
ABSTRACT
Immunotherapies against solid tumors face additional challenges compared with hematological cancers. In solid tumors, immune cells and antibodies need to extravasate from vasculature, find the tumor, and migrate through a dense mass of cells. These multiple steps pose significant obstacles for solid tumor immunotherapy and their study has remained difficult using classic in vitro models based on Petri dishes. In this work, a microfluidic model has been developed to study natural killer cell response. The model includes a 3D breast cancer spheroid in a 3D extracellular matrix, and two flanking lumens lined with endothelial cells, replicating key structures and components during the immune response. Natural Killer cells and antibodies targeting the tumor cells were either embedded in the matrix or perfused through the lateral blood vessels. Antibodies that were perfused through the lateral lumens extravasated out of the blood vessels and rapidly diffused through the matrix. However, tumor cell-cell junctions hindered antibody penetration within the spheroid. On the other hand, natural killer cells were able to detect the presence of the tumor spheroid several hundreds of microns away and penetrate the spheroid faster than the antibodies. Once inside the spheroid, natural killer cells were able to destroy tumor cells at the spheroid periphery and, importantly, also at the innermost layers. Finally, the combination of antibody-cytokine conjugates and natural killer cells led to an enhanced cytotoxicity located mostly at the spheroid periphery. Overall, these results demonstrate the utility of the model for informing immunotherapy of solid tumors.
Introduction
In recent years, there has been a growing body of literature demonstrating the great potential of the immune system in fighting cancer.Citation1−Citation4 Some immunotherapies are based on modulating the immune system (i.e. immunomodulation), injecting exogenous molecules like cytokines (e.g. IL-2) or antibodies targeting the tumor-immune cell interaction (e.g. PD-1/PD-L1 or HER-2).Citation5 Other immunotherapies rely on the direct injection of naïve or engineered immune cells into the bloodstream (cell-based immunotherapy) to engage and destroy the tumor cells. Most of the cell-based immunotherapies rely on cytotoxic T cells (also known as CD8+ T cells) and natural killer (NK) cells given their cytotoxic capacity.Citation6,Citation7 Both cell types destroy tumor cells by opening pores in the tumor cell membranes (by secreting perforins) or secreting different apoptosis-inducing proteins (e.g. granzymes, TRAIL).Citation8 Additionally, NK cells express antibody receptors (i.e. CD16), allowing them to recognize antibody-coated tumor cells as a target in a process known as antibody-dependent cell cytotoxicity (ADCC).Citation9,Citation10 Therefore, antibodies targeting molecules that are overexpressed in tumor cells are broadly used in the clinic (e.g. trastuzumab, cetuximab, pembrolizumab), combining both immunomodulation and cell-based immunotherapy for optimal results.Citation9
Although these approaches have worked well for hematological cancers, their success in treating solid tumors has been modest. Multiple studies have highlighted different factors that may dampen the immune response in solid tumors.Citation11 In this context, during the development of a solid tumor, immune cells and antibodies need to extravasate from the blood vessels; diffuse and migrate through the matrix; penetrate through the solid tumor; and finally destroy the tumor cells.Citation12 However, solid tumors are dense structures that hinder drug penetration and cell migration; raising the question about whether NK cells and antibodies can reach the innermost layers of the tumor. Multiple studies have shown the presence of NK cells in solid tumors (e.g. melanoma, colon and breast cancer).Citation13-Citation16 However, the number of NK cells and their penetration capacity in the tumor is very heterogeneous among patients. Furthermore, different NK cell populations have been observed in solid tumors (e.g. CD56 bright vs CD56 dim NK cells), showing a positive or negative correlation with patient outcome depending on the specific NK cell population observed.Citation16 In addition, solid tumors generate a very harsh microenvironment characterized by hypoxia, nutrient starvation, waste product accumulation and pH gradients.Citation17,Citation18 These factors affect NK cell cytotoxic capacity as well as tumor cell sensitivity to NK cells.Citation19 More specifically, hypoxia has been shown to reduce the expression of activating receptors on NK cell surface (e.g. NKG2D, NKp46, NKp30 and NKp44).Citation20,Citation21 Likewise, hypoxia also downregulates the expression of NK cell ligands on the tumor cell membrane (e.g. MICA); rendering the tumor cells invisible to NK cells.Citation22 Interestingly, hypoxia does not affect the ADCC response; CD16 expression levels remain constant under hypoxic conditions and NK cells continue destroying tumor cells if they are coated with antibodies.Citation21 However, recent evidence suggests tumor cells have the potential to remove the antibody ligands as a camouflage mechanism.Citation23 Finally, NK cells can be genetically engineered to thrive in the tumor microenvironment.Citation24 Some studies are transfecting NK cells with tumor-associated chemokine receptors, growth factors (e.g. IL-2) or modified receptors to make them more cytotoxic.Citation24-Citation28 In conclusion, the immune-tumor interaction turns into an extremely complex process, where the interplay between tumor, immune cells, and tumor microenvironment (TME) will determine whether immunotherapy will be successful. Therefore, relevant models are needed to mechanistically study these interactions and inform the most practical strategies.
Unfortunately, classical in vitro models to study immune cytotoxicity and ADCC rely on 2D culture on Petri dishes, where the 3D structure and microenvironment of the solid tumor is completely lost. In order to improve the efficacy of immunotherapies, there is an urgent need for models that can reliably mimic the 3D structure and complexity of solid tumors. In this context, microfluidics offers great potential to mimic physiological structures as well as the TME.Citation29-Citation31 Different microfluidic models have been used to recreate the tumor microenvironment and key processes including tumor-induced angiogenesis during cancer metastasis.Citation32-Citation35 Recently, some models have been proposed to study the interaction between immune cells and solid tumors; focusing on the effect of hypoxia on immune migration or T cell receptor modification.Citation36-Citation38 In this work, we present a microfluidic model to study NK cell immunotherapies and ADCC. Breast cancer cells (i.e. MCF7) were grown as spheroids and embedded in a collagen hydrogel. Two flanking lateral lumens were seeded with endothelial cells, and culture media was perfused through them in order to mimic blood vessels. NK cells alone or in combination with modified antibodies were included in the model to study NK cell migration, cytotoxicity, and ADCC in a complex 3D structure. Using the model, we observed that antibody penetration into the spheroid is hindered by cell-cell junctions and tumor cells were able to endocytose the antibodies in intracellular lipid vesicles. NK-92 cells exhibited a chemotactic migration towards the spheroid and penetrated into the tumor within a few hours. Finally, ADCC-induced cytotoxicity was limited to the spheroid surface, probably because of the limited antibody penetration into the tumor.
Results
Development of the multi-component microfluidic model for tumor-NK cell interaction
In order to evaluate NK cell cytotoxicity and ADCC, a microfluidic model was developed ()). The model included a 3D hydrogel with two lateral lumens coated with endothelial cells (i.e. HUVECs), mimicking the vasculature ()). MCF7 cells were grown in hanging drops to generate tumor spheroids and they were embedded in the hydrogel alone or in combination with human NK cells (i.e. NK-92 or the CD16-positive NK-92 variant, named NK-92.CD16V) ()). MCF7 spheroids showed a hypoxic core that triggered a hypoxia response in the cancer cells () and Supporting Figures 1 and 2). Finally, antibodies were perfused through the lateral blood vessels or directly embedded in the hydrogel to study antibody dynamics and their effect on NK cytotoxic capacity.
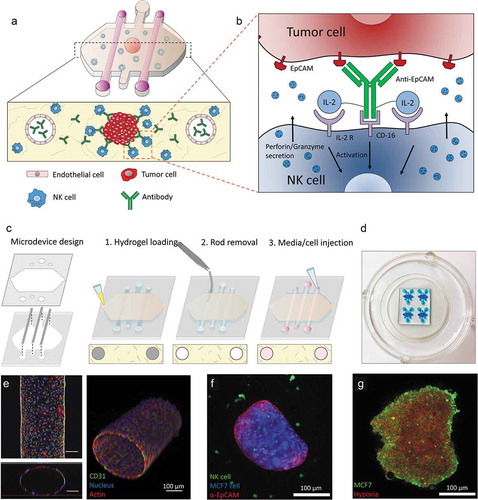
Figure 1. Conceptual scheme. (a) A microfluidic device was fabricated to study ADCC in NK cells. Collagen hydrogel is injected in the microdevice chamber with a tumor spheroid. Two flanking lateral lumens can be covered with endothelial cells to mimic blood vessels. Finally, NK cells and/or antibodies can be embedded in the hydrogel or perfused through the lateral lumens. (b) Immunocytokines are modified antibodies that are coupled to other co-stimulating molecules (e.g. IL-2) in order to enhance NK cell cytotoxicity. (c) Microdevice fabrication scheme. (d) Image of a microdevice array. The microdevices were filled with a blue dye for visualization purposes. (e) Confocal image of the HUVEC-coated lateral lumen. HUVECs were fixed and stained with α-CD31 (labelling cell membrane), phalloidin (actin) and Hoechst (nucleus). Lower panel: orthogonal cross-sectional view. Right panel: 3D reconstruction. (f) Confocal image of a MCF7 spheroid (blue) coated with an antibody targeting EpCAM (red) molecules on the tumor cell surface and NK-92 cells (green) in the collagen hydrogel. (g) The MCF7 spheroid (in green) was cultured in the presence of a hypoxia-sensing dye (in red), showing a more intense signal at the spheroid core.
Antibody dynamics: extravasation, diffusion and penetration
Once the microfluidic model was prepared, we set out to study the penetration capacity of therapeutic antibodies and the role of the vasculature. An empty collagen hydrogel (i.e. no tumor, endothelial nor NK cells embedded) was polymerized within the microdevice. After rod removal, a fluorescently-labeled anti-EpCAM antibody was perfused through one of the lateral lumens. Antibody penetration was evaluated using time-lapse microscopy ()). The results demonstrated that the antibody easily penetrated through the collagen hydrogel, reaching a linear profile after 4 hours. In another experiment, a MCF7 spheroid was included in the hydrogel. The time-lapse results showed the antibody reached the sphere after 4 hours and attached to the sphere ()). The antibody coating around the sphere was asymmetric, i.e. the spheroid half closer to the perfused lumen was more intensely labeled compared with the other half. Finally, when the lumen was covered with endothelial cells (i.e. HUVECs), the antibody penetration was delayed; demonstrating the barrier effect exerted by endothelial cells ()).
Figure 2. Antibody diffusion. MCF7 cells were labeled with cell tracker green and grown as 1000 cells/spheroids. MCF7 spheroids were embedded in a collagen hydrogel within the microdevices. After 24 hours in culture, a fluorescent antibody (anti-EpCAM) was perfused through one of the lateral lumens to study antibody penetration. (a) In the absence of the MCF7 sphere, the antibody penetrated through the matrix and after 4 hours the diffusion profile exhibited an evident linear gradient. (b) When the MCF7 spheroid was present, the antibody attached to the spheroid periphery. (c) the presence of endothelial cells coating the lumen wall delayed the antibody penetration. (d) Higher magnification of the condition shown in (b). Penetration of the antibody within the spheroid was much slower compared with its diffusion through the hydrogel. After 1 day, the antibody remained at the periphery of the sphere and even after 3 days the core was not labeled. MCF7 cells in the spheroid were stained with cell tracker green to discard light scattering was completely blocking the light coming from the spheroid core. (e) Graphs show the diffusion profile across the delimited region (yellow rectangle). (f) MCF7 spheroids were stained with the fluorescent lipid DiD and labelled with the antibody (anti-EpCAM in red) for 3 days. Confocal images at higher magnification revealed after 3 days some cells had internalized the antibody in intracellular vesicles.
Given antibody penetration within solid tumors remains poorly understood, we decided to evaluate its penetration within the spheroid. Four hours post-antibody injection, the antibody was clearly attached to those cells forming the outer spheroid layer. As mentioned above, the side of the spheroid closer to the perfused lumen showed a higher antibody fluorescence. However, after 24 hours, this gradient across the surface disappeared and the surface of the spheroid was homogeneously labeled (). Interestingly, antibody penetration within the spheroid showed no change; suggesting the antibody penetration was severely hindered by the tumor cell-cell junctions that maintain the spheroid together. When the antibody profile was analyzed after 3 days, only a moderate penetration within the sphere could be observed. Tumor cells were labeled with cell tracker prior spheroid formation in order to demonstrate that fluorescence coming from the center of the sphere could be still detected despite the high cell density within the spheroid (). Additionally, the fact that cells were still labeled within the core of the sphere after 3 days suggested there was not a necrotic core that could explain the absence of antibody signal at the core. Interestingly, after 3 days, antibody-labeled vesicles were observed within some tumor cells (). MCF7 cells were labeled with a fluorescent lipid before culturing them in the hanging drop, revealing these antibody-positive vesicles were made of lipids coming from the cell membrane. This observation indicated MCF7 may endocytose these antibodies to remain hidden from immune surveillance. MCF10A cells (non-cancerous breast cells) were also grown as spheroids and a similar experiment was performed to explore whether these observations were specific of MCF7 cells (Supporting Figure 1). The results showed that MCF10A spheroid also generated a barrier effect, delaying antibody penetration. However, no internalization in intracellular vesicles was observed. These results suggested that spheroids generated a barrier effect regardless the specific cell type. However, the capacity to endocytose the antibody into membrane vesicles seemed to depend on the specific cell type; which could inform the type of therapy used for each specific tumor depending on the behavior observed in the model.
NK cell dynamics: migration and tumor penetration
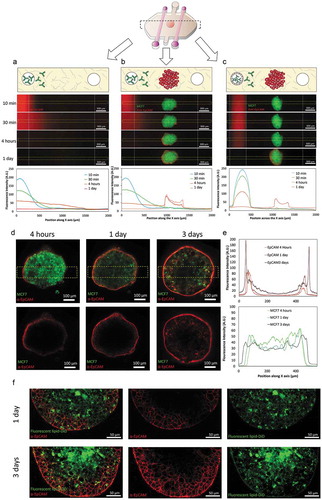
After evaluating the penetration of antibodies, the migration of NK cells, as well as their capacity to find a tumor mass in a 3D environment, was evaluated. NK and tumor cells were labeled in different colors and cell migration was monitored. During the 5 hours time-lapse experiments, NK cells exhibited an intense directional migration towards the spheroid ( and Supporting Movie 1). NK cells penetrated into the sphere at different focal planes during the course of the experiment (, Supporting Figure 4 and Supporting Movie 2). Interestingly, this directional migration was more intense on those NK cells located closer to the sphere (), suggesting NK cell migration was guided by a chemotactic signal secreted by the tumor spheroid. In the MCF7 spheroids, the cell-cell junctions were tight enough to hinder the penetration of antibodies (molecular weight 150 kDa). However, the time-lapse experiment showed NK cells, which are much bigger than antibodies; were able penetrate the spheroid in a matter of hours. A time-lapse experiment at higher magnification (40X) ( and Supporting movie 3) revealed NK cells were able to penetrate the spheroid squeezing the cell body through the tumor cell-cell junctions.
Figure 3. NK-92 directional migration towards MCF7 spheroid. (a) 5 hours time-lapse experiment showing NK-92 (labeled in green) migration towards the MCF7 spheroid (labeled in red). (b) Rose diagram shows the directionality of the NK-92 cell trajectories. (c) Graph indicating the area within the MCF7 cell spheroid occupied by NK-92 cells. NK-92 cell penetration into the spheroid followed a linear dynamic during the experiment. (d) Chemotaxis analysis of a sub-section of the images showed in “A”. The field of view was divided in three regions: distal (I), central (II) and proximal (III) and NK cell migration was analyzed. (e) The rose diagrams indicate the different chemotaxis observed in the three different regions (I, II and III), demonstrating the chemotactic response was more intense the closer to the spheroid the cells were located. (f) NK-92 cells were able to migrate within the sphere using an ameboid movement (white arrow), squeezing through the spaces between tumor cells. (g) The expression of different chemokines secreted by the MCF7 spheroid was analyzed by q-PCR. The expression of multiple chemokine receptors was also analyzed in NK-92 cells. Gene expression was normalized to the reference genes. Chemokine-Receptor matching pairs found in the system are shown in the table.
Next, we set out to study MCF7 spheroid-NK cell communication. MCF7 spheroids and NK cells were cultured individually in separated microdevices and after 24 hours the chemokine secretion as well as the chemokine receptor expression was analyzed by q-PCR in MCF7 spheroids and NK cells respectively ()). The results showed NK cells expressed multiple receptors for different chemokines secreted by the MCF7 spheroid (e.g. CXCL12-CXCR4, CCL28-CCR3, CXCL8-CXCR2 and CXCR1, CXCL5-CXCR2); supporting the hypothesis of a chemotaxis-guided migration.
Cytotoxicity and ADCC in 2D
In order to study ADCC, NK cytotoxicity was tested in a 2D environment against the target cells. MCF7 cells were cultured in 2D on a flat Petri dish and NK-92 cells were added on top 24 later. MCF7 cell viability was evaluated 3 days later, showing a significant reduction in the amount of viable MCF7 cells. NK cell cytotoxicity was more intense as the NK: MCF7 ratio was increased (Supporting Figure 5A). The specificity of NK cell cytotoxicity against non-cancer cells was tested co-culturing NK cells with HUVECs (Supporting Figure 5B). After 3 days in co-culture, HUVECs viability was evaluated, showing a moderate toxicity compared with the MCF7 results (50% viability reduction in HUVECs vs more than 95% in MCF7) at the highest NK ratio. This off-target behavior could be explained by the graft-versus-host effect, where immune cells from one donor can react against normal cells from different donor with a mismatching haplotype.
ADCC requires the expression of antibody receptors (CD16) in NK cells and NK-92 cells lack this receptor. Therefore, for ADCC experiments, the CD16-positive NK-92 cell variant (i.e. NK-92.CD16V) was used (Supporting Figure 6). 24 hours after seeding MCF7, the immunocytokine (i.e. IL2-conjugated anti-EpCAM antibody) was added. 4 hours later, NK-92.CD16V cells were added on top and cell viability was evaluated after only 16 hours to minimize the potential effect of the antibody-bound IL-2 on NK cell proliferation; which could alter the NK: MCF7 ratio. The addition of the immunocytokine significantly increased NK cell cytotoxicity in 2D assays (Supporting Figure 7), validating the NK-92.CD16V cells and the immunocytokine for the 3D experiments within the microdevice.
ADCC in 3D
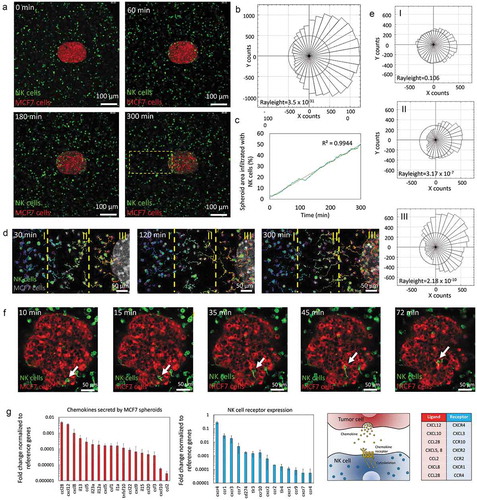
As commented in the introduction, NK cytotoxicity in 2D can be very different compared with a more complex 3D system. Thus, the NK-92 cytotoxicity against the MCF7 spheroid was assessed. MCF7 cells were labeled with cell tracker green prior to forming the sphere and then were co-cultured with NK-92 in 3D collagen hydrogels at different ratios. After 3 days, PI was added, and viable and dead cells were visualized by confocal microscopy. In the absence of NK cells, the MCF7 spheroid remained viable (cell viability greater than 95%) ()). The presence of NK cells induced MCF7 mortality in a dose-response manner. However, at lower NK cell ratios (i.e. 3 NK: 1 MCF7) cytotoxicity was not homogenous across the spheroid; in fact, the most intense cytotoxicity was located at the outer layers of the spheroid as well as the invading tumor cells ()). On the other hand, the innermost regions of the sphere remained viable and unaffected by the presence of the NK-92 cells. Interestingly, at a higher ratio (i.e. 15 NK: 1 MCF7), NK cell-induced cytotoxicity was observed throughout the entire spheroid () and Supporting Figure 8). This observation raised the question whether NK cells needed to destroy the outer layer of the tumor before killing the inner regions. Interestingly, a high magnification time-lapse experiment (40X magnification) revealed NK cells were able to directly penetrate the spheroid and destroy multiple cells located even at the spheroid core in a matter of few hours () and Supporting Movie 4).
Figure 4. 3D ADCC. 1000 green-labeled MCF7 cells spheroids were embedded in a 4.5 collagen hydrogel and co-cultured in the absence/presence of NK-92 cells at different ratios. After 3 days in culture, dead cells were stained, and viability was evaluated. (a) When the spheroids were cultured in the absence of NK cells, the spheroid showed a high viability (>95%). (b) When NK cells were added at 3:1 ratio, a clear necrotic perimeter appeared surrounding the sphere, whereas the intermediate region and the core remained viable. (c) At 15:1 ratio, tumor necrosis appeared across all the spheroid area. (d) Time-lapse experiment at 15:1 ratio showing NK-92 killing within the spheroid. The images show an NK cell patrolling around the core of the spheroid and inducing apoptosis on the surrounding tumor cells. (e) MCF7 spheroids were embedded in the collagen hydrogel in the presence of the immunocytokine. After 16 hours in culture, cell viability was evaluated. (f) NK-92.CD16V at a 15:1 ratio showed a moderate cytotoxicity after 16 hours in co-culture. (g) The combination of the immunocytokine with NK-92.CD16V cells lead to higher cytotoxicity, demonstrating ADCC in the 3D model. The graph shows the area covered by dead cells in the different conditions. Scale bar is 250 µm.
Finally, we assessed ADCC in the 3D model using the NK-92.CD16V cells and the immunocytokine (i.e. anti-EpCAM antibody with IL-2 molecules attached). In order to avoid undesired gradients of the immunocytokine across the hydrogel, it was added directly to the collagen mixture before hydrogel polymerization. As mentioned above, IL-2 can also induce NK cell proliferation; hence we analyzed NK cell cytotoxicity after only 16 hours in culture to minimize any possible increase in NK cell proliferation. Here, we found that NK cell-mediated cytotoxicity was significantly increased in the presence of the immunocytokine, leading to increased tumor apoptosis. ()). Interestingly, NK cell cytotoxicity was located mostly at the spheroid periphery, which agrees with our previous observation that after 24 hours antibodies remained at the first cell layers of the spheroid.
Discussion
Immunotherapies are arising as very promising tools against cancer. However, the immune cells face daunting challenges in the TME of solid tumors. Therefore, models like the one presented here could be extremely valuable tools to improve immunotherapies against solid tumors. Using this model, we have seen that antibody dynamics are a complex process where endothelial permeability, tumor penetration and antibody clearance by tumor cells are deeply involved. Recent therapies are targeting the tumor shedding mechanism that remove antibody ligands from the tumor cell surface, enhancing the effect of therapeutic antibodies.Citation23 In this context this model could be applied to evaluate the efficiency of this strategy. Regarding immune homing and migration, the model revealed NK cells are capable of detecting the presence of the tumor several hundreds of microns away. The q-PCR suggested some potential chemokine-receptor pairs that could explain this chemotactic process. However, those chemokines or factors secreted by the tumor whose receptor was not detected in the NK cells could be more important; since we could engineer NK cells to express those receptors, making them more efficient tumor hunters.Citation24 Penetration into solid tumors has been also a controversial topic. Although NK cells are present in solid tumors, the mechanisms they use to navigate through the tumor and how they survive within the harsh tumor microenvironment remain poorly understood.Citation39,Citation40 In this work, we saw NK cells can penetrate the tumor spheroid remarkably fast compared with antibodies, and NK cells were able to move through the tumor cell-cell junctions. Using this model, the machinery employed by NK cell to open the cell-cell spaces could be studied; generating new potential NK cell modifications that improve tumor penetration.Citation24 Finally, some studies are addressing whether NK cell cytotoxicity is affected within the solid tumors due to hypoxia or acidic pH, as well as a continuous activation of the NK cells.Citation19 In this paper, NK cells within the spheroid remained capable of killing tumor cells; suggesting any possible NK cell exhaustion is not immediate, but a slow process. In future studies, NK cell density could be reduced in order to maintain a low NK: tumor cell ratio, keeping most of the tumor spheroid viable for a long period of time. This configuration would allow the study of NK cell exhaustion within the solid tumor; once again enabling the identification of potential therapeutic targets to improve NK cytotoxicity. Cancer immune response is a very complex process where many different cell types are involved (e.g., tumor-associated macrophages, tumor-associated fibroblasts, Myeloid-derived suppressor cells, T regs or T effector cells, etc.).Citation41 Thus, these other cell types could be included in the model (e.g., stromal cells could be embedded in the matrix, randomly mixed into the spheroid with cancer cells, or even an organized multi-layer spheroid could be generated to mimic the stromal barrier) to study different aspects of the cancer immune response. In conclusion, the presented microfluidic model could be used to identify new therapeutic approaches to improve immunotherapy against solid tumors. Traditionally, microfluidic models have been developed and used mainly in engineer labs. Commonly, these models required an in-depth expertise and sophisticated equipment to operate (e.g. clean-room facilities, pumps, sensors, tubing), making them less appealing to conventional cellular and molecular biology labs.Citation29,Citation42 Here we presented a model that does not require clean room processing and can be operated with no other additional equipment rather than micropipettes. Currently, most of these models, like the one presented here, are fabricated in labs at research institutions. However, there is an increasing effort to generate simple models that could be mass-produced like any other traditional labware (e.g. well-plates, Petri dishes). We think these simpler models could bridge the gap between engineer and biology labs and finally implement microfluidic models in the biomedical research community.
Materials and methods
Reagents
The Natural Killer cell line NK-92 was purchased from the ATCC and cultured in X-VIVO 10 (Lonza, 04-380Q) supplemented with 10% fetal bovine serum (FBS), 10% Horse serum, 0.02 mM folic acid (Sigma, F8758) dissolved in 1 N NaOH, 0.2 mM myo-inositol (Sigma, I7508) and 100 units/ml IL-2 (Peprotech, 200-02). NK-92 show cytotoxic capacity in vitro and has been used in multiple studies and clinical trials.Citation43-Citation46 However, NK-92 cells are negative for CD16, therefore a variant transformed with CD16 high affinity (NK-92.CD16V), kindly provided by Nantkwest®, was also used in this work and cultured in the same media as the parental NK-92 cell line.
The breast cancer cell line MCF7 was purchased from the ATCC and cultured in 4.5 mg/ml glucose DMEM (Lonza, 12-604F) supplemented with 10% FBS. HUVECs (Lonza) were grown in EGM-2. (Lonza, CC-3162) and only cells below 8 passages were used.
Microdevice design and fabrication
Microdevice fabrication is described in more detail in.Citation33 Briefly, the template was designed in illustrator and fabricated using SU-8 based lithography. Next, PDMS-based microdevices were fabricated using the SU-8 template and the microdevices were treated with oxygen plasma and bonded to a 60mm glass bottom Petri dish. Microdevices were sterilized by UV exposure for 15 min prior to cell culture. The final microdevice was comprised of a central microchamber to inject a 3D hydrogel and three parallel 340 µm-diameter PDMS rods. To increase hydrogel attachment to the PDMS, the microdevices were treated 10 min with poly(ethyleneimine) (Sigma-Aldrich, 03880) diluted at 1%; followed by 30 min with glutaraldehyde (Sigma-Aldrich, G6257) diluted at 0.1% in water; and finally washed three times with water.
Cell culture
Tumor spheroids were generated by the hanging drop method. Briefly, MCF7 cells were trypsinized and resuspended at 40 cells/ml in media supplemented with 20% 12g/l methylcellulose dissolved in media. 25 µl droplets were placed on top of a Petri dish lid and distilled water was added to the bottom of the dish to reduce evaporation during the spheroid formation. After 2 days in the incubator, one single spheroid per droplet was formed.
In order to embed the spheroid/NK cells in the microdevice, a 4.0 mg/ml collagen hydrogel was prepared and injected within the central microchamber. The hydrogel was prepared as follows: 5 µl of 10X PBS, 1.12 µl of 1N NaOH; 45 µl of 8.90 mg/ml collagen type I; and 48.88 µl of culture media w/wo cells depending on the experiment. Collagen was polymerized at room temperature for 20 min to allow the formation of longer collagen fibers. Next, PDMS rods were removed, generating a cylindrical tunnel through the collagen hydrogel (i.e. lumen). Endothelial cells (HUVECs) were injected through the lateral lumens at 20 million cells/ml to mimic the vasculature. Depending on the experiment, NK-92 cells at different concentrations and/or antibodies were embedded in the matrix or perfused through the lateral blood.
HRE-GFP transfection
5HRE/GFP was a gift from Martin Brown & Thomas Foster (Addgene, plasmid # 46926). The plasmid contains a neomycin-resistance gene and the GFP gene controlled by the minimal promotor of the cytomegalovirus (CMV) including five copies of the VEGF hypoxia response element (HRE). The plasmid was purified using the QIAprep Spin Miniprep Kit (Quiagen, 27104) and transfected in MCF7 using Xfect transfection reagent (Takara, 631317). Briefly, 5 µg of the plasmid was combined with 1.5 µl of Xfect transfection reagent in 88.5 µl of Xfect transfection buffer. The mixture was incubated 10 min at room temperature and then added to a 6 well-plate containing 5 × 105MCF7. Cells were incubated for 4 hours with the mixture and the washed twice and left in the incubator for 48 hours. Media was then supplemented with 750 ng/ml G418, which killed all the non-transfected cells. After two weeks in selection media, only transfected cells were observed.
Cell staining
MCF7 cells were stained with the infrared-fluorescent lipid DiD (Thermo Fisher, V22889) following supplier instructions prior culturing the cells in the hanging drop. NK-92 or NK-92.CD16V cells were stained in red with the fluorescent lipid Dil (Thermo Fisher, V22889) in PBS for 10 min. Cell apoptosis was detected using the green-fluorescent CellEVENT Caspase-3/7 detection reagent (Thermo Fisher, C10423).
Flow cytometry
NK92 and NK92-176V cells were collected and resuspended in cold PBS supplemented with 1% FBS and 1ug/mL mouse IGG and incubated for 5 minutes before incubating with CD16-PE (clone 3G8; BD Biosciences) at 4°C for 30 minutes. The stained cells were then washed and resuspended in PBS/1% FBS, DAPI was added, and data were acquired on a Thermo Fisher Attune NxT cytometer.
Diffusion assays
MCF7 cells were stained with cell tracker green or blue following supplier instructions. Anti-EpCAM conjugated to Phycoerythryn (PE anti-EpCAM) (AbCAM, VU-1 D9) was used at 1:100 to evaluate the diffusion and penetration of therapeutic antibodies. For antibody diffusion experiments, the microdevices were placed in a IX81 fluorescence microscope with a cell incubation system and diffusion was visualized at different time points.
mRNA extraction, cDNA retrotranscription and q-PCR
To study the chemotactic response, the expression of a panel of chemokines and chemokine receptors was analyzed by q-PCR following supplier instructions (Qiagen, PAHS-181Z). Briefly, NK-92 cells or MCF7 spheroids were cultured individually in the microdevices. mRNA was extracted after 24 hours a Dynabeads™ mRNA DIRECT™ Purification Kit (Thermo Fisher, 61011). mRNA was retrotranscribed to cDNA using the RT2 First strand kit (Qiagen, 330401). cDNA was analyzed for q-PCR and data was analyzed using the Qiagen online software (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php).
2D and 3D cytotoxicity assays
NK-92 cell cytotoxicity in 2D was evaluated co-culturing them with MCF7 cells and HUVECs. 7000 MCF7 cells or HUVECs were plated and 24 hours later NK cells were added. After the co-culture, NK-92 cells and dead floating MCF7 cells or HUVECs were washed and viable attached cells were stained with 10 µM calcein acetomethyl ester (CAM) (Thermo Fisher, C3100MP). For 3D experiments, MCF7 cells were stained with cell tracker green before culturing them in hanging drops as described above. Once the spheroid was completely formed, MCF7 spheres were embedded in a collagen hydrogel in the presence of NK-92 cells. After the co-culture, 4 µg/ml propidium iodide (PI) (Thermo Fisher, P1304MP) was added to the system and 30 minutes later cell viability was evaluated by fluorescence/confocal microscopy.
Anti-EpCAM IL-2 fusion protein preparation
The anti-EpCAM immunocytokine, IC65, was constructed as reported earlier.Citation47 The variable regions of the anti-EpCAM antibody, C215 were used to construct the IC65 immunocytokine.Citation48
Confocal microscopy and image analysis
Cells were visualized in a Leica SP8 3X STED super-resolution confocal microscope equipped with a super-continuum white-light laser for fluorescent excitation from 470nm to 670nm, a separate 405nm diode laser. The unit is equipped with 3 PMTs and 2 high-sensitive HyD detectors for image collection. For live-cell experiments, a stage top incubator set at 37°C and 5% CO2 was used. For NK-92 chemotaxis and cytotoxicity experiments images were taken every 30 seconds during 5 hours at a single focal plane. For antibody diffusion experiments, images were taken every 5 min. Z-stacks were perform using a 1 and 10µm step at 40X and 10X respectively. NK-92 cell migration was analyzed using an automatic cell tracking plugin (TrackMate) running on Fiji® (https://fiji.sc/). A Laplacian of Gaussian filter was applied to identify the NK-92 cells and cell estimated size and threshold were set at 10µm and 0.9 respectively.
Statistics
All experiments were repeated at least three times. Data were analyzed using Graphpad software, and statistical significance was set at p-value < 0.05. Results are presented as mean ± standard error. The normal distribution was tested by the Kolmogorov-Smirnov test. For parametric comparisons, one-way ANOVA with Bonferroni post hoc tests were performed. For nonparametric comparisons, a Kruskal-Wallis test was performed followed by the U test of Mann-Whitney.
Conflict of interests
David J. Beebe is a board member and stockowner of Tasso, Inc. and a stockowner of Bellbrook Labs, LLC. David J. Beebe is a founder, stockowner, and consultant of Salus Discovery LLC. David J. Beebe is an advisor and stockowner of Lynx Biosciences, LLC, Onexio Biosystems, LLC, and Stacks to the Future, LLC. David J. Beebe holds equity in Bellbrook Labs, LLC, Tasso Inc., Salus Discovery LLC, Stacks to the Future, LLC and Onexio Biosystems, LLC.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (66.7 MB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10:94. doi:10.1186/s13045-017-0453-8.
- Jeanbart L, Swartz MA. Engineering opportunities in cancer immunotherapy. Proc Natl Acad Sci USA. 2015;112:14467–14472. doi:10.1073/pnas.1508516112.
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi:10.1126/science.aar6711.
- Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–1360. doi:10.1126/science.aar7112.
- Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi:10.1126/science.aar4060.
- Stagg J, Smyth MJ. NK cell-based cancer immunotherapy. Drug News Perspect. 2007;20:155–163. doi:10.1358/dnp.2007.20.3.1092096.
- Ye B, Stary CM, Gao Q, Wang Q, Zeng Z, Jian Z, Gu L, Xiong X. Genetically modified T-Cell-Based adoptive immunotherapy in hematological malignancies. J Immunol Res. 2017;2017:5210459. doi:10.1155/2017/5974574.
- Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.01124.
- Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, Levy R, Brody J. Combination strategies to enhance antitumor ADCC. Immunotherapy-Uk. 2012;4:511–527. doi:10.2217/imt.12.38.
- Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6. doi:10.3389/fimmu.2015.00368.
- Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27:1482–1492. doi:10.1093/annonc/mdw168.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi:10.1016/j.immuni.2013.07.012.
- Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci USA. 2017;114:E9271–E9. doi:10.1073/pnas.1703921114.
- Sandel MH, Speetjens FM, Menon AG, Albertsson PA, Basse PH, Hokland M, Nagelkerke JF, Tollenaar RAEM, van de Velde CJH, Kuppen PJK. Natural killer cells infiltrating colorectal cancer and MHC class I expression. Mol Immunol. 2005;42:541–546. doi:10.1016/j.molimm.2004.07.039.
- Wang B, Wang Q, Wang Z, Jiang J, Yu S-C, Ping Y-F, Yang J, Xu S-L, Ye X-Z, Xu C, et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014;74:5746–5757. doi:10.1158/0008-5472.CAN-13-2563.
- Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, Rachmilewitz J. Characterization of tumor infiltrating natural killer cell subset. Oncotarget. 2015;6:13835–13843. doi:10.18632/oncotarget.3453.
- Carmona-Fontaine C, Bucci V, Akkari L, Deforet M, Joyce JA, Xavier JB. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc Natl Acad Sci USA. 2013;110:19402–19407. doi:10.1073/pnas.1311939110.
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi:10.1038/nm.3394.
- Bi JC, Tian ZG. NK Cell exhaustion. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.00760.
- Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris J-H, Noman MZ, Chouaib S. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. 2015;6. doi:10.3389/fimmu.2015.00482.
- Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, Varesio L, Moretta L, Bosco MC, Vitale M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. 2013;43:2756–2764. doi:10.1002/eji.201343448.
- Yamada N, Yamanegi K, Ohyama H, Hata M, Nakasho K, Futani H, Okamura H, Terada N. Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1 alpha-dependent manner. Int J Oncol. 2012;41:2005–2012. doi:10.3892/ijo.2012.1630.
- de Andrade LF, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, Tsoucas D, Franz B, May KF, Harvey CJ, et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science. 2018;359:1537–1542. doi:10.1126/science.aao0505.
- Carlsten M, Childs RW. Genetic manipulation of NK Cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. 2015;6:266. doi:10.3389/fimmu.2015.00266.
- Inngjerdingen M, Rolstad B, Ryan JC. Activating and inhibitory Ly49 receptors modulate NK cell chemotaxis to CXC chemokine ligand (CXCL) 10 and CXCL12. J Immunol. 2003;171:2889–2895.
- Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, Klingemann H. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53:958–965. doi:10.3109/10428194.2011.634048.
- Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol. 2008;180:6392–6401.
- Konstantinidis KV, Alici E, Aints A, Christensson B, Ljunggren H-G, Dilber MS. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp Hematol. 2005;33:159–164. doi:10.1016/j.exphem.2004.11.003.
- Sackmann EK, Fulton AL, Beebe DJ. The present and future role of microfluidics in biomedical research. Nature. 2014;507:181–189. doi:10.1038/nature13118.
- Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32:760–772. doi:10.1038/nbt.2989.
- Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi:10.1126/science.1188302.
- Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc Natl Acad Sci USA. 2015;112:214–219. doi:10.1073/pnas.1417115112.
- Jimenez-Torres JA, Peery SL, Sung KE, Beebe DJ. LumeNEXT: a practical method to pattern luminal structures in ECM gels. Adv Healthc Mater. 2016;5:198–204. doi:10.1002/adhm.201500608.
- Sobrino A, Phan DTT, Datta R, Wang X, Hachey SJ, Romero-López M, Gratton E, Lee AP, George SC, Hughes CCW. 3D microtumors in vitro supported by perfused vascular networks. Sci Rep-Uk. 2016;6.
- Kurokawa YK, Yin RT, Shang MR, Shirure VS, Moya ML, George SC. Human induced pluripotent stem cell-derived endothelial cells for three-dimensional microphysiological systems. Tissue Eng Part C-Me. 2017;23:474–484. doi:10.1089/ten.tec.2017.0133.
- Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, Miccolis C, Ceccarello E, Adriani G, Raimondi MT, et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. Jci Insight. 2017;2.
- Adriani G, Pavesi A, Tan AT, Bertoletti A, Thiery JP, Kamm RD. Microfluidic models for adoptive cell-mediated cancer immunotherapies. Drug Discov Today. 2016;21:1472–1478. doi:10.1016/j.drudis.2016.05.006.
- Christakou AE, Ohlin M, Onfelt B, Wiklund M. Ultrasonic three-dimensional on-chip cell culture for dynamic studies of tumor immune surveillance by natural killer cells. Lab Chip. 2015;15:3222–3231. doi:10.1039/c5lc00436e.
- Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and NK-Cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4:522–526. doi:10.1158/2159-8290.CD-13-0985.
- Hoogstad-van Evert JS, Cany J, van Den Brand D, Oudenampsen M, Brock R, Torensma R, Bekkers RL, Jansen JH, Massuger LF, Dolstra H. Umbilical cord blood CD34(+) progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rg(null) mice. Oncoimmunology. 2017;6:e1320630. doi:10.1080/2162402X.2017.1320630.
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–550. doi:10.1038/s41591-018-0014-x.
- Berthier E, Young EW, Beebe D. Engineers are from PDMS-land, biologists are from polystyrenia. Lab Chip. 2012;12:1224–1237. doi:10.1039/c2lc20982a.
- Klingemann HG, Miyagawa B. Purging of malignant cells from blood after short ex vivo incubation with NK-92 cells. Blood. 1996;87:4913–4914.
- Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–658.
- Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, Suttorp M, Seifried E, Ottmann OG, Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. doi:10.1016/j.jcyt.2013.06.017.
- Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–632. doi:10.1080/14653240802301872.
- Gillies SD. A new platform for constructing antibody-cytokine fusion proteins (immunocytokines) with improved biological properties and adaptable cytokine activity. Protein Eng Des Sel. 2013;26:561–569. doi:10.1093/protein/gzt045.
- Larson LN, Johansson C, Lindholm L, Holmgren J. Mouse monoclonal-antibodies for experimental immunotherapy promotes killing of tumor-cells. Int J Cancer. 1988;42:877–882.