
ABSTRACT
Eradication of tumors by the immune system relies on the efficient activation of a T-cell response. For many years, the main focus of cancer immunotherapy has been on cytotoxic CD8 T-cell. However, stimulation of CD4 helper T cells is critical for the promotion and maintenance of immune memory, thus a good vaccine should evoke a two-dimensional T-cell response. The invariant chain (Ii) is required for the MHC class II heterodimer to be correctly guided through the cell, loaded with peptide, and expressed on the surface of antigen presenting cells (APC). We previously showed that by replacing the Ii CLIP peptide by an MHC-I cancer peptide, we could efficiently load MHC-I. This prompted us to test whether longer cancer peptides could be loaded on both MHC classes and whether such peptides could be accommodated in the CLIP region of Ii. We here present data showing that expanding the CLIP replacement size leads to T-cell activation. We demonstrate by using long peptides that APCs can present peptides from the same Ii molecule on both MHC-I and -II. In addition, we present evidence that antigen presentation after Ii-loading was superior to an ER-targeted minigene construct, suggesting that ER-localization was not sufficient to obtain efficient MHC-II loading. Finally, we verified that Ii-expressing dendritic cells could prime CD4+ and CD8+ T cells from a naïve population. Taken together our study demonstrates that CLIP peptide replaced Ii constructs fulfill some of the major requirements for an efficient vector for cancer vaccination.
Introduction
Cancer immunotherapy represents an attractive strategy to defeat cancer; it relies on the activation of the patients’ own immune system and its ability to identify and eradicate neoplastic cells.1,Citation2 One approach, so-called active immunotherapy, is through vaccination of patients by the delivery of cancer antigens together with an adjuvant. The desired outcome is to evoke a tumor-specific T cell immune response in vivo. Because of their special properties as professional antigen presenting cells (APCs), dendritic cells (DCs) represent a natural vehicle for antigen delivery in therapeutic cancer vaccines.Citation3 These cells have a unique ability to efficiently process and present antigenic peptides to effector cells in the context of Major Histocompatibility Complex (MHC or HLA, Human Leukocyte Antigen, in humans) molecules. For a long time it was believed that stimulation of cytotoxic CD8+ T cells was sufficient to induce the antitumor response, and therefore, vaccination campaigns have been focused on MHC-I restricted peptides.Citation4,Citation5 The rationale behind this was that all cells, including tumor cells, are MHC-I positive, whereas MHC-II expression is restricted to APCs, and therefore MHC-I presentation should be sufficient for cancer surveillance and for cancer immunotherapy. However, several studies have shown that CD4+ Helper T cells play a critical role in the promotion and maintenance of memory CD8+ T cells, and also in tumor control,Citation6 suggesting that both CD8+ and CD4+ T cell responses should be evoked through vaccination strategies.Citation6,Citation7 Consequently, in order to obtain efficient stimulation, antigenic peptides should be loaded on both MHC-I and MHC-II molecules. Ideally, the CD8+ and CD4+ T cell responses should be directed against nested epitopes as high avidity memory responses are obtained through specific rather than bystander CD4+ T-cell help.Citation8,Citation9
The classical view of antigen loading is that endogenous proteins are degraded by the proteasome and retro-transported to the ER for MHC-I loading, whereas exogenous proteins are taken up by endocytosis and loaded on MHC-II in specialized endosomal compartments.Citation10,Citation11 In addition, alternative models have been suggested for MHC-I loading including cross-presentation pathwaysCitation12 where MHC-I are loaded with peptides from exogenous proteins. Both MHC-I and II are synthesized in the ER, but while MHC-I is normally loaded in this compartment, MHC-II is most efficiently chaperoned by Invariant Chain (Ii) to proteolytic late endosomal peptide-loading compartments where peptides generated from exogenous proteins may bind. Peptide loading of MHC-II requires an additional molecule, HLA-DM, to catalyze the process.Citation13 Methods have been proposed to improve both MHC- I and II peptide loadingCitation14,Citation15 and many of these focus on ER targeting; suggesting that when peptides are routed to the ER, both MHC molecules can be loaded.
Several studies have demonstrated that the MHC-II scaffolding partner Ii is able to load MHC-II molecules with specific antigens when the coding sequence of Class II-associated Invariant chain peptide (CLIP) region is replaced with known MHC-I or II epitopes.Citation16–Citation19 In addition, Ii and MHC-I are reported to bind each other.Citation20-Citation22 Based on this, we recently tested the use of Ii-CLIP replaced constructs to load MHC-I and exchanged Ii-CLIP peptide with validated Class I epitopes for which we possess the cognate T cell receptors (TCRs). We presented evidence that MHC-I could be loaded by modified Ii and further demonstrated that presentation through Ii could evoke specific activation of both memory and naïve CTLs when the modified li was expressed in DCs.Citation23 We showed that the use of CLIP-exchanged Ii constructs could be a straightforward way of loading antigenic peptides without using the classical loading machinery necessary for MHC-I presentation. Thereby, Ii-loading could avoid proteasome cleavage, ER retro-translocation or any of the more complicated cross-presentation mechanisms.
In the present study, the aim was to extend the spectrum of Ii-based loading peptides for MHC-I and, in addition, test simultaneous loading of MHC-II. We used a previously validated antigenic peptide from the transforming growth factor b receptor II (TGFbRII) frameshift mutant.Citation24 We took advantage of the following points: (i) the peptide had previously been used in the clinic, (ii) from immunomonitoring studies we had isolated both Class-I and Class-II restricted TCRs specific for overlapping peptides of the vaccination peptideCitation25 and unpublished data. We here show that CLIP replacement with a long peptide indeed enabled peptide presentation on both MHC types in the same type of APCs. We further screened for the size of the CLIP-replacement insert and demonstrated that a replacement of up to 34 amino acids was tolerated for antigen loading. Interestingly, the fusion of the full-length frameshift TGFbRII to Invariant chain was retained in the ER and not presented on MHC-II, supporting the advantage of an invariant chain-based CLIP-oriented design over a simple ER-targeting one. We found that all our invariant chain constructs resided in the ER, but only those that were transported to the endosomal pathway could load both MHC-I and II suggesting that ER targeting is not sufficient for proper MHC loading. Finally, we show that not only model cells but also DCs could be loaded by the CLIP replaced Ii-vector and stimulate both CD4+ and CD8+ T cells.
Results
CLIP region of Ii can accommodate long peptide
We previously found that CLIP-exchanged Ii could deliver MHC-I peptides.Citation23 In order to expand the range of HLA loading, we tested the possibility of including a longer peptide in the Ii construct. As a target, we used a relevant antigenic peptide corresponding to the common frameshift mutation in TGFbRII.Citation26 When mutated a neo-antigen stretch of 34 amino acids is generated ()), from which we previously defined a peptide of 9 amino acids (9AA) that was later shown to be HLA-A2 restricted.Citation24,Citation27 In addition, we recently published on a TCR, Radium-1, which is specific for this peptide-MHC (pMHC) combination.Citation25 This TCR was isolated from a vaccinated cancer patient.Citation26 We thus selected the original vaccination peptide, a 19-mer peptide (), 19AA) and subcloned it into Ii by CLIP replacement (), IiTGF19AA) in order to study the presentation of longer peptides on MHC-I. A construct using the validated 9-mer was also prepared and used as a control (IiTGF9AA).
Figure 1. Antigen loading by CLIP-replaced invariant chain results in efficient activation of both CD4+ and CD8+ T cells. (a) representation of the TGFbRII protein and the specific domains (Wild-type, WT, upper panel and frameshift mutant lower panel). The red arrowhead points at the frameshift site and the amino acid sequence is depicted. In blue the WT sequence and in red the mutant sequence resulting from the frameshift mutation. The invariant chain constructs are compared to the WT and the position of the epitope replacements are indicated. (b) Activation of CD8+ T cells was monitored by CD107a surface expression. In vitro expanded T cells were electroporated with Radium-1 TCR mRNA and co-incubated for 5 h with autologous EBV-LCL (APC) previously electroporated with the indicated construct (SCT = single chain trimer; frameshift mutant = full-length frameshift mutant construct; Ii wild-type (p35 isoform); IiTGF19AA; IiTGF9AA), loaded with the 19AA peptide or untreated. Graphs are representative of two independent experiments. (c) Same experiment as in (b) where the T cells were electroporated with Radium-6 TCR and TNF-α accumulation was detected after O/N incubation. Data are shown as mean ± S.D. (n = 4), ****P < 0.0001, *** P < 0.001 by ANOVA and Dunnett’s multiple comparison test, N = 2, except for SCT where N = 1 and n = 2.
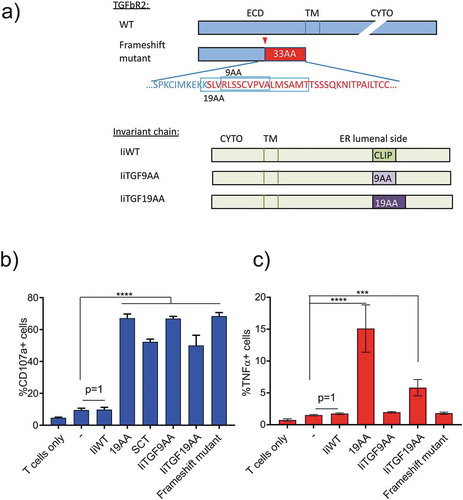
First, we measured the ability of Radium-1 to recognize the different Ii constructs in a functional assay where CD107a was used as an activation marker. As shown, healthy donor redirected T cells were only stimulated when the cognate TGF-antigenic peptide was presented, independently of the size: IiTGF19AA was as efficient as IiTGF9AA, suggesting that when inserted into Ii the same peptide was, after processing, loaded on MHC-I. In addition, we compared the peptide loading efficiencies of Ii constructs to those of other loading vectors. As expected, the vaccination peptide (19AA) as well as transfection with full-length TGFbRII frameshift protein or with a single chain trimer constructCitation28 displaying the 9AA peptide (SCT9AA) were equally potent to present the 19AA vaccination peptide ()). From this, we see that presentation of a 9-mer by the Invariant chain can be expanded to peptides longer than the short CLIP sequence which opens the possibility to combine MHC-I and MHC-II presentation. We recently isolated MHC-II restricted TCRs from another TGFbRII-vaccinated patient (Inderberg, Wälchli et al. unpublished data). One of them, Radium-6 TCR, is HLA-DR4 restricted and was used in a functional assay against EBV-LCL HLA-DR4+ cells presenting the 19AA peptide. We monitored TNF-α released from CD4+ T cells as an activation marker. As shown in ), only the vaccination peptide (19AA) and the IiTGF19AA construct were able to significantly stimulate Radium-6 redirected T cells. As expected the IiTGF9AA did not activate Radium-6 TCR since the peptide sequence was too short and neither did the full-length frameshift mutant protein when overexpressed in APCs. This is in agreement with our previous report showing that CLIP-replaced Ii constructs efficiently transferred epitopes to the luminal side of the ER, and then transported to the endosomal pathway for processing and loading while cytosolic proteins were not presented. In order to confirm the potency of our design, we prepared another construct from a validated tumor antigen, CEA. We used the HLA-A2 CEA peptide691-699 and expanded its sequence to create a longer peptide678-702 similar to a vaccination peptide (Sup. Figure 1(a)). Similar to the experiments using TGFbRII, these antigenic peptides were inserted instead of the CLIP sequence of Ii. We confirmed our first observation and showed that the short and the long sequences could both be presented to T cells expressing the specific TCR; suggesting that the long peptide was correctly trimmed in the cell (Sup. Figure 1(b)). Taken together these data show that CLIP replaced Ii with a long peptide antigen is functional in terms of peptide loading and it can further load both MHC-I and MHC-II molecules.
Optimization of peptide length insert for Ii delivery
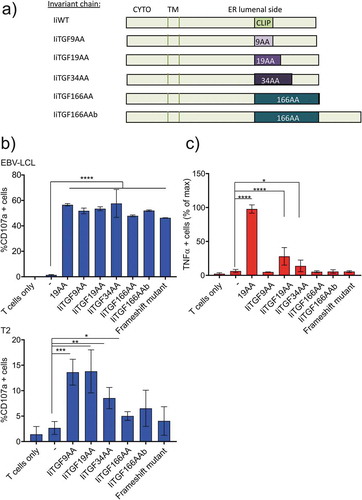
We next tested, using the initial target, the optimal size of the peptide for antigen presentation by Ii. We designed three additional constructs ()): IiTGF34AA contains the full-length neo antigen at the CLIP site, IiTGF166AA is a fusion between Ii until the CLIP peptide with the full-length TGFbRII frameshift and IiTGF166AAb is the same construct but where the frameshift sequence replaces CLIP. EBV-LCL HLA-A2+/HLA-DR4+ were electroporated with the indicated constructs. A functional assay was then performed using autologous redirected T cells. As shown, when Radium-1 TCR was expressed, all Ii-constructs were as stimulatory as the 19AA peptide, suggesting that they were all expressed, degraded and loaded with an equivalent efficiency () top). However, this observation did not provide any indication about the origin of the peptides. In order to understand this, we tested if the antigenic peptides were produced by the proteasome-dependent degradation machinery (in the cytosol) or if they were loaded in the ER bypassing the cytosolic degradation as we previously showed for CLIP exchanged peptide.Citation23 In order to restrict our analysis to the ER-dependent loading, we used T2 cells which are unable to transfer peptide from the cytosol to the ER due to their deficiency in a peptide transporter associated with antigen processing (TAP). We have previously shown with Melan-A antigen that T2 could load MHC-I from CLIP-modified Ii but not from full-length cytosolic protein.Citation23 As expected, in T2 cells, the full-length TGFbRII frameshift construct was barely presented () bottom). Conversely, the Ii constructs carrying antigenic peptides of 34 amino acids or shorter could bypass the TAP deficiency and were successfully presented. Insertion of the longest constructs (166AA) resulted in no loading of the antigenic peptide, suggesting that these constructs were mainly degraded in the cytosol when tested in EBV-LCL cells. We also repeated the experiment in T2 cells using IiCEA constructs and demonstrated that the trimming of the long peptide (CEA678-702) was not by cytosolic degradation but rather by trimming in the lumen of ER or later in the endomembrane system (Sup. Figure 1(c)). Interestingly, when we monitored the Ii-TGFbRII presentation on MHC-II with Radium-6 TCR we observed a similar pattern of presentation ()). Indeed, EBV-LCL cells only presented the Ii constructs containing 19 and 34 amino acids on MHC-II ()). These results were confirmed by using a TCR of a different MHC-II restriction, the Radium-5 TCR (Sup. Figure 2). Thus, MHC-II loading was only observed with IiTGF19AA and IiTGF34AA constructs, suggesting that either the long fusions were not correctly inserted into the ER, or that peptides were not efficiently transferred to the ER.
Figure 2. Loading of antigen by invariant chain is dependent on the length of antigenic insert. (a) Representation of the different Ii constructs used. (b) Functional assay of redirected T cells expressing Radium-1 TCR and incubated with APCs expressing the indicated constructs for 5 h. T cell activation was measured as percent of CD107a positive cells. APCs were autologous EBV-LCL (top) and T2 cell line (bottom) either electroporated with the indicated constructs or loaded with the 19AA peptide or untreated (-). Data are shown as mean ± S.D. (n = 2 for EBV-LCL and n = 4 for T2), *P < 0.05, **P < 0.01, ***P < 0.001 by ANOVA and Dunnett’s multiple comparison test, N = 2 for T2 and 1 for EBV-LCL. (c) Same experiment as in (b) where T cells were electroporated with Radium-6 TCR. T-cell activation was measured as the percent of TNF-α producing T cells. Normalized data where + peptide (19AA) was set at 100% are shown as mean ± S.D. (n = 4), *P < 0.05 and ****P < 0.0001 by ANOVA and Dunnett’s multiple comparison test, where N = 2–3 and n = 6–8.
Subcellular distribution of Ii-constructs
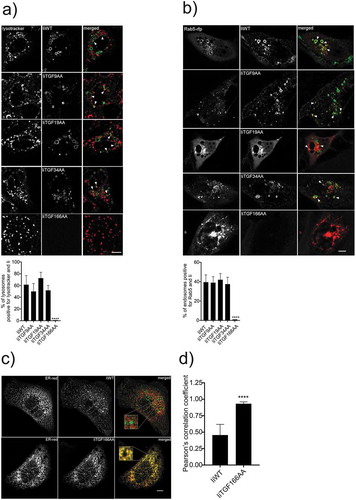
To verify that all the Ii constructs could egress from the ER and follow the conventional endocytic pathway, we performed colocalization studies using confocal microscopy on MDCK cells expressing different constructs (). As a marker for Ii, we added anti-Ii fluorescent antibodies to the medium. These bound to Ii at the plasma membrane and were rapidly internalized,Citation29 showing that the Ii constructs traffic via the plasma membrane to the endosomal pathway. We first analyzed the vesicular transport of the internalized antibody to lysotracker positive vesicles ()) and found that all constructs except IiTGF166AA trafficked to the lysosomal pathway without any marked differences. We next analyzed the colocalization of Ii with Rab-5 positive vesicles and observed the same distribution as with lysosomes ()). The immunofluorescence showed that the late endosomal membranes were labeled with the antibody indicating that the constructs themselves could traffic to late proteolytic compartments. Thus, CLIP exchange does not seem to significantly influence the trafficking of Ii up to 34 amino acids in size. We finally investigated the localization of the IiTGF166AA construct and found that it was retained in the ER (,)). Consequently, as expected, processing and loading of peptides onto MHC-II is dependent on transport to the endosomal pathway. From these data, we conclude that CLIP can be replaced by larger peptides, but only to a defined extent, as it was dependent on the ability to traffic to the endosomal pathway.
Figure 3. Intracellular trafficking of different CLIP-replaced constructs. a and b) Indicated Ii and dsRed-ER constructs were expressed in MDCK cells. 16–20 h later, cells were incubated in medium containing fluorochrome-labeled anti-Ii antibody (MB-741). The internalized antibodies were imaged in live cells, and the degree of co-localization between the early endosomal protein Rab5-RFP and the low pH marker lysotracker was quantified. After 1 h of incubation live cells were analyzed for colocalization between Ii and lysotracker in double positive lysosomes (a, arrows) Live cells were also analyzed for colocalization between Ii and Rab5 (red) positive endosomes (b, arrows). All co-localization values were obtained from independent experiments (N = 3) where at least 10 cells per experiment were analyzed (n > 10). Error bars are S.D. and significance was analyzed by Student’s t-test where ****P < 0.0001. (c) One representative example of sub-cellular localization of wild-type Ii (IiWT) and IiTGF166AA (upper and lower panel, respectively). These cells were fixed and permeabilized before staining with an anti-Ii antibody. (d) Quantification of the observed degree of co-localization in (c). The colocalization data were generated using ImageJ plugins. At least 10 cells were analyzed in five different optical regions (N = 3). Error bars are S.D and significance was analyzed by Student’s t-test where ****P < 0.0001. Bars 10 µm.
Quality of ii-driven MHC-II loading
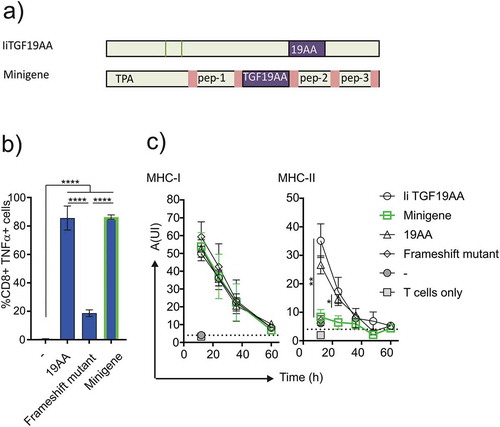
Although more difficult to establish in a clinical setting, vaccination through endogenous routes such as mRNA electroporation,Citation30 DNA transfectionCitation31 or viral transductionCitation32 have some advantages over direct peptide loading of MHC molecules. We have previously shown that MHC-I loading by CLIP replaced Ii construct was independent of cytosolic proteolysis and transport to the ER.Citation23 Most of the recent literature describes the use of an ER-targeted minigene vector for vaccination. Although minigenes most commonly are adapted to present on MHC-I, we wanted to compare loading by Ii to that of a minigene for both MHC classes. We therefore generated and validated a minigene-based construct composed of an ER-targeting sequence and a fusion of antigenic peptides separated by furin cleavage sitesCitation33 in which we placed the 19AA peptide from the TGFbRII frameshift mutant ()). In order to validate the correct transport of the minigene in a TAP-independent manner, we performed a functional assay using T2 cells as antigen presenting cells. T2 cells were transfected with either the minigene or the frameshift full-length TGFbRII protein or loaded with 19AA and incubated with Radium-1 expressing T cells ()). T cell stimulation was monitored by TNF-α release and showed efficient minigene-dependent stimulation, suggesting that the construct was functional and could traffic antigenic peptide to the ER independently of TAP. We then compared the minigene to the Ii-construct side-by-side in electroporated EBV-LCL cells compatible to Radium-1 and Radium-6 TCRs. To obtain data on loading dynamics using both constructs we measured equivalent T cell stimulation over time. As shown in ), there was no difference between constructs or peptide pulsing when comparing MHC-I loading. However, MHC-II loading showed stricter requirements: IiTGF19AA was as efficient as peptide loading in terms of complex stability and presentation, but minigene was unable to load MHC-II molecules. Taken together, these data show that Ii is superior to minigene for MHC-II loading.
Figure 4. Comparison of minigene and Ii constructs stimulation. (a) Schematics showing the design of minigene and Ii constructs. (b) Functional assay of antigen loading efficiencies obtained when loading T2 cells with minigene. T2 cells were incubated with Radium-1 redirected T cells for 5 h prior to stimulation analysis. TNF-α+ cells were used as a readout for the frequency of stimulated T cells. Data are shown as mean ± S.D. (N = 2, n = 6), ****P < 0.0001, *** P < 0.001 by ANOVA and Dunnett’s multiple comparisons test. (c) Comparison of antigen loading between constructs shown in (a) in a functional assay. Post-transfection EBV LCL cells were incubated for increasing intervals of time, before aliquots were used to stimulate cognate T cells. Antigen loading was measured for both MHC-I and MHC-II using the same batch of EBV-LCL cells. Points show mean ± S.D. Multi-variated bi-directional Student t-test was used to compare invariant chain and minigene constructs for the two first time points (n = 3 for each condition).
Ii loading by professional APCs
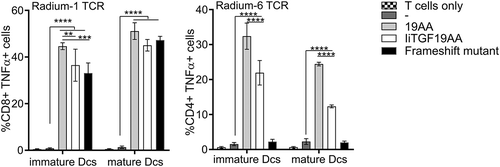
To further validate the efficiency of Ii as cancer vaccine solution, we evaluated Ii potency in DCs which are professional APCs. One crucial aspect contributing to antitumor vaccine efficacy is the capability to generate strong and durable tumor-specific CD8+ and CD4 + T cell immunity, and DCs have been shown to activate both naïve and memory T cells. We selected the IiTGF19AA construct since it was the most efficient loading vector for stimulating both CD4+ and CD8+ T cells. We selected a healthy donor carrying the MHC alleles corresponding to our reactive TCRs, namely HLA-A2, -DR7 and -DR4, for Radium-1, −5 and −6, respectively. We first validated our assay by transfecting donor T cells with the TCR constructs and incubated them with autologous DCs either at the stage of mature DCs or immature DCs; in the latter case immature DCs were further stimulated with the maturation cocktail to achieve a fully mature state (Sup. Figure 3(a)), because mature DCs are less efficient to present peptide.Citation34,Citation35 The strategy was performed to supply the tumor antigen to the immature DCs and then achieve a fully DC maturation by using a clinical approved maturation cocktail. DCs were either loaded directly by peptide incubation, or mRNA electroporated with IiTGF19AA construct or the full-length TGFbRII frameshift construct. T-cell stimulation was monitored by TNF-α-presence in both CD8+ and CD4+ T cells. As shown in , both mature and immature DCs could stimulate through MHC-I (Radium-1) or MHC-II (Radium-6 and Radium-5, Sup. Figure3(b)), thus T cells could be stimulated by DCs electroporated to express li constructs, independently of their maturation state for antigen loading. In the case of MHC-II, Radium-6 and Radium-5 differed in their preference: Radium-6 TCR was more strongly stimulated by exogenous peptides in contrast to Radium-5 TCR which was more sensitive to the Ii construct (Sup. Figure 3(b)). This suggests that MHC-II capacity to be loaded might also be allele dependent; thus, quantitative conclusions on the presented data would be meaningless at this stage. In addition, the amount of peptide used to load DCs is many folds higher than the peptide generated by the Ii construct, restricting us to qualitative conclusions. Finally, the full-length TGFbRII frameshift protein was only able to stimulate Radium-1 TCR upon mRNA transfection (, black) which is in agreement with the MHC-II-dependent exogenous delivery model.
Figure 5. Antigen presentation by messengerRNA electroporated DCs. Monocyte-derived DCs isolated from healthy donors were differentiated and provided with antigen either at the mature or immature state. These DCs were either loaded with 19AA peptide (19AA) or electroporated with the indicated constructs (IiTGF19AA or the frameshift mutant construct). Control cells (-) were mock electroporated. In parallel, autologous T cells were electroporated with mRNA encoding the indicated TCR (Radium-1 or Radium-6). The electroporated cells (DCs and T cells) were finally co-incubated for 5 h and activation of T cells was monitored by TNF-α intracellular expression. Data are shown as mean ± S.D. of two separate experiments (N = 2, n = 3–5). Only significant variations are shown, **P < 0.01, ***P < 0.001, ****P < 0.0001 by ANOVA.
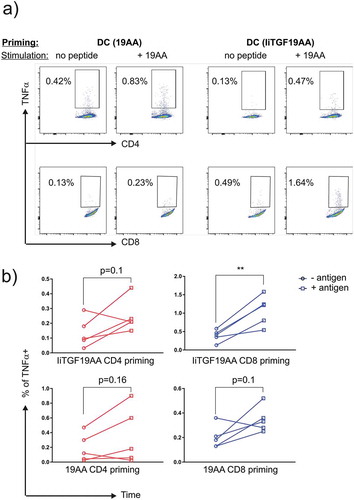
We then tested whether mature DCs could prime naïve T cells ()). We isolated monocyte-derived DC, which were matured and loaded with TGF peptide either by incubating with peptides or by electroporation. The experiments were performed for the 19AA and IiTGF19AA construct. We then incubated these DCs with autologous T cells. A boost was given on day 12 using either peptide or mRNA. After 22 days of co-culture, peptide-reactive T cells were detected using a functional assay where autologous EBV-LCL cells were loaded with the 19AA peptide, and representative flow plots are shown in ). For our analysis, we separated CD4 and CD8 T cells in order to study the efficiency in MHC-I and MHC-II loading between the peptide and Ii. We then pooled data from two independent experiments (n = 5) in order to obtain a quantitative view of the antigen loading efficacy. As shown, significance was reached only when CD8+ T cells were primed by IiTGF19AA electroporated DC ()). The fact that even the clinically tested 19AA peptide did not reach significance indicating that a higher number of DC stimulations should have been analyzed to increase the statistical power; however, it also suggests that Ii is an efficient vaccination tool despite the suboptimal analysis conditions. In addition, a marked tendency was observed in all the conditions, we thus believe that the number of experiments performed and donors tested was too low considering the fact that the expected frequencies of specific T cells in healthy donors is very low and the sensitivity of the assay (ICS by flow cytometry) might not be sufficient. Furthermore, the multimer technology, that represents the most sensitive tool for such purposes, could not be performed because the MHC-multimer production failed upon multiple attempts. From these data we conclude that the replacement of CLIP with a large peptide could be expressed in DCs and presented on both MHC Class I and Class II, leading to an efficient priming of CD8+ T cells and a tendency for enhanced priming of CD4+ T cells.
Figure 6. Priming of CD4+ and CD8 + T cells by DCs expressing IiTGF19AA. PBMC from a healthy donor were co-cultured with autologous DCs, either loaded with 19-mer TGF peptide (19AA) or expressing IiTGF19AA. On day 12, T-cell cultures were restimulated in the same conditions. On day 22, T cells were harvested and tested for the presence of peptide reactivity. This was done by performing a functional assay where TNF-α expression was detected by flow cytometry analysis. Here, autologous EBV-LCL were loaded or not with 19AA peptide and incubated for 12 h with the harvested T cell culture from the priming (Day 22). Cells were stained with anti-CD8, anti-CD4, and anti-TNF-α antibodies. The presence of TNFα producing T cells was monitored by flow cytometry. (a) Representative plot showing TNF-α signal in the indicated T-cell population (CD4 or CD8). In the top row, the priming method to obtain the T cell culture is depicted (peptide or Ii) and in the lower row the stimulation method for the detection of reactive T cells (EBV-LCL presenting cells loaded or not with peptide) is indicated. (b) Quantification of the experiment, as % of TNF-α observed in CD4 (red) and CD8 (blue) population. Signal from individual cultures was plotted (n = 5), significance was analyzed by Student’s t-test where **P < 0.01.
Discussion
Cancer vaccination can be considered as a safe therapy with a minimal toxicity, and if well designed or well combined with other drugs, it could become a standard cancer treatment. In addition, recent reports have demonstrated that DC-based vaccines are promising since they reduce tumor growth and improve survival.Citation7,Citation36 An ideal vaccine should, therefore, have the capacity to evoke an integrated immune response, where DC would induce both CD4+ and CD8+ T-cell stimulation.Citation37,Citation38 Although certain studies have shown that bystander CD4 T-cell help is very efficient,Citation39 others have shown that specific CD4 T-cell help is required.Citation9 One advantage is that CD4 T cells that specifically recognize tumor antigens could provide local help for anti-tumor CD8 T cells in the TMECitation40: CD4 T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8 T lymphocytes. Long peptides containing multiple epitopes which can be loaded by different MHC alleles will probably represent the optimal alternative. In this context, CLIP replaced Ii can be seen as a promising vaccination vector candidate.
We show that the CLIP peptide replacement could be extended to 34 amino acids without affecting the conventional mode of antigen presentation. Indeed, both short and long Ii TGFbRII vehicles promoted epitope loading on MHC class I molecule and could stimulate Class-I restricted Radium-1 TCR. Notably, responses elicited by presentation of the antigen in an Ii vector were comparable to those achieved by other strategies/routes of antigen loading such as exogenous peptide loading, which is commonly used in DC-based cancer vaccines, or APCs genetically modified to process the full-length target protein or APCs expressing SCT. Activation of Radium-1 T cells mediated by the 19-mer Ii construct (IiTGF19AA) was as potent as the response seen upon stimulation with the short Ii mutant (IiTGF9AA). These results indicate that the long 19-mer peptide loaded by Ii was properly processed and shorter peptides, with the correct length, were generated and bound to HLA-A2 molecule. This represents an extension of our previous report using Ii and short peptidesCitation23 and suggests that CD4+ T cells could be stimulated by the same construct. Therefore, we tested T cells bearing the class II-restricted TCRs, Radium-6 or Radium-5, recognizing the same frameshift mutation as Radium-1 but in the context of MHC-II molecules. These T cells responded to target cells expressing IiTGF19AA proving that the Ii construct could be trimmed both for Class I and Class II peptide loading. The strongest activation was achieved in the presence of exogenously loaded peptide; however, the beneficial effect of this MHC loading approach might be drastically reduced in an in vivo setting by the rapid degradation of the peptide.Citation41,Citation42
A precise microscopy analysis of the Ii constructs showed that all Ii constructs except the one with 166AA were transported to early and late endosomes. The uptake of antibodies recognizing the luminal domain of Ii showed that these constructs traffic via the plasma membrane like the native Ii.Citation43 The longest construct containing 166 amino acids was found to be retained in the ER, potentially by misfolding or more specific retention. Our limited number of constructs thus showed that the CLIP region could be exchanged within peptides from 9 to 34 amino acids without altering the traffic to the endocytic pathway. Potentially somewhat longer peptides could have been inserted but this was not necessary for the combined MHC-I and MHC-II antigen presentation in our experiments. The influence of inserted peptides are likely dependent on the specific sequence as well as its length; therefore, the insertion of a new peptide should be tested in an intracellular trafficking assay. We found that the 166AA construct was retained in ER and could not load MHC-I and II, showing that ER localization was not sufficient to ensure proper loading. This was confirmed when we used a published minigene construct targeted to ER: although the MHC-I epitope was properly trimmed to be presented by MHC-I as previously reported,Citation33 MHC-II molecules could not be loaded. Thus, the Ii constructs provided an ideal environment not only for targeting to the endosomal pathway but also for proper antigen loading of both MHC-I and MHC-II. This is surprising as others have shown that co-expression of MHC-II and Ii had rather an inhibitory effect on the T cells; however, the expression was performed on cancer cell with the wild-type molecules.Citation44 It would, however, be interesting to understand the mechanism behind Ii-dependent activation and inhibition of immune response. It could reside in the loading mechanism which is still unclear: it was recently shownCitation45 how protein antigen could be processed through an autophagy- and proteasome-dependent pathway after endocytosis and how CD8 T-cell epitopes are loaded onto MHC-I molecules within the autophagolysomal compartment rather than the conventional secretory pathway, which requires transporters associated with antigen processing-dependent transport. This could also be the case for long peptides, but we do not know the precise mechanism for the cross-presentation of long peptides used for CLIP replacement in our system. This would be a mechanistic study of antigen presentation that is outside the scope of this paper, but we have provided functional proof of T-cell stimulation that this cross-presentation does indeed exist.
Finally, we used DCs to test the priming and boosting potency of Ii construct. DCs have been extensively investigated as antigen delivery vehicles in cancer immunotherapy via vaccination. In order to demonstrate the priming capacity of Ii-electroporated DCs, we used autologous naive T cells and showed that positive T cell population could be observed. Although we reached statistical significance only with IiTGF19AA priming of CD8+ T cells and not with the original vaccination peptide, we believe that the experiment was an underestimation of the vaccination potential for the following reasons: (i) the detection method (functional assay) might not be ideal to distinguish lower affinity clones or low frequency antigen-specific T-cell clones; unfortunately multimer-based assessment of TGFbRII-specific T-cell populations could not be performed because of instability of the multimers, (ii) primed TGFbRII-specific T cells might have peaked earlier than day 22 and became barely detectable by the day of the functional assay, (iii) in vitro culture conditions may not be ideal for efficient priming and (iv) more experimental time points, and more experiments and donors, would be needed. However, although significant only for the CD8 T cells primed with IiTGF19AA, each T cell subtype shows a tendency to increase their reactivity to the TGF peptide after priming. This strongly suggests that Ii constructs can evoke an immune response, in a clinical setting, and our study demonstrates the great potential of CLIP replaced Ii construct for cancer vaccination.
Materials and methods
Cell culture
Leukapheresis of non-mobilized healthy donor was performed on site and both monocytes and lymphocytes were harvested. T cells were thawed and expanded using Dynabeads CD3/CD28. In brief, T cells were cultured with Dynabeads (Dynabeads®ClinExVivo™ CD3/CD28, Thermo Fischer Scientific) at a 3:1 ratio in complete CellGro DC Medium (CellGenix, Freiburg, Germany) with 100 U/mL recombinant human IL-2 (Novartis, Emeryville, CA) for 10 days. The cells were frozen and aliquots were thawed and rested in complete medium before transfection and antigen presentation assay. Frozen autologous monocytes were used to generate dendritic cells. Briefly, monocytes were thawed and cultured 2 days in CellGro DC medium supplemented with IL4 (20 ng/ml) and GM-CSF (100 ng/mL) followed by 24h in the presence of IL4 (20 ng/mL), GM-CSF(100 ng/mL), IL1-b (10 ng/mL), TNF-α (20 ng/mL), IFN-γ (5.000 IU/mL), R848 (1 µg/mL) and PGE2 (250ng/mL) in order to provide the necessary signals for DC maturation. Epstein-Barr virus-transformed lymphoblastoid cell lines (EBV-LCL) were prepared in house by immortalization with EBV using supernatants from the Mamorset cell line B95-8.
Human cell lines were routinely checked for mycoplasma using (Venor® GeM; Minerva Biolabs, Berlin, Germany). T2 cells (ATCC CRL-1992) were cultured in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FCS (Gibco, Thermo Fisher Scientific) and gentamycin 0.05 mg/mL (Thermo Fisher Scientific). Madin Darby Canine Kidney (MDCK) cells (ATCC CCL-34) were cultured in DMEM cell culture medium (Whittaker, Walkersville, MD, USA) supplemented with 10% FCS, L-glutamine (Gibco, Thermo Fisher Scientific) and gentamycin 0.05 mg/mL. All cells were grown at 37ºC in 5% CO2.
DNA manipulation and cloning
The full-length construct encoding the TGFbRII frameshift mutant was ordered as a synthetic DNA. The construct encoding TGF9AA was cloned by site-directed mutagenesis of an Ii WT constructCitation23 using the following primers: 5ʹ-CTG TGA GCA AGC GTC TGT CAT CAT GCG TTC CTG TGG CTC AGG CGC TGC CC-3ʹ and 5ʹ-GGG CAG CGC CTG AGC CAC AGG AAC GCA TGA TGA CAG ACG CTT GCT CAC AG-3ʹ. Single Chain trimer encoding for HLA-A2 with the TGF9AA peptide was prepared by site-directed mutagenesis using our previously reported constructsCitation46 as a template and the following primers: 5ʹ-GGC CTC GAG GCT CGT CTG TCA TCA TGC GTT CCT GTG GCT GGC TGT GGC AGC-3ʹ and 5ʹ-GCT GCC ACA GCC AGC CAC AGG AAC GCA TGA TGA CAG ACG AGC CTC GAG GCC-3ʹ. A construct containing human Ii coding sequence (p33 isoform) was modified by site-directed mutagenesis to replace the CLIP region by two restriction sites KpnI/XhoI (Addgene construct: 104634). The construct was opened, dephosphorylated and the coding sequence for TGF19AA peptides () was designed as sense and complement primers with sticky-ends (underlined) for the restriction sites: 5ʹ-CAA GAG CCT GGT GAG ACT GAG CAG CTG CGT GCC CGT GGC CCT GAT GAG CGC CAT GAC CC-3ʹ and 5ʹ-TCG AGG GTC ATG GCG CTC ATC AGG GCC ACG GGC ACG CAG CTG CTC AGT CTC ACC AGG CTC TTG GTA C-3ʹ. Primers were ordered 5ʹ-phosphorylated and were subcloned into the Ii vector after annealing using a PCR cycler set to the following conditions: 95ºC for 30 s, 72ºC for 2 min, 37ºC 2 min, 25ºC 2 min. The insert was then kept on ice and used at 0.5 µM final concentration. Construction of TGF34AA was done by site-directed mutagenesis using primers adding the coding sequence of 15 amino acids in addition the previous 19 amino acids using these primers: CAC CAG CAG CAG CCA GAA GAA CAT TAC TCC TGC CAT ACT GAC TTG CTG CGA ATT CCA GAA TGC CAC CAA G and CTT GGT GGC ATT CTG GAA TTC GCA GCA AGT CAG TAT GGC AGG AGT AAT GTT CTT CTG GCT GCT GCT GGT G. The TGF166AA and TGF166AAb coding sequence were inserted by amplifying it from the full-length coding sequence (our collection) and ligating it to the Ii construct. For TGF166AA, one primer pair was designed to remove the C-terminal part of the Ii from translation by inserting two STOP codons after the peptide coding sequence: tat GGT ACC ATG GGT AGA GGC TTG CTT C and TAC TCG AGT CAT CAG CAG CAA GTC AGT ATG G. TGF166AAb was prepared with the same forward primer as for TGF166AA cloning, but a reverse primers without STOP codons: TAC TCG AGG CAG CAA GTC AGT ATG G. Amplification was carried out using Pfx polymerase (Invitrogen, Carlsbad, CA, USA) following these cycling conditions: 94°C 4 min, 20 cycles of (94°C 30 sec, 53°C 30 sec, 68°C 45 sec) 68°C 7 min, 4°C ∞. Purified product (Nucleospin Gel and PCR clean-up, Macherey-Nagel, Düren, GE) was cut with KpnI and XhoI and ligated into Ii-containing plasmid. Ii constructs containing CEA691-699 and CEA678-702 were constructed by primer annealing using the following pairs: CTG TGA GCA AGA TCA TGA TCG GCG TCC TGG TGG GGG TGC AGG CGC TGC CC and GGG CAG CGC CTG CAC CCC CAC CAG GAC GCC GAT CAT GAT CTT GCT CAC AG, and CGG CAC CAG CCC CGG CCT GAG CGC CGG CGC CAC CGT GGG CAT CAT GAT CGG CGT GCT GGT GGG CGT GGC CCT GAT CG and TCG ACG ATC AGG GCC ACG CCC ACC AGC ACG CCG ATC ATG ATG CCC ACG GTG GCG CCG GCG CTC AGG CCG GGG CTG GTG CCG GTA C, respectively. The CEA TCR was previously publishedCitation47 and its sequence was kindly shared by Dr. M. Parkhurst (NCI, Bethesda, MD, USA). A synthetic DNA was ordered as codon-optimized.
All constructs were cloned into pENTR/TOPO following manufacturer’s protocol (ThermoFisher) and were, after sequence confirmation, subcloned into expression pCIpA102 or pMP71 vectors as described in.Citation48 Primer synthesis, artificial DNA preparation (minigene) and sequencing were performed by Eurofins-MWG (Ebersberg, Germany).
mRNA synthesis was performed as follows: pClpA102 plasmids containing the gene of interest were linearized by cutting at unique MfeI restriction site situated at the end of the polyA tail. The mRNA was prepared from the linearized template using RiboMAX™ Large scale T7 (Promega, Madison, WI, USA) adding ARCA-cap (TriLink BioTechnologies, San Diego, CA, USA). Finally, mRNA was purified using RNeasy Midi kit (Qiagen, Hilden, Germany) and stocked at −20ºC until further use.
Electroporation of cells was performed in a 2 mm gap electroporation cuvette at 250 V for 2 ms (5x106 EBV LCLs) or 4 mm gap cuvette at 500V for 2 ms (40x106 T cells and 20 × 106 DCs) using an Square Wave Electroporator (BTX Technologies Inc., Hawthorne, NY, USA). Cells were prepared for electroporation by harvesting and washing them twice in cold RPMI 1640. Cells were then mixed with mRNA at a final concentration of 100 ng/µL in either 200 µL (2 mm gap cuvette) or 800 µL (2 mm gap cuvette). Immediately after electroporation, cells were transferred to 2 mL complete medium and kept at 37°C in 5% CO2 overnight.
Retroviral manipulation and transduction was performed as previously described.Citation48
Functional assay
TCR expressing T cells were stimulated for either 5 h (CD8+ T cell stimulation) or 12 h (CD4+ T cell stimulation) with autologous DCs, EBV immortalized LCL cells or T2 cells. DCs were loaded with the tumor-derived peptide either at the stage of mature DCs or immature DCs; in the latter case, the immature DCs were further incubated for 24 h in the presence of the maturation cocktail as described above in Material and Methods. Antigen-presenting cells were either loaded with 10 µM of 19-mer TGFbRII frameshift peptide or electroporated to express IiTGF constructs or the full-length TGFbRII frameshift protein. Control samples (-) were mock electroporated with dH2O and were used as a negative stimulation condition. T cells and APCs were mixed at an effector-to-target ratio of 1:2 and in the presence of anti-CD107a antibody, BD GolgiPlug and BD Golgistop (BD Biosciences, Chicago, IL, USA) at a 1/1000 dilution. Post stimulation, cells were stained with anti-CD8, anti-CD4, anti-TNF-α antibodies (BD Biosciences, Chicago, IL, USA). The functional assay over time ()) was performed as follows: EBV-LCL (antigen presenting cells) were electroporated with liTGF19AA, Minigene, full-length TGFbRII frameshift protein or loaded with the corresponding peptide. The cells were then grown for 12, 24, 36, 48 or 60 h before being mixed with the effector T cells and stained as depicted above. Degranulation or cytokine production signals were acquired on a BD FACSCanto II flow cytometer (BD biosciences) and the data were analyzed using FlowJo (TreeStar, Ashland, OR, USA).
In vitro generation of DC for antigen presentation and priming assay
Monocytes from a healthy donor were cultured 2 days in CellGro DC medium supplemented with IL-4 and GM-CSF. Immature monocyte-derived DCs were supplied with the tumor antigen; briefly immature DCs were electroporated with either mRNA encoding for Ii carrying TGF19AA peptide (liTGF19AA) or loaded with TGF19AA peptide (19AA, 10 µM). The immature DCs were then cultured for 24 h in the presence of a maturational cocktail containing IL4, GM-CSF, IL1-b, TNF-α, IFN-γ, R848, and PGE2. Mature DCs were then used in T cell priming experiment. The two distinct DC populations (liTGF19AA and 19AA) were cultured with autologous PBMCs at 1:10 or 1:20 DC:PBMC cell ratio in CellGro DC medium. On day 3, T cell cultures were supplemented with hIL2 (20 U/mL) and hIL7 (5 ng/mL). On day 12, T cell cultures were restimulated under same DC conditions and, on day 22, T cell cultures were screened for the presence of TGFbRII frameshift-specific T cells by measuring the frequency of T cells producing TNF-α cytokine by flow-cytometry
Confocal laser scanning microscopy
MDCK cells were grown to 70% confluency in 35 mm microwell dishes (MarTek, Ashland, MA, USA). The cells were then transiently transfected with appropriate constructs using Fugene 6 (Roche) as described by the manufacturer. One hour prior to imaging, the cell medium was exchanged with complete DMEM without phenol red, and cells were incubated with anti-Ii (M-B741-Alexa 647) to a final concentration of 1 µg/mL. The Rab5-RFP construct was a gift of Frode Skjeldal at the Normic Oslo imaging platform. Confocal images were acquired on an Olympus FluoView 1000 inverted microscope equipped with Plan/Apo 60/1.10 NA oil objective (Olympus, Hamburg, Germany). Constant temperature was set to 37°C and CO2 to 6% by an incubator enclosing the microscope stage. Fluorochromes were exited with 488nm, 543nm, and 647nm lasers. All image acquisition was done by sequential line scanning to eliminate bleed-through.
Disclosure of potential conflicts of interest
GG, GK, OB, EMI, and SW are inventors of the patent WO2015189267. NM, AG, NBP, PD, and MRM disclose no competing interests.
Supplemental Material
Download MS Word (70.8 KB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Shang N, Figini M, Shangguan J, Wang B, Sun C, Pan L, Ma Q, Zhang Z. Dendritic cells based immunotherapy. Am J Cancer Res. 2017;7(10):2091–2102.
- Garg AD, Coulie PG, Van Den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017;38(8):577–593. doi:10.1016/j.it.2017.05.006.
- Garg AD, Vara Perez M, Schaaf M, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: dendritic cell-based anticancer immunotherapy. Oncoimmunology. 2017;6(7):e1328341. doi:10.1080/2162402X.2017.1328341.
- Slingluff CL, Petroni GR, Chianese-Bullock KA, Smolkin ME, Hibbitts S, Murphy C, Johansen N, Grosh WW, Yamshchikov GV, Neese PY, et al. Immunologic and clinical outcomes of a randomized phase II trial of two multipeptide vaccines for melanoma in the adjuvant setting. Clin Cancer Res. 2007;13(21):6386–6395. doi:10.1158/1078-0432.CCR-07-0486.
- Slingluff CL, Petroni GR, Chianese-Bullock KA, Smolkin ME, Ross MI, Haas NB, von Mehren M, Grosh WW. Randomized multicenter trial of the effects of Melanoma-associated helper peptides and cyclophosphamide on the immunogenicity of a multipeptide Melanoma vaccine. J Clin Oncol. 2011;29(21):2924–2932. doi:10.1200/JCO.2010.33.8053.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696. doi:10.1038/nature14426.
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi:10.1038/nature23003.
- Bristol JA, Orsini C, Lindinger P, Thalhamer J, Abrams SI. Identification of a ras oncogene peptide that contains both CD4(+) and CD8(+) T cell epitopes in a nested configuration and elicits both T cell subset responses by peptide or DNA immunization. Cell Immunol. 2000;205(2):73–83. doi:10.1006/cimm.2000.1712.
- Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL, Ye L, Sharpe AH, Freeman GJ, Ahmed R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci USA. 2011;108(52):21182–21187. doi:10.1073/pnas.1118450109.
- Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836. doi:10.1038/nri3084.
- Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31(1):443–473. doi:10.1146/annurev-immunol-032712-095910.
- Cruz FM, Colbert JD, Merino E, Kriegsman BA, Rock KL. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu Rev Immunol. 2017;35:149–176. doi:10.1146/annurev-immunol-041015-055254.
- Mellins ED, Stern LJ. HLA-DM and HLA-DO, key regulators of MHC-II processing and presentation. Curr Opin Immunol. 2014;26:115–122. doi:10.1016/j.coi.2013.11.005.
- Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, Huber C, Tureci O, Sahin U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2008;180(1):309–318. doi:10.4049/jimmunol.180.1.309.
- Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, Tureci O, Sahin U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108(13):4009–4017. doi:10.1182/blood-2006-04-015024.
- Gregers TF, Fleckenstein B, Vartdal F, Roepstorff P, Bakke O, Sandlie I. MHC class II loading of high or low affinity peptides directed by Ii/peptide fusion constructs: implications for T cell activation. Int Immunol. 2003;15(11):1291–1299.
- Malcherek G, Wirblich C, Willcox N, Rammensee HG, Trowsdale J, Melms A. MHC class II-associated invariant chain peptide replacement by T cell epitopes: engineered invariant chain as a vehicle for directed and enhanced MHC class II antigen processing and presentation. Eur J Immunol. 1998;28(5):1524–1533. doi:10.1002/(SICI)1521-4141(199805)28:05<1524::AID-IMMU1524>3.0.CO;2-T.
- Fujii S, Senju S, Chen YZ, Ando M, Matsushita S, Nishimura Y. The CLIP-substituted invariant chain efficiently targets an antigenic peptide to HLA class II pathway in L cells. Hum Immunol. 1998;59(10):607–614.
- Carstens C, Newman DK, Bohlen H, Konig A, Koch N. Invariant chains with the class II binding site replaced by a sequence from influenza virus matrix protein constrain low-affinity sequences to MHC II presentation. Int Immunol. 2000;12(11):1561–1568.
- Vigna JL, Smith KD, Lutz CT. Invariant chain association with MHC class I: preference for HLA class I/beta 2-microglobulin heterodimers, specificity, and influence of the MHC peptide-binding groove. J Immunol. 1996;157(10):4503–4510.
- Powis SJ. CLIP-region mediated interaction of invariant chain with MHC class I molecules. FEBS Lett. 2006;580(13):3112–3116. doi:10.1016/j.febslet.2006.04.060.
- Basha G, Omilusik K, Chavez-Steenbock A, Reinicke AT, Lack N, Choi KB, Jefferies WA. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nat Immunol. 2012;13(3):237–245. doi:10.1038/ni.2225.
- Walchli S, Kumari S, Fallang L-E-E, Sand KMKM, Yang W, Landsverk OJBJ, Bakke O, Olweus J, Gregers TFF. Invariant chain as a vehicle to load antigenic peptides on human MHC class I for cytotoxic T-cell activation. 2014;44(3):774–784. doi:10.1002/eji.201343671.
- Saeterdal I, Gjertsen MK, Straten P, Eriksen JA, Gaudernack G. A TGF betaRII frameshift-mutation-derived CTL epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol Immunother. 2001;50(9):469–476. doi:10.1007/s002620100222.
- Inderberg EM, Wälchli S, Myhre MR, Trachsel S, Almåsbak H, Kvalheim G, Gaudernack G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology. 2017;6(4):e1302631. doi:10.1080/2162402X.2017.1302631.
- Saeterdal I, Bjorheim J, Lislerud K, Gjertsen MK, Bukholm IK, Olsen OC, Nesland JM, Eriksen JA, Moller M, Lindblom A, et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci USA. 2001;98(23):13255–13260. doi:10.1073/pnas.231326898.
- Linnebacher M, Gebert J, Rudy W, Woerner S, Yuan YP, Bork P, von Knebel Doeberitz M. Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int J Cancer. 2001;93(1):6–11. doi:10.1002/(ISSN)1097-0215.
- Yu YY, Netuschil N, Lybarger L, Connolly JM, Hansen TH. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. J Immunol. 2002;168(7):3145–3149. doi:10.4049/jimmunol.168.7.3145.
- Bremnes B, Madsen T, Gedde-Dahl M, Bakke O. An LI and ML motif in the cytoplasmic tail of the MHC-associated invariant chain mediate rapid internalization. J Cell Sci. 1994;107(Pt 7):2021–2032.
- Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood. 2001;98(1):49–56.
- Mahvi DM, Sondel PM, Yang N-S, Albertini MR, Schiller JH, Hank J, Heiner J, Gan J, Swain W, Logrono R. Phase I/IB study of immunization with autologous tumor cells transfected with the GM-CSF gene by particle-mediated transfer in patients with Melanoma or Sarcoma University of Wisconsin, Madison, Wisconsin. Hum Gene Ther. 1997;8(7):875–891. doi:10.1089/hum.1997.8.7-875.
- Lopes L, Fletcher K, Ikeda Y, Collins M. Lentiviral vector expression of tumour antigens in dendritic cells as an immunotherapeutic strategy. Cancer Immunol Immunother. 2006;55(8):1011–1016. doi:10.1007/s00262-005-0095-5.
- Aurisicchio L, Fridman A, Bagchi A, Scarselli E, La Monica N, Ciliberto G. A novel minigene scaffold for therapeutic cancer vaccines. Oncoimmunology. 2014;3(1):e27529. doi:10.4161/onci.27529.
- Wagner CS, Cresswell P. TLR and nucleotide-binding oligomerization domain-like receptor signals differentially regulate exogenous antigen presentation. J Immunol. 2012;188(2):686–693. doi:10.4049/jimmunol.1102214.
- Gil-Torregrosa BC, Lennon-Duménil AM, Kessler B, Guermonprez P, Ploegh HL, Fruci D, van Endert P, Amigorena S. Control of cross-presentation during dendritic cell maturation. Eur J Immunol. 2004;34(2):398–407. doi:10.1002/eji.200324508.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi:10.1038/nature22991.
- Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–647. doi:10.1038/s41577-018-0044-0.
- Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4 + and CD8 + T cells during the effector phase. J Exp Med. 2010;207(11):2469–2477. doi:10.1084/jem.20092450.
- Di Genova G, Savelyeva N, Suchacki A, Thirdborough SM, Stevenson FK. Bystander stimulation of activated CD4 + T cells of unrelated specificity following a booster vaccination with tetanus toxoid. Eur J Immunol. 2010;40(4):976–985. doi:10.1002/eji.200940017.
- Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70(21):8368–8377. doi:10.1158/0008-5472.CAN-10-1322.
- Mathur D, Prakash S, Anand P, Kaur H, Agrawal P, Mehta A, Kumar R, Singh S, Raghava GPS. PEPlife: a repository of the half-life of peptides. Sci Rep. 2016;6(1):36617. doi:10.1038/srep36617.
- Moynihan KD, Holden RL, Mehta NK, Wang C, Karver MR, Dinter J, Liang S, Abraham W, Melo MB, Zhang AQ, et al. Enhancement of peptide vaccine immunogenicity by increasing lymphatic drainage and boosting serum stability. Cancer Immunol Res. 2018 Jun 18:canimm.0607.2017. doi:10.1158/2326-6066.CIR-17-0607.
- Roche PA, Teletski CL, Stang E, Bakke O, Long EO. Cell surface HLA-DR-invariant chain complexes are targeted to endosomes by rapid internalization. Proc Natl Acad Sci USA. 1993;90(18):8581–8585. doi:10.1073/pnas.90.18.8581.
- Armstrong TD, Clements VK, Martin BK, Ting JP, Ostrand-Rosenberg S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc Natl Acad Sci USA. 1997;94(13):6886–6891. doi:10.1073/PNAS.94.13.6886.
- Dasari V, Rehan S, Tey S-K, Smyth MJ, Smith C, Khanna R. Autophagy and proteasome interconnect to coordinate cross-presentation through MHC class I pathway in B cells. Immunol Cell Biol. 2016;94(10):964–974. doi:10.1038/icb.2016.59.
- Walseng E, Wälchli S, Fallang L-E, Yang W, Vefferstad A, Areffard A, Olweus J. Soluble T-cell receptors produced in human cells for targeted delivery. PLoS One. 2015;10(4). doi:10.1371/journal.pone.0119559.
- Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosenberg SA. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res. 2009;15(1):169–180. doi:10.1158/1078-0432.CCR-08-1638.
- Walchli S, Loset GA, Kumari S, Johansen JN, Yang W, Sandlie I, Olweus J. A practical approach to T-cell receptor cloning and expression. PLoS One. 2011;6(11):e27930. doi:10.1371/journal.pone.0027930.