leonard JH, Dash P, Holland P, Kearsley JH, Bell JR. Characterisation of four merkel cell carcinoma adherent cell lines. Int J Cancer. 1995;60:100–107.
- Yodoi J, Teshigawara K, Nikaido T, Fukui K, Noma T, Honjo T, Takigawa M, Sasaki M, Minato N, Tsudo M. TCGF (IL 2)-receptor inducing factor(s). I. regulation of IL 2 receptor on a natural killer-like cell line (YT cells). J Immunol. 1985;134:1623–1630.
- Tagawa T, Albanese M, Bouvet M, Moosmann A, Mautner J, Heissmeyer V, Zielinski C, Lutter D, Hoser J, Hastreiter M, et al. Epstein-barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J Exp Med. 2016;213:2065–2080. doi:10.1084/jem.20160248.
- Steinbruck L, Gustems M, Medele S, Schulz TF, Lutter D, Hammerschmidt W. K1 and K15 of kaposi’s sarcoma-associated herpesvirus are partial functional homologues of latent membrane protein 2A of epstein-barr virus. J Virol. 2015;89:7248–7261. doi:10.1128/JVI.00839-15.
- Hoppner M, Luhm J, Schlenke P, Koritke P, Frohn C. A flow-cytometry based cytotoxicity assay using stained effector cells in combination with native target cells. J Immunol Methods. 2002;267:157–163.