
ABSTRACT
The antigenic similarity between embryos and tumors has raised the idea of using embryonic material as a preventative vaccine against neoplastic disease. Indeed, we have previously reported that a vaccine comprises allogeneic murine embryonic stem cells (ESCs) and murine fibroblasts expressing GM-CSF (to amplify immune responses) successfully blocks the outgrowth of an implantable cancer (Lewis lung carcinoma; LLC) and lung tumors generated in mice using a combination of a mutagen followed by chronic pulmonary inflammation. However, such a vaccine is obviously impractical for application to humans. The use of fibroblasts to generate GM-CSF is needlessly complicated, and intact whole ESCs carry the hazard of generating embryomas/teratomas. Here, we report the successful application of an alternative prophylactic vaccine comprises exosomes derived from murine ESCs engineered to produce GM-CSF. Vaccination of mice with these exosomes significantly slowed or blocked the outgrowth of implanted LLC while control exosomes lacking GM-CSF were ineffective. Examination of tumor-infiltrating immune cells from mice vaccinated with the GM-CSF-expressing exosomes showed robust tumor-reactive CD8+ T effector responses, Th1 cytokine responses, and higher CD8+ T effector/CD4+CD25+Foxp3+ T regulatory cell ratio in the tumors. We conclude that a similar vaccine derived from GM-CSF- expressing human ESCs can be employed as a preventative vaccine for humans with an increased risk of developing cancer.
Introduction
A popular hypothesis of tumorigenesis suggests that mutations in undifferentiated progenitor cells give rise to malignant cells that are capable of both self-renewal and differentiation. Interestingly, this theory essentially predicted the discovery of cancer-initiating cells (CICs) that came over 100 years later. In an example of history repeating itself, in the mid-1960s, tumor cells and embryonic stem cells were shown to possess common gene products such as the oncofetal antigens. During the ensuing decade, a large number of studies confirmed these findings and revealed that embryonic antigens are re-expressed in cells from solid tumors from a number of different tissues and that vaccination of animals with fetal material can prevent the outgrowth of tumors (see review)Citation1. It now appears that most, if not all, types of neoplastic cells express certain embryonal antigens; this phenomenon has been termed “retrogenetic expression”.Citation2 Many of these embryonic gene products (also called carcinoembryonic antigens) are not expressed in the adults and thus, are not included in the repertoire of ‘self’ during the process of thymic selection that occurs near the end of gestation. Such ‘non-self’ embryonic antigens are immunogenic and can prime the immune cells to mount an anti-tumor response.Citation3 Exploiting such embryonic antigen immunogenicity, we have designed a unique stem cell-based vaccine that stimulates the immune system to recognize shared oncofetal antigens and confers protection against tumors.
In our initial attempts to produce a prophylactic vaccine we successfully combined irradiated allogeneic murine embryonic stem cells (ESCs) with murine fibroblasts expressing GM-CSF (STO-GM) as an immune stimulant (ES cell vaccine).Citation4 We discovered that ES cell vaccination was 70–100% effective in preventing both implanted and carcinogen-induced lung adenocarcinomas.Citation4 Vaccinated mice remained tumor-free over a 90-day observation period. Importantly, irradiated STO-GM cells provided no protection against the outgrowth of LLC tumors, suggesting that the observed protection with ESC/STO-GM is not due to non-specific immune responses against allogeneic whole cell antigens.Citation4 A very recent study by Kooreman et al.,Citation5 supports our earlier results albeit with a different form of pluripotent stem cells (induced pluripotent stem cells; iPSCs) and a different adjuvant (CpG) and lends further credence to the theory that embryonic material can be an effective prophylactic vaccine against “other” non-mutated neo-antigens, i.e., tumor antigens derived from proteins that are only expressed during embryonic development and not in adult tissues.
Although the ES cell vaccine holds promise for inducing anti-lung cancer immune responses, such an approach has two obvious problems in terms of application to humans: (i) The use of whole ESCs – although irradiated – raises the hazard of the generation of embryomas/teratomas. (ii) Using murine fibroblasts to generate GM-CSF is needlessly complicated. To overcome these challenges, we have developed an alternative prophylactic vaccine composed of murine ES cells engineered to produce GM-CSF in amounts similar to those produced by the aforementioned fibroblasts.Citation4 We then purified ESC-derived exosomes (ES-exo), thereby producing a self-contained, relatively stable exosome-based vaccine (and obviating the need for the administration of intact ESCs).
Exosomes are cell-derived nanovesiclesCitation6 (50–100 nm) that have recently gained renewed interest as they unveil immense potential for cancer therapy.Citation7 Within the cells, invagination and budding from the limiting membrane of late endosomes results in the formation of vesicles that contain cytosol and the extracellular domain of transferrin receptors at their surface. These internal vesicles (called exosomes) are then secreted into the extracellular environment following the fusion of multivesicular endosomes with the plasma membrane.Citation8 In vitro and in vivo studies suggest that exosomes can bind to target-cell membranes, or can fuse with target cells and, so, exchange membrane proteins and cytosol between two cell types.Citation9 Importantly, exosomes seem to also transfer nucleic acids such as mRNA and microRNA and thus, represent a new paradigm of genetic exchange between cells.Citation9,Citation10 Recent studies indicate that exosomes can operate as potential immunotherapeutic agents, with promising results in pre-clinical studies of cancer immunotherapy.Citation7 Exosomes have several advantages over cell-based therapies owing to high bio-availability, bio-stability, and lower costs.Citation11,Citation12 Since exosomes can deliver large amounts of cargo directly to target cells, and this property of exosomes can be exploited to include therapeutics as well as immunostimulatory adjuvants in the engineered exosomes.Citation13
Here we show that, in a prophylactic setting, vaccination of mice with ESC-exosomes expressing GM-CSF (ES-exo/GM-CSF) is very effective in preventing implantable lung tumors with no detectable toxicity. Importantly, anti-tumor efficacy of the ES-exo/GM-CSF combination vaccine is associated with robust CD8+ T effector responses, infiltration of CD8+ T cells into the tumor leading to increased intratumoral CD8+ T effector/T regulatory cell ratio in the tumors. Collectively, our findings provide a strong rationale for further developing this novel cell-free exosome-based vaccination strategy for the prevention of cancer.
Results
Stable expression of GM-CSF in pluripotent murine embryonic cells
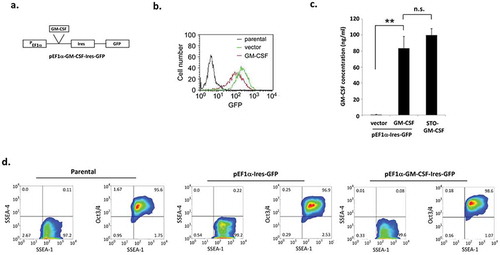
Our earlier efforts to over-express GM-CSF in murine ESCs by retroviral infection were largely unsuccessful, probably due to transcriptional suppression of endogenous and exogenous retroviruses in those cells.Citation14 Previous studies that tested different viral and cellular promoters have demonstrated that the cellular elongation factor-1α (EF1α) promoter efficiently drives exogenous gene expression in murine ESCs.Citation15,Citation16 Under the control of an EF1α promoter, GM-CSF was stably over-expressed in ES-D3 cells by transfection. As shown in , the vector employed in these studies expresses both GM-CSF and GFP from the EF1α promoter with an IRES sequence allowing us to use the expression of GFP as a marker to track GM-CSF expression. Flow cytometry analysis revealed that both GM-CSF-expressing and empty vector control ES-D3 cells express GFP at high levels in comparison to untransfected parental ES-D3 cells (). As shown in , the amounts of GM-CSF secreted by ES-D3 cells are roughly equivalent to those secreted by the STO fibroblasts employed in earlier experiments.Citation4 GM-CSF has been shown to promote differentiation of immune cells ex vivo under certain culture conditions.Citation17 Therefore, to ensure that GM-CSF-expressing ES-D3 cells maintain their pluripotent undifferentiated state, we analyzed cellular expression of multiple markers of pluripotency (SSEA-1 and Oct-3/4) and differentiation (SSEA-4).Citation18 As shown in , >95% of each of parental and transfected ES-D3 cells were positive for the expression of pluripotency markers Oct 3/4 and SSEA-1, and <1% of these cells were positive for the expression of the differentiation marker – SSEA-4. These data suggest that expression of GM-CSF in ES-D3 cells did not alter their pluripotency.
Figure 1. Murine embryonic stem cells expressing GM-CSF maintain their pluripotency.
(a) Schematic diagram of the plasmid with the EF1-α promoter driving GM-CSF expression. (b) Expression of GFP in GM-CSF-expressing ES-D3 cells and in empty vector control ES-D3 cells was evaluated by flow cytometry. (c) GM-CSF levels in transfected ES-D3 cells. ELISA measurements of GM-CSF concentrations in the medium of the indicated cells. The data are shown as mean ± standard deviations (mean ± SD) of three independements, **, p < 0.001; ANOVA with Tukey’s multiple comparison test, n.s = not significant. (d) Flow cytometry analysis of the expression of pluripotency and differentiation markers in parental, vector control and GM-CSF-expressing ES-D3 cells. The data shown are representative of three independent flow cytometry assays.
Characterization of exosomes isolated from GM-CSF-expressing murine embryonic stem cells (ESCs)
Exosomes from vector control and GM-CSF-expressing ES-D3 cells were isolated and examined by transmission electron microscopy (TEM) (). TEM images revealed the presence of exosomes of varying sizes, a common feature of exosomal preparations.Citation19 Also, expression of exosomal markers – CD81, Alix, annexin V, Flotillin-1, and CD86 – was enriched in ESC-derived exosomes when compared to that of the corresponding whole cell extracts (WCE). Furthermore, ESC-derived exosomes lacked expression of (i) endoplasmic reticulum markers – protein disulfide isomerase (PDI) and calnexin, (ii) mitochondrial markers – cytochrome C and COX IV-subunit IV, and (iii) the cytosolic marker GAPDH, demonstrating the purity of ESC-derived exosomal material. ().Citation20 We used a proteomics approach to analyze the profile of exosomal proteins. As expected, well-known exosomal protein markers, such as CD81, annexin V, and Alix, were present in exosomes obtained from ES-D3 cells [Supplemental Table 1].Citation21 Importantly, tumor antigens identified in a variety of tumor types were also localized in exosomes from ES-D3 cells,Citation22–Citation24 which is in agreement with the idea that embryos and tumors share similar antigens.Citation2,Citation25–Citation27
Figure 2. Characterization of exosomes isolated from ES-D3 cells.
(a) Exosomes were isolated from ES-D3 cells that were transfected with either the plasmid expressing GM-CSF or its empty vector. Transmission electron microscopy (TEM) imaging of the ES-D3-derived exosomes. Arrow heads indicate exosomes, Scale bar 100 nM. (b) Western blot analysis of the expression of the indicated exosomal markers, endoplasmic reticulum (ER) markers, mitochondrial markers, and cytosolic markers that are expressed either in the whole cell extracts (WCE) or in the exosomes. PDI, protein disulfide isomerase; cyto C, cytochrome C. Molecular weights markers (kD) are labeled on the left. (c) The amounts of GM-CSF in exosomes isolated from GM-CSF-expressing ES-D3 cells or from vector control ES-D3 cells were evaluated by ELISA. Exosomes pretreated with or without 0.05% Tween-20 were added to an ELISA plate. ELISA was carried out using washing buffer containing either PBS only or PBS + 0.05% Tween-20. The data are shown as mean ± SD of three independent ELISA experiments. ***, p < 0.0001; ANOVA with Tukey’s multiple comparison test.
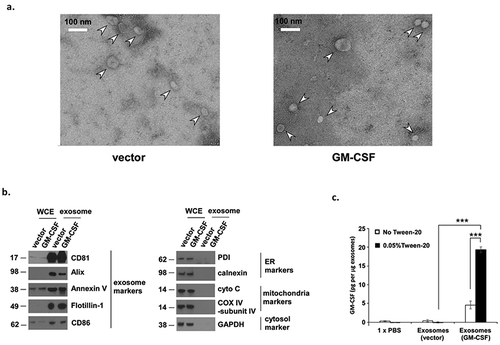
We evaluated the levels of GM-CSF in isolated exosomes using ELISA (). Exosomes isolated from vector control cells or GM-CSF-expressing cells were pre-treated with the detergent Tween-20 (0.05%) to permeabilize their membranes. Also, presence of Tween-20 in the wash buffer used during the assay significantly reduced the background GM-CSF levels detected in the control exosomes, but increased the amounts of GM-CSF detected from the exosomes isolated from GM-CSF-expressing cells (), implying that Tween-20 is permeabilizing the exosomes and thus, rendering intra-exosomal GM-CSF available for detection. These data suggest that almost all GM-CSF is in the lumen of the exosomes. We also examined the sensitivity of exosomal proteins to proteinase K treatment. While exosomal membrane-spanning proteins Annexin V and CD81 were protected from proteinase K, exosomal lumen protein Alix was largely digested, indicating that proteinase K treatment is not a reliable approach to determine the localization of an exosomal protein (Supplemental Figure 1).
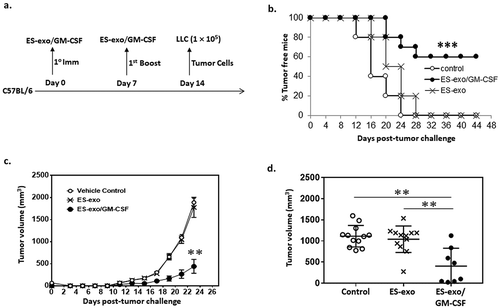
Vaccination with exosomes isolated from GM-CSF-expressing ESCs prevents the outgrowth of an implanted lung adenocarcinoma
Using a standard vaccination timing regimen (), C57BL/6 mice (n = 20) were immunized twice (days 0 and 7) with HBSS (control), exosomes alone (ES-exo) or GM-CSF-containing exosomes (ES-exo/GM-CSF). Mice were then challenged with s.c. inoculation of LLC (day 14) and monitored for tumor outgrowth. As shown in , vaccination of mice with ES-exo/GM-CSF was 60% effective in preventing tumor outgrowth whereas all non-vaccinated control animals had developed palpable tumors by day 24 post-challenge. More importantly, those LLC tumors that did develop in ES-exo/GM-CSF vaccinated mice (n = 8) were significantly smaller and tumor growth rate was greatly reduced compared to those developing in non-vaccinated control mice and in ES-exo-vaccinated mice (). Tumor volumes in individual mice measured at week 3 were significantly less in the vaccinated animals [12 mice in control and ES-exo groups and 8 mice in ES-exo/GM-CSF group ()]. We also evaluated if any toxicological effects are associated with vaccine treatment (after two once-weekly vaccinations). CBC analysis showed no significant difference in blood cell counts between control, ES-exo and ES-exo/GM-CSF-vaccinated mice (Supplemental Figure 2). Additionally, mice receiving ES-exo/GM-CSF did not display significantly altered liver or kidney enzymes when compared to control and ES-exo vaccine groups of mice (Supplemental Figure 2). These studies indicate that ES-exo/GM-CSF vaccine did not induce any toxic effects in the treated mice.
Figure 3. ESC-derived exosome vaccination prevents the outgrowth of an implanted lung adenocarcinoma.
(a) Scheme of immunization. Male C57BL/6 mice were immunized twice (days 0 and 7) with vehicle only (HBSS control) or with exosomes isolated from the vector control ES-D3 cells (ES-exo) or with exosomes isolated from ES-D3 cells over-expressing GM-CSF (ES-exo/GM-CSF) in the right flank prior to s.c. challenge with LLC on day 14. (b) C57BL/6 mice (20 mice/group) were immunized twice (days 0 and 7) with HBSS (control) or ES-exo or ES-exo/GM-CSF in the right flank prior to s.c. challenge with LLC on day 14. Tumor growth was monitored daily in all animals until sacrifice due to tumors exceeding 5% of body weight. The ES-exo/GM-CSF-vaccinated tumor-free mice remained so for up to 4 months later with no overt signs of distress. Results are representative of three independent experiments. ***, p < 0.0001; relative to control group; log-rank test. (c) Tumor growth was measured with calipers every 2nd or 3rd day and tumor volumes were plotted as indicated. The data represent the average tumor volumes of 20 mice/control group and 8 mice/ES-exo/GM-CSF group and are representative of three independent experiments. Error bars represent mean ± SD. (d) Scatter plot showing tumor volume in individual mice measured at week 3 (n = 12 mice in control and ES-exo groups and 8 mice in ES-exo/GM-CSF group, mean ± SD, **, p < 0.001; ANOVA with Tukey’s multiple comparison test).
Vaccination with GM-CSF-expressing ESC-derived exosomes induces tumor cell-specific Th1-mediated cytokine response in CD8+ T cells
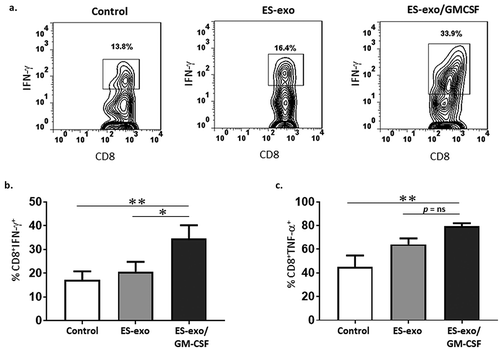
We next determined the ability of CD8+ T cells from vaccinated mice to produce effector cytokines required for effective anti-tumoral cytolytic activity. In response to re-stimulation with LLC cell lysate, a significantly higher frequency of IFN-γ and TNF-α-producing CD8+ splenocytes were obtained from ES-exo/GM-CSF- vaccinated mice when compared with the non-vaccinated control and ES-exo- vaccinated mice (–). In the absence of LLC re-stimulation, no increase in cytokine production was observed in CD8+ splenocytes from vaccinated mice when compared to unstimulated, control non-vaccinated mice (data not shown). To test whether the ES-exo/GM-CSF vaccine provides immunity against shared epitopes expressed in other cancer cells, splenocytes obtained from exosome-vaccinated mice were stimulated with lysates of B16-F10 melanoma, MC-38 colon adenocarcinoma, and E0771 medullary breast adenocarcinoma cells. Cytometry analysis showed that a significant increase in IFN-γ production in CD8+ splenocytes from ES-exo/GM-CSF-vaccinated mice in response to stimulation with two of the three cancer cell lysates – B16F10 and MC-38 – when compared with the non-vaccinated control and ES-exo-vaccinated mice (–). Importantly, analysis of the phenotype of tumor-infiltrating immune cells supports the concept that vaccination led to an immune-based suppression of tumor growth. Using tumors isolated from unvaccinated, ES-exo- and ES-exo/GM-CSF-vaccinated mice, we found substantial increases in IFN-γ- and TNF-α-producing CD8+ T cells only in mice vaccinated with exosomes prepared from GM-CSF-expressing ESCs in response to re-stimulation with LLC lysate (–).
Figure 4. ESC-derived exosome vaccination induces Th1-mediated cytokine responses in splenic CD8+ T cells.
(a–c) C57BL/6 mice (n = 6 per group) were immunized twice (days 0 and 7) with vehicle only (HBSS control) or with exosomes isolated from vector control ES-D3 cells (ES-exo) or with exosomes isolated from ES-D3 cells over-expressing GM-CSF (ES-exo/GM-CSF) in the right flank. Ten days after the boost, mice were euthanized and spleens were removed. Splenocytes from vaccinated and control mice were co-cultured with LLC lysate (50 μg/mL) for an additional 4 days. Effector cells were then restimulated for 6 h with LLC lysate (50 μg/mL) in the presence of Brefeldin A (1 µL/mL of the culture medium). After restimulation, cells were harvested, Fc receptors were blocked, and stained for surface expression of CD3, CD8 and intracellular expression of cytokines and analyzed by flow cytometry. (a) Dot plots showing IFN-γ expression in CD8+ T cells in splenocyte cultures obtained from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. Numbers in quadrants represent the percentages of each subpopulation. (b, c) Bar graphs showing percentages of CD8+IFN-γ+, and CD8+TNF-α+ in splenocyte cultures derived from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. Results are expressed as percentages of total cells (n = 6 per group, mean ± SD, **, p < 0.001; ***, p < 0.0001; ANOVA with Tukey’s multiple comparison test). (d–f) ESC-derived exosome vaccination induces Th1-mediated cytokine response in splenic CD8+ T cells when stimulated with antigens expressed by several tumor cell lines. Splenocytes from vaccinated and control mice were co-cultured with B16-F10, MC-38, E0771 lysate at 50 μg/ml for 4 days. Effector cells were then restimulated for 6 h with B16-F10 cell lysate (50 μg/mL) (d), MC-38 cell lysate (50 μg/mL) (e), and E0771 lysate (50 μg/mL) (f), in the presence of Brefeldin A (1 µL/mL of the culture medium). After restimulation, cells were harvested, Fc receptors were blocked, and stained for surface expression of CD8 and intracellular expression of IFN-γ and analyzed by flow cytometry. (d–f) Bar graphs showing percentages of CD45+CD3+CD8+IFN-γ+ in splenocytes derived from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. Results are expressed as percentages of total CD45+ cells (n = 6 per group, mean ± SD, **, p < 0.001; ***, p < 0.0001; p = ns; ANOVA with Tukey’s multiple comparison test).
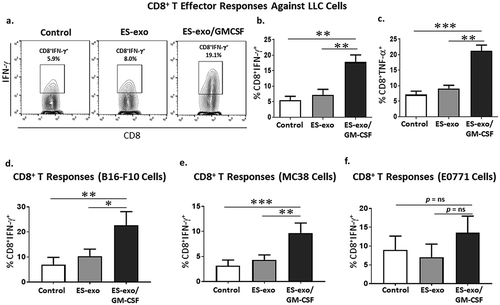
Figure 5. ESC-derived exosome vaccination induces Th1-mediated cytokine responses in intra-tumoral CD8+ T cells.
(a–c) C57BL/6 mice (n = 6 per group) were immunized twice (days 0 and 7) with vehicle only (HBSS control) or with exosomes from vector control ES-D3 cells (ES-exo) or with exosomes isolated from ES-D3 cells over-expressing GM-CSF (ES-exo/GM-CSF) in the right flank prior to s.c. challenge with LLC on day 14. Mice were euthanized 15–18 days after tumor challenge, tumors were removed and enzymatically digested. Tumor-infiltrating cells from vaccinated and control mice were stimulated with LLC lysate (50 μg/mL) for 24 h. Cells were then restimulated for 6 h with LLC lysate (50 μg/mL) in the presence of Brefeldin A (1 µL/mL of the culture medium). After restimulation, cells were harvested, Fc receptors were blocked, and stained for surface expression of CD8 and intracellular expression of cytokines and analyzed by flow cytometry. (a) Dot plots showing IFN-γ expression in CD45+CD3+CD8+ cells in tumor-infiltrating cells obtained from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. Numbers in quadrants represent the percentages of each subpopulation. (b, c) Bar graphs showing percentages of CD45+CD3+CD8+IFN-γ+ (b) and CD45+CD3+CD8+TNF-α+ (c) in tumors derived from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. Results are expressed as percentages of total CD45+ cells (n = 6 per group, mean ± SD, *, p < 0.05 **, p < 0.001; ANOVA with Tukey’s multiple comparison test).
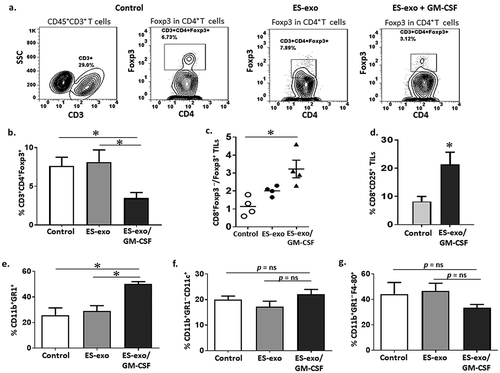
We next analyzed the effects of ES-exo/GM-CSF vaccination on CD4+CD25+Foxp3+ T regulatory cells (Tregs) and CD11b+Gr1+ myeloid-derived suppressor cells (MDSCs) – two prominent suppressor populations that hamper anti-tumoral effector responses – in the spleen.Citation28–Citation30 Our data showed that our vaccination strategy did not reduce the percentages of splenic Tregs (Supplemental Figure 3A, B) or induce any change in the ratio of CD8+ T cells to Tregs in the spleen (Supplemental Figure 3C). Also, we did not observe any significant differences in the percentages of CD4+ and CD8+ T cells (Supplemental Figure 3D) or in their absolute number (data not shown) in the spleens from vaccinated and control non-vaccinated mice. The percentage of MDSCs, however, was significantly decreased in the spleens of mice vaccinated with ES-exo/GM-CSF and challenged with LLC cells when compared to that in non-vaccinated, LLC challenged control mice (Supplemental Figure 3E). We did not observe any significant differences in the percentages of splenic CD11b+Gr-1–CD11c+ dendritic cells (Supplemental Figure 3F), and CD11b+Gr-1–F4-80+ macrophages (data not shown) between control and vaccinated groups.
Vaccination with GMCSF-expressing ESC-derived exosomes increases the ratio of CD8+ T effector cells to Tregs in the tumor
Our results thus far suggest that the ES-exo/GM-CSF vaccine-induced anti-tumor efficacy is reliant on the CD8+ T effector cells. We, therefore, analyzed the effects of our vaccination strategy on the phenotype of tumor-infiltrating CD8+ T cells, Tregs, and MDSCs. Tumors from controls and vaccinated mice (from the small numbers of ES-exo/GM-CSF- vaccinated mice that did develop LLC lesions) were harvested and used to investigate the subset profiles of tumor-infiltrating immune cells. Flow cytometry analysis showed a significant decrease in the percentage of CD4+CD25+Foxp3+ Tregs in tumor infiltrates from ES-exo/GM-CSF-vaccinated mice when compared with non-vaccinated control and ES-exo-vaccinated mice (–) and the ratio of CD8+ T cells to Tregs was significantly increased in the tumor infiltrates from ES-exo/GM-CSF-vaccinated mice (). Additionally, CD8+ cells in the ES-exo/GM-CSF tumor infiltrates had significantly elevated expression of the activation marker CD25 (). Finally, for unexplained reasons, we observed a significant increase in the percentages of CD11b+Gr-1+ MDSCs in tumors from ES-exo/GM-CSF-vaccinated mice () but there were no significant differences in the percentages of tumor-infiltrating CD11b+Gr-1–CD11c+ dendritic cells and CD11b+Gr-1–F4-80+ macrophages between control and vaccinated groups (–). Whether this reflects GM-CSF-mediated generation or attraction of MDSCs to the tumors is unclear at this time.Citation31–Citation33
Figure 6. ESC-derived exosome vaccination decreases T regulatory (Treg) cells and increases the ratio of effector CD8+ T cells to Treg in the tumors.
C57BL/6 mice (n = 4 per group) were immunized twice (days 0 and 7) with vehicle only (HBSS control) or with exosomes from vector control ES-D3 cells (ES-exo) or with exosomes isolated from ES-D3 cells over-expressing GM-CSF (ES-exo/GM-CSF) in the right flank prior to s.c. challenge with LLC on day 14. Mice were euthanized 15–18 days after tumor challenge, tumors were removed, enzymatically digested, and tumor-infiltrating cells were harvested from control and vaccinated mice and analyzed by flow cytometry. (a) Dot plots showing the percentages of CD3+CD4+Foxp3+ Tregs in CD45+ tumor-infiltrating cells obtained from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. Numbers in quadrants represent the percentages of each subpopulation. (b) Bar graphs showing the percentages of CD3+CD4+Foxp3+ Tregs sub-populations in CD45+ tumor infiltrating cells from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. Results are expressed as percentages of total CD45+ cells (n = 4 per group, mean ± SD, *, p < 0.05; ANOVA with Tukey’s multiple comparison test). (c) Bar graph showing the ratio of CD8+ Foxp3 – to CD8–Foxp3+ cells in one of two representative experiments (n = 4 per group, mean ± SD, *, p < 0.05; ANOVA with Tukey’s multiple comparison test). (d) ESC-derived exosome vaccination increases the frequency of functional CD8+ T cells in tumors. Bar graph showing the percentages of CD25+CD8+ in CD45+ tumor-infiltrating cells obtained from control and ES-exo/GM-CSF-vaccinated mice. Results are expressed as percentages of total cells. The data represent results from two independent experiments with three mice/group. *, p < 0.05; relative to control group; unpaired t test. Error bars represent mean ± SD. (e) ESC-derived exosome vaccination increases myeloid-derived suppressor cells (MDSCs) in the tumors. Bar graphs showing the percentages of CD11b+Gr-1+ MDSCs sub-populations in CD45+ tumor infiltrating cells. Results are expressed as percentages of total CD45+ cells (n = 4 per group, mean ± SD, *, p < 0.05; ANOVA with Tukey’s multiple comparison test). (f, g) Bar graphs showing the percentages of CD11b+Gr-1–CD11c+ dendritic cells (f) and percentages of CD11b+Gr-1–F4-80+ macrophages (g) in CD45+ tumor-infiltrating cells obtained from control, ES-exo- and ES-exo/GM-CSF-vaccinated mice. (n = 4 per group, mean ± SD, p = ns; ANOVA with Tukey’s multiple comparison test).
ESC-derived exosome vaccination prevents the outgrowth of an implanted mammary carcinoma
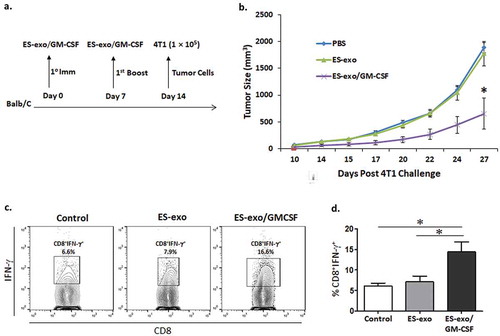
To test the effectiveness of ES-exo/GM-CSF vaccine in targeting multiple cancer types, an additional experiment was performed using the mammary carcinoma cell line 4T1, which is syngeneic to the Balb/c mouse strain. Female Balb/c mice (30 mice) were divided into HBSS, ES-exo and ES-exo/GM-CSF groups and treated for two weeks (s.c. route). Following this, 4T1 cells (1 × 105) were subcutaneously injected (). Tumor growth assessment by caliper measurement showed significantly lower tumor progression in the ES-exo/GM-CSF group (). Using splenocytes isolated from unvaccinated, ES-exo- and ES-exo/GM-CSF-vaccinated mice, we found increases in IFN-γ-producing CD8+ T cells in mice vaccinated with ES-exo/GM-CSF in response to re-stimulation with 4T1 lysate (–).
Figure 7. ESC-derived exosome vaccination slows the outgrowth of an implanted mammary carcinoma.
(a) Scheme of immunization. Female Balb/c mice were immunized twice (days 0 and 7) with vehicle only (HBSS control) or with exosomes from vector control ES-D3 cells (ES-exo) or with exosomes isolated from ES-D3 cells over-expressing GM-CSF (ES-exo/GM-CSF) in the right flank prior to s.c. challenge with syngeneic 4T1 cells on day 14. (b) Tumor growth was measured with calipers every second or third day and tumor volumes were plotted as indicated. The data represent the average tumor volumes of 20 mice/group. Error bars represent mean ± SD. (c, d) ESC-derived exosome vaccination induces Th1-mediated cytokine responses in splenic CD8+ T cells. Splenocytes from vaccinated and control mice were co-cultured with 4T1 lysate (50 μg/ml) for an additional 4 days. Effector cells were then restimulated for 6 h with 4T1 lysate (50 μg/mL) in the presence of Brefeldin A (1 µL/mL of the culture medium). After restimulation, cells were harvested, Fc receptors were blocked, and stained for surface expression of CD8 and intracellular expression of IFN-γ and analyzed by flow cytometry. (c) Dot plots showing IFN-γ expression in CD8+ cells in splenocyte cultures obtained from control, ES-exo and ES-exo/GM-CSF vaccinated mice. Numbers in quadrants represent the percentages of each subpopulation. (d) Bar graphs showing percentages of CD8+IFN-γ+ in splenocyte cultures derived from control, ES-exo and ES-exo/GM-CSF vaccinated mice. Results are expressed as percentages of total cells (n = 4 per group, mean ± SD, *, p < 0.05; ANOVA with Tukey’s multiple comparison test).
Discussion
Vaccines have vastly improved human welfare and longevity. They have led to the disappearance of smallpox and near-eradication of polio. These anti-bacterial and anti-viral vaccines are most often polyvalent, prepared from killed or attenuated organisms. Polyvalency may partially explain the effectiveness of these vaccines. In fact, truly positive results with a long-sought vaccine against malaria have been reported only with Plasmodium falciparum sporozoite preparations.Citation34 By contrast, most attempts to use monovalent vaccines as therapy or prophylaxis for various cancers have met with very limited success (such as the failure of Sipuleucel-T – a monovalent vaccine composed of a GM-CSF/prostatic acid phosphatase fusion to provide significant improvements in overall survival of patients with prostate cancer).Citation35
Our own attempts to generate a prophylactic vaccine for cancer prevention were prompted by previous observations of antigenic similarities between embryos and cancers (so-called ‘carcinoembryonic’ antigens). As reviewed in an earlier publicationCitation1, a large number of such shared antigens have been identified (and more may still be found). Research work published by Li et al.Citation36 and Dong et al.,Citation37 demonstrate that undifferentiated, pluripotent ESC delay tumor growth in implantable mouse models of transplantable colon carcinoma and lung cancer. In the study published by Dong et al., immunization of mice with ESC cells alone in the absence of any adjuvant slowed lung tumor growth; roughly 40% of mice were protected from tumor growth.Citation37 Their protocol involved a primary vaccination followed by two boosts. Under a similar vaccination strategy, we have also seen moderate protection against tumor outgrowth. However, our previously published work indicated that vaccination with irradiated, allogeneic murine ESC along with allogeneic fibroblasts expressing GM-CSF as an immunostimulatory adjuvant was 70–100% effective in preventing both implantable and carcinogen-induced lung adenocarcinomas without detectable toxicityCitation4 and the majority of the vaccinated mice remained tumor-free for over a year.Citation4 A major advantage of whole cell vaccination is that a broad range of antigens can be presented to T cells,Citation38 including known as well as unknown antigens that are shared between various tumor cells and embryonic cells. However, ethical concerns that arise from the use of whole ESCs may preclude such an approach from clinical translation.
Here, we report that exosomes derived from GM-CSF-expressing murine ESCs can be used as a vaccine for the prevention of tumor outgrowth. In those animals that did develop tumors, tumor growth was significantly slower when compared to unvaccinated control animals and those vaccinated with exosomes from ESCs which do not express GM-CSF. In the latter group of animals, tumor outgrowth did not differ from that seen in unvaccinated mice. These observations are in agreement with earlier reports of the potency of GM-CSF as an immune stimulant.Citation39 Indeed, this property has been recently exploited by Bencherif et al. for amplification of immune responses in a melanoma model.Citation40 In our experimental tumor model, ES-exo/GM-CSF combination vaccine significantly increases the ratio of CD8+ T cells to Tregs and the percentages of CD8+CD25+ and CD8+IFN-γ+ effector cells within the tumors that suggest effective vaccine-induced, tumor-reactive immune system priming. These immunophenotyping data lend additional support to the tentative conclusion that such vaccination might be a viable approach to the prevention of cancers in humans.
In recent years, exosomes have emerged as promising agents for immunotherapy of cancer. The exosome-based anti-cancer therapies have high stability, in vivo bioavailability, and an inherent ability to stimulate anti-tumor immune responses. Exosomes do not endogenously replicate, as whole cells would and exosomes can be readily bioengineered to a clinical grade and scaled up for dosing requirements.Citation11 The idea of using tumor-derived exosomes as a vaccine has been proposed earlier (see reviewCitation41) although this approach is complicated by the fact that such preparations may also be strongly immunosuppressive. Furthermore, the idea of using tumors expressing GM-CSF as a therapeutic vaccine has been explored by Dranoff and several others.Citation42,Citation43 This has shown variable success in certain animal models. In pre-clinical studies, exosomes obtained from matured dendritic cells (DCs) express more abundant MHC-I and MHC-II molecules as well as co-stimulatory molecules (e.g., CD40, CD80, and CD86) and induce potent antigen-specific anti-tumor T effector responses shown by CTL and NK cells both in vitro and in vivo.Citation44–Citation47 Similarly, ES-exo comprises stable vesicles harboring protein contents that can be tailor-manufactured from human cell lines in clinical grade (cGMP) quality.Citation11 Furthermore, ES-derived exosomes can be produced in large quantities and cryo-preserved for more than 6 months at −80°C with their functional activity intact.Citation19
It is both a strength and weakness of the current experiments that we used intact exosomes from GM-CSF expressing murine ESCs. The obvious weakness is that we are presently unable to identify the antigens involved in the cross-recognition of tumor cells which presumably involves CD8+ cytotoxic T lymphocytes (as reported previouslyCitation4). However, the strength of this approach may reside in the fact that this is a polyvalent vaccine resulting in immune recognition of a number of antigens shared by early embryos and tumors. Our preliminary proteomics studies described here (Supplemental Table 1) suggest that ESC-derived exosomes harbor multiple tumor antigens [e.g. Alpha-fetoprotein, CD151, and Ephrin type-A receptor 2 (EPHA2); Supplemental Table 1]. Based on the strong in vivo anti-tumor T cell effector responses that we have observed with this vaccine, it is very likely that at least one or more of the tumor-cell expressed embryonal antigens will be involved in eliciting anti-tumor immune responses.
Since the anti-tumor activity of the vaccine depends on the ESC-induced recognition of relevant antigens expressed in the tumor, our future studies will be aimed at identifying the cross-reactive lung tumor antigens using ES-exo and lung tumors. These studies will employ a high through-put proteomics-based screening platform in combination with mass spectrometry techniques to identify common antigens expressed in ES-derived exosomes and lung tumors. A recent report by Dr. Tuohy’s group showed that targeting the extracellular domain of anti-Mullerian hormone receptor II (AMHR2-ED) is an effective strategy for immunoprevention of ovarian cancer.Citation48 AMHR2-ED is a so-called “retired antigen” that is required for tissue-specific functions and its expression decreases with aging.Citation48,Citation49 Similar to testis-associated antigens (e.g. NY-ESO-1), AMHR2-ED antigen is specifically overexpressed in ovarian cancer cells but shows limited expression in normal tissues and hence forms an appealing target for ovarian cancer vaccine development with minimal risk for inducing autoimmunity.Citation48,Citation49 Another such example of a “retired antigen” is alpha-lactalbumin which is a breast-specific differentiation protein conditionally expressed in high levels in breast carcinomas only during lactation.Citation50 Tuohy and colleagues reported that immunoreactivity against alpha-lactalbumin provides protection against the growth of breast tumors without any detectable inflammation in normal non-lactating breast tissue.Citation50 Finally, Kooreman et al. in a very recent publication show that human and murine iPSCs express tumor-associated antigens and that irradiated autologous iPSCs, in a prophylactic setting, prevent tumor growth in syngeneic murine breast cancer, mesothelioma, and melanoma models.Citation5 These recent studies lend further credence to the concept that embryonic material can be an effective immunopreventive cancer vaccine.
Materials and methods
Generation of cell lines
Murine embryonic stem cell line ES-D3, syngeneic to C57BL/6 mice, was acquired from ATCC (CRL-11632), independently verified to be pathogen-free. KnockOut™ Dulbecco’s Modified Eagle’s Medium (DMEM; 10-829-018), β-mercaptoethanol (21-985-023), non-essential amino acids (SH3023801), G418 (10-131-035), gelatin (ES006B), and Leukemia Inhibitory Factor (LIF; ESG1106) were all purchased from Thermo Fisher. Proteinase K (P2308) and polybrene (H9268) were obtained from Sigma-Aldrich. Penicillin/streptomycin (sc45000-652), L-glutamine (VWRL0131-0100) and trypsin (45000-660) were all obtained from VWR. Fetal bovine serum was purchased from ATCC (SCRR-30-2020). ES-D3 cells were cultured in DMEM supplemented with 15% fetal bovine serum, 50 U/mL penicillin, 50 µg/mL streptomycin, 0.1 mM non-essential amino acids, 2 mM L-glutamine, 0.1 mM β-mercaptoethanol and 100 units/mL LIF. ES-D3 cells were cultured without a feeder layer in dishes pre-coated with 0.1% gelatin under standard conditions in a 5% CO2 humidified incubator at 37°C. ES-D3 cells were authenticated by ATCC cell bank using the Short Tandem Repeat (STR) profiling. To generate GM-CSF-expressing ES-D3 cells, pEF1α-GM-CSF-IRES-GFP or the empty vector was transfected into ES-D3 cells along with the plasmid pBabe-Neo using Lipofectamine 2000® transfection reagent (11668019; Thermo Fisher). Stably transfected cells were acquired by culturing the cells in medium containing 0.5 mg/mL G418, and GFP-positive ES-D3 cell clones were subsequently selected by flow cytometry sorting (MoFlo; Beckman Coulter) into a 96-well tissue culture plate pre-coated with 0.1% gelatin. Sorted empty vector control and GM-CSF-expressing ES-D3 clones were cultured under standard conditions in a 5% CO2 humidified incubator at 37°C and further expanded for isolation of exosomes.
Cancer cell lines
Lewis lung carcinoma (LLC) cells, B16-F10 melanoma, MC-38 colon adenocarcinoma, and E0771 medullary breast adenocarcinoma were all derived from C57BL6 mice. The 4T1 mammary carcinoma cell line is syngeneic to Balb/c mice. All the five cell lines were obtained from ATCC and cultured under standard conditions in DMEM supplemented with 10% fetal bovine serum in a 5% CO2 humidified incubator at 37°C. We do not culture cell lines longer than 6–8 weeks and all of our stocks come from thawed vials that were frozen at passage two after receipt from ATCC and were authenticated by ATCC cell bank using the Short Tandem Repeat (STR) profiling.
Plasmids
The transfection expression vector pEF1α-IRES-GFP was generated by removing FD3ER cDNA from the plasmid pEF1a-FD3ER-IRES-hrGFP acquired from Addgene (37270). Murine GM-CSF cDNA was cloned into pEF1α-IRES-GFP to generate pEF1α-GM-CSF-IRES-GFP. The plasmid identities were validated by sequencing.
Exosomes
Fetal bovine serum was centrifuged at 100,000 × g for 16 h at 4°C using a 45Ti rotor in an ultracentrifuge (OptimaTm L-100XP, Beckman Coulter) to remove exosomes. Serum supernatant acquired after centrifugation was used as an exosome-free fetal bovine serum to culture ES-D3 cells for exosome preparation. ES-D3 cells (5 × 106) were cultured in 15 cm tissue culture dishes pre-coated with 0.1% gelatin in 15 mL medium for 72 h. The medium was collected and centrifuged at 5,000 × g for 60 min at 4°C using a JA-10 rotor in a centrifuge (Avanti® J-26XPI, Beckman Coulter). The supernatant was then collected and centrifuged again at 100,000 × g for 90 min at 4°C using a 45Ti rotor in an ultracentrifuge (OptimaTm L-100XP, Beckman Coulter). Following removal of the supernatant, the tight pellets at the bottom of the centrifuge tubes were gently rinsed twice with 1 mL of PBS to remove any traces of the culture supernatant. The pellets were then resuspended in PBS, and protein concentration of exosomes was measured using BCA (bicinchoninic acid) assay (23223; Thermo Fisher). The yield of exosomes from ES-D3 cells was about 4 μg protein per mL of culture medium. To prepare samples for electron microscopy, exosomes were fixed by incubation with 2% EM grade paraformaldehyde (15710; Electron Microscopy Sciences) at room temperature for 2 h. Fixed exosomes were loaded on Cu grids with carbon support film (FF200-Cu; Electron Microscopy Sciences) and stained with UranyLess staining solution (22409; Electron Microscopy Sciences) following manufacturer's protocol. The electron microscopy images were acquired on a transmission electron microscope (HT7700; Hitachi) at Kentucky Biomedical Research Infrastructure Network (KBRIN) of the University of Louisville. For proteinase K treatment, exosomes (2 mg/mL) were incubated with 0.8 μg/mL proteinase K at 37°C for 30 min. The reactions were stopped by putting the samples on ice.
ELISA
The concentrations of GM-CSF in the ES-D3 cell supernatants were determined using a murine GM-CSF ELISA kit (88733422; Thermo Fisher) following manufacturer’s protocol. GM-CSF levels in exosomes were determined using the same ELISA protocol but with slight modifications. Briefly, exosomes (0.6 µg) were pre-treated in 100 µL of PBS with or without 0.05% Tween-20 at room temperature for 30 min following which samples were added to an ELISA plate coated with capture antibody and washed with either PBS alone or PBS + 0.05% Tween-20 buffer. Following incubation with detection antibody and avidin-HRP, the absorbance at 450 nM was determined using a microplate reader (PowerWave XS; BioTek).
Western blot analysis
Samples were separated on a 4–12% Bis-Tris gel (3450125; Bio-Rad) and transferred onto PVDF membranes (IPVH00010; Millipore EMD). The membranes were incubated with appropriate primary and secondary antibodies in blotting buffer (PBS with 0.2% Tween-20) supplemented with 10% (w/v) non-fat dry milk (NC9022655; Thermo Fisher). Proteins were detected using an enhanced chemiluminescence detection system (32106, Thermo Fisher). Antibodies (Abs) for western blot were: anti-Alix mAb (clone AC-15, sc-53540); anti-Annexin V mAb (clone H-3, sc-74438); anti-CD81 mAb (clone B-11, sc-166029); anti-Flotillin-1 (clone C-2; sc-74566); anti-CD86 mAb (clone D-6, sc-28347), anti-cytochrome c mAb (clone A-8, sc-13156) from Santa Cruz Biotechnology, anti-calnexin pAb (ADI-SPA-860); anti-protein disulfide isomerase (PDI) pAb (ADI-SPA-890) from Enzo, anti-GAPDH pAb (600–401-A33S) from Rockland, anti-Oxphos COX IV-subunit IV mAb (clone 20E8C12; A21348); peroxidase-conjugated goat anti-rabbit IgG (31460); peroxidase-conjugated goat anti-mouse IgG (31430) from Thermo Fisher.
Evaluation of pluripotency
The pluripotency of ES-D3 cells was evaluated using a StemflowTM Human and Mouse Pluripotent Stem Cell Analysis Kit (BDB560477; Beckon Dickinson) following manufacturer’s protocol. The expression levels of SSEA-1, Oct-3/4 and SSEA-4 were measured using flow cytometry (FACScalibur; Beckon Dickinson).
Proteomics analysis
Proteins in ES-D3 cell-derived exosomes were extracted using a buffer containing 50 mM Tris (pH7.4), 150 mM NaCl, 1% NP-40, and 0.05% SDS at 4°C, and analyzed at the Genome Center, University of California at Davis. Following a standard procedure of protein digestion by Trypsin/Lys-C (V5071; Promega), samples were desalted using C18 Microspin columns (SEMSS18V; Nest Group) and lyophilized by vacuum centrifugation. Liquid chromatography (LC) separation was carried out using an LC system (Easy-nLC 1000; Thermo Scientific). The digested peptides were reconstituted in 2% acetonitrile/0.1% trifluoroacetic acid and 3 µg of the sample was loaded onto a 100 micron × 25 mm Magic C18 100Å 5U reverse phase trap where they were desalted online before being separated on a 75 micron × 150 mm Magic C18 200Å 3U reverse phase column. Peptides were eluted using a gradient of 0.1% formic acid (A) and 100% acetonitrile (B), which was run with 5% to 35% B (45 minutes), 35% to 80% B (8 minutes), 80% B (1 minute), 80% to 5% B (1 minute), and 5% B (10 minutes). Mass spectra were collected on a mass spectrometer (Orbitrap Q Exactive Plus; Thermo Fisher Scientific) in a data-dependent mode with one MS precursor scan followed by 15 MS/MS scans. A dynamic exclusion of 15 s was used. All MS/MS samples were analyzed using X! Tandem (CYCLONE (2013.02.01.1); The GPM) to search the uniprot mouse proteome plus an equal number of reverse decoy sequences (142010 entries). The software Scaffold (Scaffold_4.8.2; Proteome Software) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 98.0% probability by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 5.0% probability to achieve an FDR less than 5.0% and contained at least one identified peptide.
Mice
Male C57BL/6 and female Balb/c mice (20–22 g body weight, 6–8 weeks of age) were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed under standard conditions. Mice were handled in accord with AAALAC international guidelines. The experiments were approved by the Institutional Animal Care and Use Committee of the University of Louisville (ID # 09021).
Vaccination and tumor challenge
C57BL/6 and Balb/c mice were immunized twice (days 0 and 7) with vehicle only (HBSS), ES-exo (225 μg of exosomes isolated from vector control ES-D3 cells) or ES-exo/GM-CSF (225 μg of exosomes isolated from ES-D3 cells stably-expressing GM-CSF). Exosomes were injected subcutaneously (s.c.) in the right flank. Exosomal protein was adjusted to be equivalent to that of the previously used whole ESC vaccine (1 × 106 cells). On day 14, mice were challenged with LLC cells or 4T1 cells (1 × 105) injected s.c. into the left flank. Tumor growth was measured with calipers every 2nd day and tumor volumes were calculated using the following formula: V = (width, mm2 × length, mm)/2.
Flow cytometric analysis
Single cell suspensions from spleen were stained with relevant antibodies (CD3, CD4, CD8, CD44, CD62L, CD69, CD25, CD43, CD11b, Ly6G, Ly6C) for 30 min after blocking with CD16/CD32 antibody (2.4G2) for 15 min at 4°C. After washing, cell surface and intracellularly stained cells were analyzed on a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ) and results were analyzed with FlowJo software (TreeStar, Inc., Ashland, OR). Details of used antibody clones are provided in Supplemental Table 2.
Intracellular cytokine staining
Spleens were isolated from different treatment groups 10 days after the last vaccination. Splenocytes were stimulated with either LLC lysate (50 μg/mL), B16F10 lysate (50 μg/mL), MC-38 lysate (50 μg/mL), E0771 lysate (50 μg/mL) or 4T1 lysate (50 μg/mL) for 4 days. For TNF-α and IFN-γ production, effector cells were harvested and restimulated for 6 h with the indicated tumor cell lysates in the presence of Golgiplug (555029; BD Biosciences) at a concentration of 1 μL/mL of culture medium. After restimulation, cells were harvested, Fc receptors were blocked, and cells were stained for surface expression of CD8 and intracellular expression of cytokines using Cytofix/Cytoperm kit (555029; BD biosciences) according to the manufacturer’s instructions and analyzed by flow cytometry.
Analysis of tumor-infiltrating T cells
Vaccinated and control mice bearing LLC tumors were euthanized 18–21 days after tumor challenge. Solid tumors were dissected and chopped into small pieces before incubation with a mixture of enzymes dissolved in HBSS [400 U/mL collagenase type IV (C9891), 0.025 mg/mL hyaluronidase (H6254), 0.01 mg/mL DNase I (D5025) purchased from Sigma-Aldrich] for 2 h at 37°C with occasional shaking. The resultant cells were washed and passed through a Ficoll gradient to eliminate dead cells. Tumor-infiltrating lymphocytes (TILs) were then analyzed by flow cytometry for the expression of CD4, CD8, and CD25 markers. T regulatory cells (Treg; Foxp3+) were analyzed using the anti-mouse Foxp3 staining kit (00-5523-00; eBioscience). The same number of cells (based on side-scatter and forward-scatter analyses) was acquired in all samples. Anti-CD45 antibody was used to selectively exclude CD45− tumor cells from analysis. For intracellular IFN-γ analysis, TILs were stimulated with LLC lysates (50 μg/mL) for 24 h and restimulated with LLC lysates for 6 h in the presence of Brefeldin A. After restimulation, cells were harvested, Fc receptors were blocked, and cells were stained for surface expression of CD8 and intracellular expression of cytokines using Cytofix/Cytoperm kit according to the manufacturer’s instructions (555029; BD Biosciences) and analyzed by flow cytometry.
Toxicological analysis
C57BL/6 mice (n = 4 per group) were immunized twice (days 0 and 7) with vehicle only (HBSS), ES-exo (225 μg) or ES-exo/GM-CSF (225 μg) via s.c. route in the right flank. On day 14, whole blood was drawn from mice by cardiac puncture immediately following CO2 asphyxiation. Whole blood (250 μL) was collected in EDTA coated tubes (02-669-33, BD Biosciences) and analyzed by the Research Resource Center (RRC) facility at the University of Louisville for complete blood count (CBC) analysis. Simultaneously, whole blood was also collected in serum separator tubes (02-675-185, BD Biosciences). After 1 h post-collection, samples were centrifuged for 10 min at 10,000 × g and serum (100 μL) was collected from each sample and analyzed by RRC for non-steroidal anti-inflammatory drug (NSAID) toxicological analysis. Liver damage was assessed by levels of alanine aminotransferase (ALT) and alkaline phosphatase (ALKP). Kidney damage was assessed by levels of blood urea nitrogen (BUN) and creatinine.
Statistical analysis
StatView version 5.0.1 software (Windows version; SAS Institute, Cary, NC) or GraphPad Prism 5.0 software (GraphPad Prism Software, Inc., La Jolla, CA) were used for all statistical analyses. Two-group comparisons between control and test samples (groups compared are indicated in the respective figures) were done by Unpaired Student’s t tests. Multiple data comparisons were derived by one-way ANOVA followed by Tukey’s post-hoc test. Kaplan-Meier curve was analyzed using the log-rank test. Statistical significance was assumed at p < 0.05.
Author Contributions
K.Y., J.E. and C.L. conceived the experiments. A.W., S.M., N.R., J.R., and A.T. conducted the experiments. K.Y., J.E. and C.L. analyzed the results. K.Y., J.E. and C.L. wrote the manuscript. All authors contributed to and reviewed the manuscript.
Conflict of interest statement
The authors declare that there is no conflict of interest regarding the publication of this manuscript.
Financial Support
This work was supported in part by grants from NIH CA198249 (K.Y.); Free to Breathe Research Grant (K.Y.); NIH AA018016-01 (J.W.E); Commonwealth of Kentucky Research Challenge Trust Fund (J.W.E.); and NIH CA106599 and CA175003 (C.L).
Supplemental Material
Download Zip (8 MB)Acknowledgments
We thank Dr. Arkadiusz Slusarczyk and Kentucky Biomedical Research Infrastructure Network (KBRIN, P20GM103436) for transmission electron microscope image acquisition. We thank Dr. Brett Phinney and Ms. Michelle Salemi (Genome Center; University of California at Davis, CA) for assistance with mass spectrometry analysis.
SUPPLEMENTAL DATA
Supplemental data for this article can be accessed at publisher’s website
Additional information
Funding
References
- Brewer BG, Mitchell RA, Harandi A, Eaton JW. Embryonic vaccines against cancer: an early history. Exp Mol Pathol. 2009;86:192–197. doi:10.1016/j.yexmp.2008.12.002.
- Stonehill EH, Bendich A. Retrogenetic expression: the reappearance of embryonal antigens in cancer cells. Nature. 1970;228:370–372.
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi:10.1038/ng.127.
- Yaddanapudi K, Mitchell RA, Putty K, Willer S, Sharma RK, Yan J, Bodduluri H, Eaton JW, Gabriele L. Vaccination with embryonic stem cells protects against lung cancer: is a broad-spectrum prophylactic vaccine against cancer possible? PLoS One. 2012;7:e42289. doi:10.1371/journal.pone.0042289.
- Kooreman NG, Kim Y, de Almeida PE, Termglinchan V, Diecke S, Shao NY, Wei -T-T, Yi H, Dey D, Nelakanti R, et al. Autologous iPSC-Based vaccines elicit anti-tumor responses in vivo. Cell Stem Cell. 2018;22:501–13 e7. doi:10.1016/j.stem.2018.01.016.
- Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi:10.1146/annurev-cellbio-101512-122326.
- Gehrmann U, Naslund TI, Hiltbrunner S, Larssen P, Gabrielsson S. Harnessing the exosome-induced immune response for cancer immunotherapy. Semin Cancer Biol. 2014;28:58–67. doi:10.1016/j.semcancer.2014.05.003.
- Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi:10.1038/nri855.
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi:10.1038/ncb1596.
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066. doi:10.3402/jev.v4.27066.
- Lener T, Gimona M, Aigner L, Borger V, Buzas E, Camussi G, Chaput N, Chatterjee D, Court FA, Portillo HAD, et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J Extracell Vesicles. 2015;4:30087. doi:10.3402/jev.v4.30087.
- Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J, Nolte-‘t Hoen EN, Piper MG, Sivaraman S, Skog J. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2(1). doi: 10.3402/jev.v2i0.20360. eCollection 2013.
- Armstrong JP, Holme MN, Stevens MM. Re-engineering extracellular vesicles as smart nanoscale therapeutics. ACS Nano. 2017;11:69–83. doi:10.1021/acsnano.6b07607.
- Schlesinger S, Lee AH, Wang GZ, Green L, Goff SP. Proviral silencing in embryonic cells is regulated by Yin Yang 1. Cell Rep. 2013;4:50–58. doi:10.1016/j.celrep.2013.06.003.
- Chung S, Andersson T, Sonntag KC, Bjorklund L, Isacson O, Kim KS. Analysis of different promoter systems for efficient transgene expression in mouse embryonic stem cell lines. Stem Cells. 2002;20:139–145. doi:10.1634/stemcells.20-2-139.
- Zeng X, Chen J, Sanchez JF, Coggiano M, Dillon-Carter O, Petersen J, Freed WJ. Stable expression of hrGFP by mouse embryonic stem cells: promoter activity in the undifferentiated state and during dopaminergic neural differentiation. Stem Cells. 2003;21:647–653. doi:10.1634/stemcells.21-6-647.
- Herbst B, Kohler G, Mackensen A, Veelken H, Lindemann A. GM-CSF promotes differentiation of a precursor cell of monocytes and Langerhans-type dendritic cells from CD34+ haemopoietic progenitor cells. Br J Haematol. 1998;101:231–241.
- Zhao W, Ji X, Zhang F, Li L, Ma L. Embryonic stem cell markers. Molecules. 2012;17:6196–6236. doi:10.3390/molecules17066196.
- Bosch S, de Beaurepaire L, Allard M, Mosser M, Heichette C, Chretien D, Jegou D, Bach J-M. Trehalose prevents aggregation of exosomes and cryodamage. Sci Rep. 2016;6:36162. doi:10.1038/srep36162.
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968–977. doi:10.1073/pnas.1521230113.
- Mignot G, Roux S, Thery C, Segura E, Zitvogel L. Prospects for exosomes in immunotherapy of cancer. J Cell Mol Med. 2006;10:376–388.
- Gutschner T, Hammerle M, Pazaitis N, Bley N, Fiskin E, Uckelmann H, Heim A, Groβ M, Hofmann N, Geffers R, et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology. 2014;59:1900–1911. doi:10.1002/hep.26997.
- Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev. 2012;38:68–75. doi:10.1016/j.ctrv.2011.04.002.
- Glas AS, Roos D, Deutekom M, Zwinderman AH, Bossuyt PM, Kurth KH. Tumor markers in the diagnosis of primary bladder cancer. A Systematic Review J Urol. 2003;169:1975–1982.
- Kornberg A, Polliack A. Serum lactic dehydrogenase (LDH) levels in acute leukemia: marked elevations in lymphoblastic leukemia. Blood. 1980;56:351–355.
- Baldwin RW, Glaves D, Vose BM. Embryonic antigen expression in chemically induced rat hepatomas and sarcomas. Int J Cancer. 1972;10:233–243.
- Baldwin RW, Glaves D, Pimm MV, Vose BM. Tumour specific and embryonic antigen expression of chemically induced rat tumours. Ann Inst Pasteur (Paris). 1972;122:715–728.
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi:10.1038/nri1806.
- Ortiz ML, Lu L, Ramachandran I, Gabrilovich DI. Myeloid-derived suppressor cells in the development of lung cancer. Cancer Immunol Res. 2014;2:50–58. doi:10.1158/2326-6066.CIR-13-0129.
- Chesney JA, Mitchell RA, Yaddanapudi K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol. 2017;102:727–740. doi:10.1189/jlb.5VMR1116-458RRR.
- Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010;123:39–49. doi:10.1007/s10549-009-0622-8.
- Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546–2553. doi:10.1200/JCO.2006.08.5829.
- Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, Shono Y, Kitabatake M, Kakimi K, Mukaida N, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–5466. doi:10.1182/blood-2008-01-136895.
- Mordmuller B, Surat G, Lagler H, Chakravarty S, Ishizuka AS, Lalremruata A, Gmeiner M, Campo JJ, Esen M, Ruben AJ, et al. Sterile protection against human malaria by chemoattenuated PfSPZ vaccine. Nature. 2017;542:445–449. doi:10.1038/nature21060.
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi:10.1056/NEJMoa1001294.
- Li Y, Zeng H, Xu RH, Liu B, Li Z. Vaccination with human pluripotent stem cells generates a broad spectrum of immunological and clinical responses against colon cancer. Stem Cells. 2009;27:3103–3111. doi:10.1002/stem.234.
- Dong W, Du J, Shen H, Gao D, Li Z, Wang G, Mu X, Liu Q. Administration of embryonic stem cells generates effective antitumor immunity in mice with minor and heavy tumor load. Cancer Immunol Immunother. 2010;59:1697–1705. doi:10.1007/s00262-010-0899-9.
- Palena C, Abrams SI, Schlom J, Hodge JW. Cancer vaccines: preclinical studies and novel strategies. Adv Cancer Res. 2006;95:115–145. doi:10.1016/S0065-230X(06)95004-0.
- Dranoff G. GM-CSF-based cancer vaccines. Immunol Rev. 2002;188:147–154.
- Bencherif SA, Warren Sands R, Ali OA, Li WA, Lewin SA, Braschler TM, Shih T-Y, Verbeke CS, Bhatta D, Dranoff G, et al. Injectable cryogel-based whole-cell cancer vaccines. Nat Commun. 2015;6:7556. doi:10.1038/ncomms8556.
- Kunigelis KE, Graner MW. The dichotomy of tumor exosomes (TEX) in cancer immunity: is it all in the conTEXt? Vaccines (Basel). 2015;3:1019–1051. doi:10.3390/vaccines3041019.
- Borrello I, Pardoll D. GM-CSF-based cellular vaccines: a review of the clinical experience. Cytokine Growth Factor Rev. 2002;13:185–193.
- Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S, Tanabe K, Duda R, Mentzer S, Jaklitsch M, et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J Clin Oncol. 2003;21:3343–3350. doi:10.1200/JCO.2003.07.005.
- Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, Raposo G, Amigorena S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999;147:599–610.
- Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172.
- Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S, Escudier B, Robert C, Caillat-Zucman S, Tursz T. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One. 2009;4:e4942. doi:10.1371/journal.pone.0004942.3
- Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, Ricciardi-Castagnoli P, Raposo G, Amigorena S. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4:594–600.
- Mazumder S, Johnson JM, Swank V, Dvorina N, Martelli E, Ko J, Tuohy VK. Primary immunoprevention of epithelial ovarian carcinoma by vaccination against the extracellular domain of anti-mullerian hormone receptor II. Cancer Prev Res (Phila). 2017;10:612–624. doi:10.1158/1940-6207.CAPR-17-0154.
- Shoemaker RH, Forsthuber TG. Targeting “retired antigens” for cancer immunoprevention. Cancer Prev Res (Phila). 2017;10:607–608. doi:10.1158/1940-6207.CAPR-17-0188.
- Jaini R, Kesaraju P, Johnson JM, Altuntas CZ, Jane-Wit D, Tuohy VK. An autoimmune-mediated strategy for prophylactic breast cancer vaccination. Nat Med. 2010;16:799–803. doi:10.1038/nm.2161.