
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Neutrophil extracellular trap (NET) formation results in the expulsion of granulocyte proteins and DNA into the extracellular space. This process is mediated by the enzyme peptidyl arginine deiminase 4 (PADI4) and translocation of elastase to the nucleus. NET formation, marked by increased levels of extracellular DNA, promotes pancreatic cancer proliferation and metastasis. Mice deficient in Padi4 demonstrate decreased pancreatic tumor growth, associated with a reduction in circulating extracellular DNA levels, diminished pancreatic stromal activation and improved survival in murine orthotopic pancreatic adenocarcinoma. Transplantation of Padi4−/− bone marrow into genetically engineered mice with Kras driven pancreatic adenocarcinoma (Pdx1-Cre:KrasG12D/+, KC mice) limits the frequency of invasive cancers when compared with syngeneic controls. DNA from neutrophils activates pancreatic stellate cells that form dense, fibrous stroma which can promote and enable tumor proliferation. DNase treatment diminishes murine tumor growth and stromal activation to reverse the effect of NETs within the tumor microenvironment. Furthermore, deletion of the receptor for advanced glycation end products (RAGE) in pancreatic stellate cells abrogates the effects of DNA in promoting stellate cell proliferation and decreases tumor growth. Circulating neutrophil-derived DNA correlates with the stage in patients with pancreatic ductal adenocarcinoma, confirming the role of NETs in human pancreatic cancer. These findings support further investigation into targeting of NETs, PADI4 and extracellular DNA as a potential treatment strategy in patients with pancreatic cancer.
Trial Registration: This study reports correlative data from a clinical trial registered with clinicaltrials.gov, NCT01978184 (November 7, 2013).
Introduction
Neutrophil extracellular traps (NETs) were first described as a microbial defense pathway by which neutrophils expel intracellular contents including histones, HMGB1, actin, reactive oxygen species, and DNA, forming an adhesive matrix to trap and kill bacteria.Citation1 NETs have also been implicated in sterile inflammation including rheumatoid arthritisCitation2 and lupus erythematosus,Citation3 as well as murine models of lung and mammary cancer.Citation4 We have previously shown that mice bearing orthotopic pancreatic tumors have an increased propensity to form NETs through activation of the MHC Class III-encoded DNA receptor RAGE (receptor for advanced glycation end products). Markers of NET formation are elevated in pancreatic tumor-bearing mice, and patients with pancreatic adenocarcinoma (PDA).Citation5 The impact of NETs on the pathogenesis of pancreatic cancer and the effects of NETs in the pancreatic tumor microenvironment have yet to be elucidated.
Peptidyl arginine deiminase 4 (PADI4) is a critical enzyme involved in NET formation.Citation6 PADI4 belongs to a family of post-translational modifying enzymes, catalyzing the deimination of arginine on histones H3 and H4 leading to chromatin decondensation and subsequent expulsion during NET formation.Citation7–Citation9 As a result, citrullinated histone H3 (CitH3) is an important marker of NET formation.Citation10
Extracellular DNA has been implicated in pancreatic cancer invasion and metastasis, but its origin has not been definitively determined.Citation11 Given that extracellular DNA is the principal component released from NETs, we hypothesized the neutrophil-derived DNA released during NETs may have effects on pancreatic tumor growth and in the tumor microenvironment. Inhibition of NETs mediated by direct small molecule PADI4 inhibition or depletion of extracellular DNA with DNase treatment diminishes liver metastasisCitation12 and has demonstrable antitumor effects.Citation13 Furthermore, pancreatic cancer is characterized by a dense, fibrotic stroma as a result of pancreatic stellate cell (PSC) activation.Citation14,Citation15 DNA in the form of bacterial CpG motifs activates pancreatic stellate cells (PSCs) to become fibrogenic and promote epithelial proliferation.Citation16 Based on these findings, we hypothesized that extracellular DNA derived from NETs promotes activation of PSC within the tumor microenvironment.
Results
Neutrophil extracellular trap (NET) DNA promotes pancreatic cancer cell proliferation
Neutrophils were isolated and stimulated to form NETs using phorbol 12-myristate 13-acetate (PMA), a known inducer of the NET formation.Citation17 Supernatant was collected and co-cultured with Panc02 murine pancreatic cancer cells. NET supernatant resulted in a significant, dose-dependent increase in cell proliferation as measured by CCK8 assay compared with media control (). Treatment of NET supernatant with DNase to lyse cell-free DNA prior to co-culture with Panc02 cells ameliorated the NET induced proliferative effect, suggesting a role for NET released DNA in tumor cell growth ().
Figure 1. NET-deficient Padi4−/− mice demonstrate decreased pancreatic tumor burden, decreased NETs, decreased DNA and increased survival. Addition of NET supernatant to Panc02 cells results in a dose-dependent increase in cell proliferation compared with media control (A). Treatment of NET supernatant with DNase reverses this proliferative effect, while it has minimal effect on media control (B). Immunofluorescence imaging of resected orthotopic pancreatic tumors demonstrates the absence of NETs in KO tumors (Citrullinated Histone H3:green, Gr-1:red, C). Plasma from orthotopic tumor-bearing WT mice demonstrates increased DNA compared with Padi4−/− consistent with upregulation of NETs (D, 695 ± 102 ng/ml vs. 406 ± 28.2 ng/ml, p = .03). Orthotopic tumor-bearing mice demonstrate increased tumor burden at sacrifice (E, 451 ± 55 mg vs. 266 ± 32 mg, p = .01; n = 4) and increased tumorigenesis by IVIS imaging (F, 2.4e7 photon flux vs. 1.6e6 photon flux, p = .05, n = 5). Survival of Padi4-deficient tumor-bearing mice was markedly improved (G, median 118 days vs 41 days; p = .02). DNase treatment of orthotopic tumor-bearing mice decreases tumor burden in NET bearing but not NET deficient mice as assessed by tumor weight (H, 336 mg vs. 206 mg, p = .05; 195 mg vs. 217 mg, p = .29, n = 5), and IVIS (I, 9.9e5 photon flux vs. 3.8e5 photon flux, p = .05).
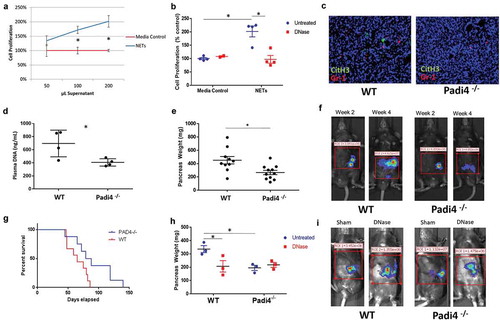
Padi4 mediated NET formation releases circulating DNA that drives the growth of orthotopic transplantable murine pancreatic tumors
To assess the impact of NET formation on pancreatic tumor growth, we grafted a transplantable murine pancreas tumor by injecting Panc02 cells into the pancreatic tail of Padi4−/− or strain-matched WT control mice. To confirm that genetic knockdown of Padi4 resulted in decreased NETs, we performed immunofluorescence staining of tumor specimens for citrullinated histone H3 (Cit H3), which is formed during the release of NETs when PADI4 citrullinates histones to allow for unwinding and subsequent expulsion from the neutrophil.Citation18 We also assessed levels of circulating DNA, which is a marker of NET formation since DNA is released from neutrophils when NETs are formed.Citation5 Padi4−/− tumors had decreased NET formation, demonstrated by diminished Cit H3 in the tumor (), and reduced circulating cell-free DNA levels (406 ± 28.2 ng/ml in Padi4−/− vs 695 ± 102 ng/ml in WT, p = .03) (). Tumorigenesis was decreased in Padi4−/− as demonstrated by decreased pancreatic weights compared with WT (266 ± 32 mg in Padi4−/− vs 451 ± 55 mg in WT, n = 4, p = .005, ) and bioluminescence imaging of luciferase transfected pancreatic cancer cells prior to sacrifice (2.4x107 photon flux vs. 1.6 × 106 photon flux, p = .05, ). Padi4−/− orthotopic mice demonstrate improved survival compared with WT orthotopic tumor-bearing mice (median survival 118 days vs 41 days, n = 10, p = .02, ).
Based on our in vitro data, we hypothesized that extracellular DNA released from NETs was driving increased tumor growth; therefore, we treated the orthotopically transplanted mice with DNase to lyse circulating cell-free DNA. DNase (5mg/kg IP daily) treatment of WT mice lead to significantly decreased tumor growth compared with sham-treated controls (pancreas weight: 336 ± 26.4 mg vs. 206 ± 31.6 mg, p = .05, ; IVIS: 9.9 × 105 photon flux vs. 3.8 × 105 photon flux, p = .05, ). There was no significant change in tumor growth in Padi4−/− animals treated with DNase (pancreas weight: 195 ± 19.3 mg vs. 217 ± 18.5 mg, p = .29; IVIS: 8.5 × 105 photons vs. 6.9 × 105 photons, p = .43).
Padi4 expression on BM-derived cells promotes stromal activation and accelerated tumorigenesis in a genetically engineered murine model of pancreatic cancer
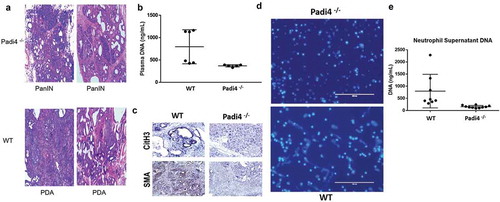
To assess the importance of Padi4-mediated NETs in a spontaneous model of pancreatic cancer, we performed transplantation of Padi4−/− and WT bone marrow (BM) into the genetically engineered Pdx1-Cre:KrasG12D/+ (KC) model. This model has previously been reported to be an excellent surrogate for human pancreatic carcinogenesis.Citation19 Seven weeks following BM transplant, pancreata from these animals were harvested, fixed, stained and scored by an expert pancreatic pathologist (ADS). Mice that received Padi4−/− BM demonstrated statistically less invasive cancers than those receiving WT BM (23% vs 67%, p = .02, ). This decreased tumorigenesis was associated with significantly diminished plasma DNA levels (787 ng/mL vs. 385 ng/mL; p = .001, ) at the time of harvest, consistent with NET inhibition. Compared with WT BM transplanted animals, pancreata from mice receiving Padi 4 deficient BM demonstrated diminished NET formation and stromal activation which were represented by an expression of CitH3 and α-SMACitation20 respectively in the tumor microenvironment (). Bone marrow neutrophils were isolated at the time of sacrifice and stimulated ex vivo to undergo NET formation using phorbol myristate acetate (PMA).Citation17 Hoechst staining of isolated neutrophils demonstrated decreased NETs from the Padi4−/− recipients (). Supernatant from these stimulated neutrophils demonstrated significantly less DNA from neutrophils harvested from Padi4−/− BM compared with WT BM (), consistent with inhibition of NETs as visualized by Hoechst staining in .
Figure 2. NET-derived DNA promotes accelerated tumorigenesis, DNA release and pancreatic stellate cell activation. Genetically engineered Pdx1-Cre:KrasG12D/+ (KC) mice received bone marrow transplantation for either WT or Padi4−/− donors. KC mice were harvested 7 weeks after bone marrow transplantation and tumor acceleration with cerulean. Mice that received BM deficient in Padi4 demonstrated statistically less invasive cancers (normal pancreas or PanIN lesions) than those receiving WT BM (A, 23% vs 67%, p = .02). Plasma from transplanted KC mice contained higher DNA levels (B, 787 ng/mL vs. 385 ng/mL; p = .001). Additionally, these mice demonstrated less NETs measured by CitH3 staining and less PSC activation by SMA staining (C). Neutrophils isolated from the bone marrow at the time of harvest were plated and stimulated to form NETs. Immunofluorescence imaging revealed increased NETs for WT BM (lower panel) compared to Padi4−/− donors (upper panel) as assessed by Hoechst staining of DNA (D). Additionally, the supernatant from the stimulated neutrophils demonstrated increased DNA from WT compared with Padi4−/− donors consistent with increased NETs (E, 796 ng/mL vs. 197 ng/mL, p = .001).
Neutrophil DNA activates pancreatic stellate cells through interaction with the receptor for advanced glycation end productions (RAGE)
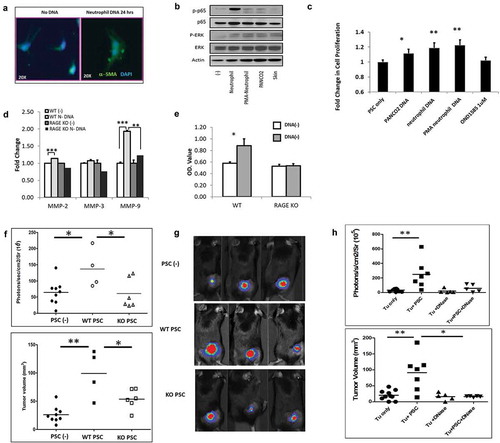
To better assess the effect of circulating DNA in the tumor microenvironment, pancreatic stellate cells (PSC) were harvested from the pancreas of 8 to 12 week-old mice and stimulated in culture with exogenous DNA. PSC from WT mice demonstrate increased activation when stimulated by neutrophil DNA as indicated by increased α–SMA immunostainingCitation20 () and, increased phospho-NFκB p65 and p-ERK on western blot analysis (). NET-derived DNA significantly increased PSC proliferation (). Furthermore, NET DNA enhanced production of MMP2 and MMP9 (), which are secreted by PSCs and enhance pancreatic cancer progression and metastases.Citation21,Citation22 In contrast, PSC derived from RAGE−/− mice, lacking this important DNA receptor,Citation5,Citation23,Citation24 did not respond to exogenous neutrophil DNA, demonstrated by no change in cell proliferation and MMP2 and MMP9 production (, ). These results suggest that RAGE may play a critical role in PSC activation by DNA.
Figure 3. Endogenous DNA activates pancreatic stellate cells and promotes tumor growth. Isolated mouse pancreatic stellate cells (PSCs) were stimulated by 5 μg/ml neutrophil DNA for 24 h in vitro and then assessed with immunofluorescence staining for α–SMA in green and DAPI in blue; demonstrating in an increase in α–SMA following neutrophil DNA exposure (A). Western blot analysis of p-p65 and p-ERK in PSCs after stimulation by various sources of DNA (5μg/ml) for 30 min in vitro (B). Co-culture with 5μg/ml of DNA for 24 h resulted in increased PSC proliferation as measured by CCK-8 assay (C). PSCs were cultured with 5μg/ml neutrophil DNA for 24 h, and then culture supernatant was collected and extracellular matrix protein production was measured, demonstrating increased MMP-2 and MMP-9 in WT PSCs but not in RAGE knockout PSCs (D). WT PSCs underwent increased proliferation in response to neutrophil DNA as measured by CCK-8 assay, but RAGE KO PSCs had no response (E). WT mice injected subcutaneously with WT PSCs (0.5 × 106) and Panc02 cells (1 × 106) had a significant increase in tumor growth assessed by bioluminescence imaging (upper panel) and caliper measurements (bottom panel). Tumor volume was calculated using formulations V = (L x W x W)/2, where V is tumor volume, W is tumor width, L is tumor length. Co-injection of RAGE KO PSCs did not have a significant effect on tumor growth (F). Bioluminescence imaging demonstrating an increase in tumor growth with an injection of WT PSCs and cancer cells, but not RAGE KO PSCs (G). Treatment with DNase reversed the ability of PSCs to promote tumor growth with a dramatic reduction in IVIS fluorescence (upper panel) and tumor volume (bottom panel) in DNase treated mice (H). Tumor volume on day 6 post-inoculation is presented. Data is represented one of 2 ~ 3 independent experiments and expressed as mean ± SEM. statistical analysis was performed using Student’s t-test for comparisons of two groups. *: p < .05, **: p < .01, ***: p < .001.
DNA activates pancreatic stellate cells to drive pancreatic tumor growth in a murine subcutaneous tumor model
In order to assess the significance of PSCs in the tumor microenvironment, we also utilized a subcutaneous tumor model with the injection of cells into the hind limb. Murine PSCs from WT mice were combined with Panc02 tumor cells and inoculated into WT mice. Mice with co-inoculation of WT PSC and Panc02 demonstrated enhanced tumor growth compared to those receiving tumor alone (, ). On day 14 post-tumor inoculation, tumor volume in Panc02 plus PSC group was 75.4 ± 17.0 compared to 20.4 ± 4.86 in tumor cell alone group, p = .002. Co-injection of Panc02 cells with RAGE knockout PSCs does not promote tumor growth (, ). DNase (5mg/kg IP daily for 3 weeks) treatment of WT tumor-burdened mice reversed the ability of WT PSC to promote tumor growth (, ). Tumor volume was significantly reduced in DNase treated mice compared to untreated, supporting the role of DNA in PSC activation.
Table 1. Treatment with DNase limits the ability of PSCs to promote tumor growth.
Total and neutrophil-specific DNA levels in human pancreatic cancer patients correlate with clinical stage in potentially resectable patients
To assess for the clinical relevance of circulating neutrophil DNA in human PDA; human samples were acquired from a recently completed clinical trial protocol of patients with pancreatic adenocarcinoma. Pretreatment serum samples were analyzed for total DNA content by QuantiT™Picogreen© and NET-specific DNA according to MPO-DNA quantification. DNA levels were then correlated with the clinical stage. Overall circulating DNA and MPO-DNA both correlated well with the clinical stage of disease at presentation (Supplemental Figure 1), suggesting a role for NETs in disease progression and identifying markers as potentially significant for prognosis.
Discussion
Here we demonstrate that DNA released from NETs plays a role in the driving progression of pancreatic tumors in both a transplantable and a genetically engineered murine model of pancreatic cancer. Moreover, total circulating serum DNA and MPO-DNA levels correlate with the clinical stage at diagnosis in human pancreatic cancer patients. While this certainly does not prove a causal mechanism, it is a supportive correlative association in human disease that strengthens our findings. These findings support a growing body of literature that suggests that NETs and more specifically, DNA can serve as a damage associated molecular pattern (DAMP) and drive carcinogenesis.
We have previously shown that RAGE-mediated autophagy, which is permissive in pancreatic cancer progression, is required for neutrophils to undergo NET formation.Citation5 The receptor for advanced glycation end products (RAGE) promotes inflammatory responses to DNA,Citation21 pancreatic carcinogenesis, and accumulation of myeloid-derived suppressor cells (MDSCs).Citation23 High-mobility group box (HMGB1), the prototypical damage-associated molecular pattern (DAMP) molecule, is a chromatin-binding protein that binds DNA located in the nucleus.Citation24 HMGB1 has been demonstrated to function as a chemoattractant for monocytes and smooth muscle cells.Citation25,Citation26 HMGB1-DNA immune complexes activate TLR9 through a RAGE-dependent pathway.Citation27 Herein, we extend these findings to demonstrate that DNA derived from sterile activation of neutrophils through the Padi4 pathway leads to the release of DNA and activates RAGE.
Though initially studied in models of sepsis, NETs have been implicated in numerous murine cancer models including mammary, lung, liver and pancreatic carcinoma.Citation1,Citation4,Citation5,Citation12,Citation28 Demers et al. described NETs as providing a “seeding soil” for cancer cells.Citation29 Tohme et al. demonstrated that increased levels of NETs, measured by serum MPO-DNA, following major liver resection for colorectal liver metastases correlated with an increase in early metastatic disease.Citation12 Our work supports this growing body of evidence, and for the first time suggests that Padi4 is critical to activation of NETs in the setting of sterile inflammation in the pancreatic tumor microenvironment. Thus, pharmacological intervention in the PADI4 pathway might be clinically important in patients with pancreatic cancer. There are several inhibitors of PADI4 that have been shown to inhibit mouse and human PADI4 and NET formation that warrant further exploration.Citation30
Extracellular, or circulating cell-free DNA levels correlate with cancer development and progression.Citation31,Citation32 As early as 1977, Leon et al. demonstrated that extracellular DNA levels were elevated in cancer patients and that reduction in DNA levels following radiotherapy correlated with clinical response.Citation33 Levels of circulating DNA in cancer patients were initially thought to derive from dead and dying tumor cells; however, a high degree of variability between tumor-specific DNA and extracellular DNA sequences challenges the assumption that a majority of circulating DNA originates from tumor cells.Citation34–Citation36 Extracellular DNA promotes pancreatic cancer cell invasiveness.Citation11 Our findings suggest NET-specific MPO-DNA levels mirror overall circulating serum DNA levels, consistent with an alternative to tumor-derived DNA as the primary source of cell-free DNA within the circulation. Padi4−/− mice, incapable of NET formation, have dramatically diminished levels of circulating DNA compared with wild-type mice. We have also previously shown that neutrophil depletion results in decreased DNA in tumor-bearing mice.Citation5 While this suggests that most of the circulating DNA is derived from neutrophils, this could be further elucidated in future studies by evaluating neutrophil-specific Padi4−/− mice. Importantly, Padi4−/− mice still have circulating DNA suggesting at least some of the DNA comes from a source distinct from NETs, most likely tumor cell necrosis. Our results suggest that DNA is the principle component of NETs that drive tumor progression, as treatment with DNase partially reverses tumor growth and ameliorates the impact of NETs in pancreatic cancer.
Pancreatic cancer is characterized by an excess of fibrous stroma and cellular elements, primarily composed of activated PSCs.Citation37 Quiescent, lipid- and vitamin A-laden, PSCs transform into activated myofibroblast-resembling cells upon stimulation by either direct contact with tumor or stimulation by growth factors secreted from other cells.Citation38–Citation40 PSC activation, an autophagy-dependent process, results in the production of extracellular matrix molecules and cytokines that promote tumor aggressiveness.Citation41 Zambirinis et al. showed that bacterial DNA could activate PSCs to become fibrogenic and promote epithelial proliferation.Citation14 In this model, PSC activation appeared to mediate their effect through the exclusion of T cells. We show here that neutrophil-DNA strongly activates PSCs through interaction with RAGE in vitro. Moreover, it appears that the ability of PSC to support tumor growth in vivo is dependent upon DNA. We have previously hypothesized that release of DAMPs, including DNA itself, within the tumor microenvironment, can drive maladaptive immune responses. This work supports this paradigm as well as identifying several novel targets, including PADI4, circulating DNA, and NETs in pancreatic carcinogenesis.
The activation of PSCs by neutrophil DNA has profound implications for the immune response to pancreatic adenocarcinoma that warrant future investigation. Because activated PSCs sequester T cells outside of the tumor,Citation42,Citation43 targeting NETs may decrease in PSC activation to allow for more T cell infiltration. DAMPs, RAGE, and the NET inhibitor chloroquine have been independently implicated in tumor-associated macrophage differentiation; therefore, NETs and DNA released from them may increase the M1 phenotype in infiltrating macrophages.Citation44–Citation46 NET mediated activation of pancreatic stellate cell fibrosis may be a critical component of the immunosuppressive tumor microenvironment that allows for tumor growth unabated by an effective adaptive immune response. Alternatively, DNA is known to activate stimulator of interferon genes (STING) to promote anti-tumor immunity,Citation47–Citation52 therefore further study is required to fully evaluate the impact of NET inhibition on the complex immune interaction in the pancreatic tumor microenvironment.
In summary, our findings demonstrate the importance of Padi4-mediated NETs in pancreatic cancer progression. DNA release from NETs appears to activate PSCs to promote stromal proliferation. PADI4 inhibition and DNase administration may be novel therapeutic interventions in pancreatic cancer patients.
Materials and methods
Mouse strains
Wild-type (WT) C57BL/6 mice age 6–8 weeks were purchased from Taconic Farms (Hudson, NY). Padi4 knockout (Padi4−/−) mice were obtained as a kind gift from Dr. Kerri Mowen.Citation53 Pdx1-Cre:KrasG12D/+ (KC)Citation17 mice were obtained from the Mouse Models of Human Cancers Consortium/National Cancer Institute Mouse Repository. RAGE knockout (RAGE−/−.GFP) mice (SVEV129xC57BL/6) were kindly gifted by the late Dr. Angelika Bierhaus.Citation54 All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (Protocol # 14084123) and performed in accordance with guidelines for University of Pittsburgh Division of Laboratory Animal Services, the American Veterinary Medical Association and the Guide for the Care and Use of Laboratory Animals.
Cell culture
The murine pancreatic adenocarcinoma cell line Panc02 (NCI Repository, 2008) was cultured in RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin antibiotic solution in a humidified incubator with 5% CO2.
Luciferase transfection and bioluminescence imaging
Transduced Panc02-luciferase tumor cells expressing firefly luciferase gene were generated using lentiviral transfection of the pGL4 Luciferase Reporter Vector (Promega, #E6751) then selected with puromycin. Phenotype of transfected cells was compared to parental strains in vitro to confirm the absence of effects attributable to retroviral transfection.
For imaging, mice were anesthetized with inhaled isoflurane followed by intraperitoneal injection of Potassium Luciferin (300mg/kg, Gold Biotechnology, Luck-100). After 10 min to allow for luciferin distribution, mice were imaged using the IVIS 200 System (Xenogen Corporation) according to the manufacturer’s instructions. Analysis of resultant data was performed using Living Image software. Regions of interest were manually selected and quantified for average photon flux (photons/second/cm2/steradian).
Murine tumor models
Our orthotopic pancreatic cancer model was performed by injecting 1 × 106 Panc02-luciferase cells into the murine pancreatic tail through a limited laparotomy.
Bone marrow transplantation was performed in KC mice 8 weeks of age. Mice were lethally irradiated with10.0 Gy. Bone marrow from C57BL/6 wild type and Padi4−/− mice was harvested and prepared by gradient centrifugation and red cell lysis as previously described.Citation55 Within 4 h, KC recipient mice were injected with 1 × 106 bone marrow cells via tail vein. Four weeks elapsed for bone marrow reconstitution. Cerulein acceleration of tumorigenesis was performed with 7 hourly intraperitoneal injections of 50 mg/kg and repeated after 48 h.
Immunohistochemistry and immunofluorescence
For immunolabeling of pancreatic specimens, resected mouse pancreata were embedded in formalin. Slides were deparaffinized and rehydrated two times in xylene for 5 min, two times in 100% ethanol for 5 min, two times 95% ethanol for 1 min then 5 min in running H20. Antigen retrieval was performed by heat-induction in Citra (Biogenix, HK0809K) using a microwave. Endogenous peroxidases were quenched, and slides were blocked in 1% bovine serum albumin for 30 min at 25°C. Slides were incubated in primary antibody-SMA (1:500 and CitH3 1:200-overnight at 4°C). Following two washes in PBS, slides were incubated in secondary antibody 45 min at 25°C. After ABC and DAB with Dako, slides were counterstained with hematoxylin. Negative controls included secondary antibodies alone.
Murine pancreatic specimens were frozen in Tissue-Tek® Optimal Cutting Temperature (OCT) compound, frozen, and stored at −80°C. Cryostat (8μm sections) were used for immunofluorescence staining. Tissue was fixed in 2% paraformaldehyde for 30 min at room temperature, and incubated with anti-Histone H3 antibody (Abcam, ab5103) at 1:50 dilution and anti-mouse Ly6G antibody (RB6-85C, eBioscience, 16–5931) overnight at 4°C. Following three washes with PBS, slides were incubated with fluorescent-conjugated secondary antibody for 45 min followed by Hoechst nuclear staining. Negative controls included antibody isotypes and secondary antibody alone. Imaging was performed using Olympus Fluoview 1000 microscope with a Plan Apo N (×60).
Neutrophil isolation and neutrophil extracellular trap (NET) formation
Under sterile conditions, bone marrow neutrophils were isolated from the femur and tibia of euthanized mice by the previously described protocol.Citation56 Bones were cleaned with 70% ethanol and rinsed with ice-cold PBS. Bone marrow was flushed from bones using RPMI supplemented with 10% heat-inactivated fetal bovine serum and 2mM EDTA using a 27.5 gauge needle. Cells were collected into a 50 mL conical tube through a 100um filter. Following centrifugation at 1400 rpm for 7 min at 4°C red blood cells were subjected to hypotonic lysis with 0.2% NaCl for 20 s. Cells were then collected by centrifugation, washed in supplemented RPMI, centrifuged, and then resuspended in ice-cold PBS. Density gradient centrifugation was performed with Histopaque 1119 and 1077. Neutrophils were collected from the gradient interface, washed twice, and counted.
Neutrophils were plated in Hank’s Balanced Salt Solution (HBSS) and stimulated with 500 nM phorbol 12-myristate 13-acetate (PMA, Sigma, St. Louis, MO, USA) for 2 h. Cells were fixed with 3% paraformaldehyde and stained for DNA with Hoechst 33342 (H-3570, Molecular Probes, Grand Island, NY, USA) to visualize NETs. Supernatant was collected, centrifuged at 14,000 x g for 10 min and the level of DNA measured using Quant-iT Picogreen (MP07581, Invitrogen, Grand Island, NY, USA). Supernatant was also collected and added cultured Panc02 cells to assess the impact on cell proliferation, measured using CCK8 kit following the manufacturer’s protocol. Media control included the addition of equivalent volumes of HBSS with 500nM PMA. DNase (1 mg/mL, Sigma Aldrich, St. Louis, MO, USA) was added to NET supernatant or media control for 10 min prior to co-culture with Panc02 cells.
Pancreatic stellate cell isolation and culture
Murine pancreatic stellate cells (PSC) were generated as previously described.Citation14 Pancreata of mice were collected and pooled in Gey‘s Balanced Salt Solution (GBSS) media then washed. After finely mincing the specimens, centrifugation was utilized to remove fat. Pancreatic tissue was digested for 50 min in a solution of Collagenase P, Pronase and DNase I incubated at 37°C. Digestion was quenched with cold GBSS supplemented with 3% bovine serum albumin after straining the tissue. PSCs were isolated by density centrifugation in 28.7% Nycodenz gradient solution under GBSS. PSCs were collected from the interface, washed and resuspended in a growth medium of DMEM with 10% FBS, penicillin/streptomycin, and L-Glutamine.
DNA isolation and PSC stimulation
Panc02 cells and isolated murine neutrophils were re-suspended at 107/ml in PBS. Cells were lysed, and exogenous DNA was purified using QIAGEN blood & cell culture DNA kit (Germantown, MD) following the manufacturer’s protocol. DNA was dissolved in TE buffer, the concentration was measured by Quant-iTTM PicoGreen DNA Reagent and kits (Invitrogen, MP07581) follow the manufacturer’s protocol and diluted to a desired concentration in culture medium. In proliferation assay, PSCs were added in 96 well culture plate, 104/well in the presence of different sources of DNA (5 μg/ml), cultured at 37°C, 5% CO2 for 24, 48 and 72 h. Cell proliferation was measured by Cell Proliferation/Cytotoxicity Assay Kit (CCK8) following the manufacturer’s protocol.
Western blot
Cell lysates from treated PSCs were run on a 4 ~ 12% Bis-Tris gel then transferred to a 0.2 mm nitrocellulose membrane. After blocking in 5% milk, membranes were incubated overnight at 4°C with primary antibodies for phospho-NF-κB p65 (93H1), NF-κB p65 (L8F6), phosphor-ERKd13.14.4E), ERK (137F5) (All from Cell Signaling Technology) and B-actin (Sigma, A5316). Membranes were washed then incubated with peroxidase-conjugated goat-anti-rabbit-HRP and goat-anti-mouse-HRP secondary antibodies (Fisher Thermo, 31430) for 1 h at room temperature then developed with SuperSignal® West Femto chemiluminescence kit (Thermo Scientific, 34095) and exposed on film.
Patient correlative samples
Pretreatment serum samples were obtained from patients enrolled in a randomized phase II clinical trial of pre-operative gemcitabine/nab-paclitaxel with or without hydroxychloroquine in potentially resectable pancreatic cancer under protocol # 13080444 approved by the University of Pittsburgh Institutional Review Board. The trial was reports on clinicaltrials.gov in November 2013 (NCT01978184). All patients signed informed consent and acknowledged understanding that they would remain anonymous and not be identified through the publication of study results. Coagulated blood was centrifuged at 1000 x g for 10 min then serum was collected, frozen, and stored at −80°C.
DNA and MPO-DNA quantification
After 10x dilution, DNA levels were measured in murine serum and plasma and human serum according to the manufacturer’s instructions for QuantiT™Picogreen© (Invitrogen, MP07581) fluorescent nucleic acid stain for quantifying double-stranded DNA. To identify NETs in patient serum, a capture ELISA myeloperoxidase (MPO) associated with DNA was performed as previously described.Citation57
Statistics
Data are expressed as mean ± SEM of a minimum of two independent experiments. Statistical analysis was performed using Student’s t-test for comparisons of two groups or χ2 test for correlations between multiple groups. p values <.05 were considered statistically significant. Survival analyses were conducted by the Kaplan–Meier method and log-rank test between curves.
Study approval
All animal studies were approved by the University of Pittsburgh IACUC (Protocol # 14084123) and performed within the guidelines of the University of Pittsburgh Division of Laboratory Animal Resources policies. The University of Pittsburgh Institution Review Board approved the clinical study from which correlative studies were included (Protocol # 13080444). Written informed consent was received from participants prior to inclusion in the study.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
JLM, BAB, XL, WRD, ADS conducted experiments, acquired and analyzed data. MTL, DT, RK, HJZ contributed to experimental design and data analysis. JLM, XL, BAB, MTL and HJZ contributed to writing the manuscript. All authors reviewed, edited and approved the final manuscript.
Supplemental Material
Download JPEG Image (193.7 KB){kind=link}
Acknowledgments
JLM was supported by NIH NCI T32 CA113263 and a Project Purple Fellowship grant. HJZ and MTL received support from R01 CA160417-01A1. We appreciate the assistance of the George Miller lab in developing PSC cultures.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385.
- Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, Choi JH, Choi Y, Shim S, Lyu IS, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun Rev. 2017;16(11):1160–1173. doi:10.1016/j.autrev.2017.09.012.
- Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. 2015;74(7):1417–1424. doi:10.1136/annrheumdis-2013-204837.
- Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A. 2012;109(32):13076–13081. doi:10.1073/pnas.1200419109.
- Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, Singhi AD, Kang R, Tang D, Lotze MT, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015;22(6):326–334. doi:10.1038/cgt.2015.21.
- Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–1862. doi:10.1084/jem.20100239.
- Mohanan S, Cherrington BD, Horibata S, McElwee JL, Thompson PR, Coonrod SA, Elliott PM, Christiansen M. Potential role of peptidylarginine deiminase enzymes and protein citrullination in cancer pathogenesis. Biochem Res Int. 2012;2012:895343. doi:10.1155/2012/685108.
- Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, et al. Histone deimination antagonizes arginine methylation. Cell. 2004;118(5):545–553. doi:10.1016/j.cell.2004.08.020.
- Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306(5694):279–283. doi:10.1126/science.1101400.
- Hirose T, Hamaguchi S, Matsumoto N, Irisawa T, Seki M, Tasaki O, Hosotsubo H, Yamamoto N, Yamamoto K, Akeda Y, et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS One. 2014;9(11):e111755. doi:10.1371/journal.pone.0111755.
- Wen F, Shen A, Choi A, Gerner EW, Shi J. Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis. Cancer Res. 2013;73(14):4256–4266. doi:10.1158/0008-5472.CAN-12-3287.
- Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, Wang Y, Simmons RL, Huang H, Tsung A. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016;76(6):1367–1380. doi:10.1158/0008-5472.CAN-15-1591.
- Trejo-Becerril C, Perez-Cardenas E, Gutierrez-Diaz B, De La Cruz-Siguenza D, Taja-Chayeb L, Gonzalez-Ballesteros M, García-López P, Chanona J, Dueñas-González A. Antitumor effects of systemic DNAse I and proteases in an In Vivo model. Integr Cancer Ther. 2016;15(4):NP35–NP43. doi:10.1177/1534735416631102.
- Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, Seifert L, Daley D, Greco SH, Deutsch M, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. 2015;212(12):2077–2094. doi:10.1084/jem.20142162.
- Najmeh S, Cools-Lartigue J, Giannias B, Spicer J, Ferri LE. Simplified human neutrophil extracellular traps (NETs) isolation and handling. J Vis Exp. 2015;98:52687.
- Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. 2013;110(21):8674–8679. doi:10.1073/pnas.1301059110.
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450.
- Haber PS, Keogh GW, Apte MV, Moran CS, Stewart NL, Crawford DH, Pirola RC, McCaughan GW, Ramm GA, Wilson JS. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am J Pathol. 1999;155(4):1087–1095. doi:10.1016/S0002-9440(10)65211-X.
- Schneiderhan W, Diaz F, Fundel M, Zhou S, Siech M, Hasel C, Möller P, Gschwend JE, Seufferlein T, Gress T, et al. Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J Cell Sci. 2007;120(Pt 3):512–519. doi:10.1242/jcs.03347.
- Bachem MG, Zhou S, Buck K, Schneiderhan W, Siech M. Pancreatic stellate cells–role in pancreas cancer. Langenbeck‘S Archives of Surgery. 2008;393(6):891–900. doi:10.1007/s00423-008-0279-5.
- Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, Mian A, Jiang J, Schrum J, Bossaller L, et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210(11):2447–2463. doi:10.1084/jem.20120201.
- Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17(4):666–676. doi:10.1038/cdd.2009.149.
- Vernon PJ, Loux TJ, Schapiro NE, Kang R, Muthuswamy R, Kalinski P, Tang D, Lotze MT, Zeh HJ. The receptor for advanced glycation end products promotes pancreatic carcinogenesis and accumulation of myeloid-derived suppressor cells. J Immunol. 2013;190(3):1372–1379. doi:10.4049/jimmunol.1201151.
- Stott K, Tang GS, Lee KB, Thomas JO. Structure of a complex of tandem HMG boxes and DNA. J Mol Biol. 2006;360(1):90–104. doi:10.1016/j.jmb.2006.04.059.
- Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570.
- Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–1206.
- Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. doi:10.1038/ni1457.
- Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, Drosos GI, Boumpas DT, Ritis K, Fritz JH. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS One. 2011;6(12):e29318. doi:10.1371/journal.pone.0029318.
- Demers M, Wagner DD. Neutrophil extracellular traps: A new link to cancer-associated thrombosis and potential implications for tumor progression. Oncoimmunology. 2013;2(2):e22946. doi:10.4161/onci.22946.
- Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, Bicker KL, Bingham RP, Campbell M, Chen YH, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11(3):189–191. doi:10.1038/nchembio.1735.
- Tamkovich SN, Cherepanova AV, Kolesnikova EV, Rykova EY, Pyshnyi DV, Vlassov VV, Laktionov PP. Circulating DNA and DNase activity in human blood. Ann N Y Acad Sci. 2006;1075:191–196. doi:10.1196/annals.1368.026.
- Cherepanova AV, Tamkovich SN, Bryzgunova OE, Vlassov VV, Laktionov PP. Deoxyribonuclease activity and circulating DNA concentration in blood plasma of patients with prostate tumors. Ann N Y Acad Sci. 2008;1137:218–221. doi:10.1196/annals.1448.016.
- Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37:646–650.
- Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer–a survey. Biochim Biophys Acta. 2007;1775(1):181–232. doi:10.1016/j.bbcan.2006.10.001.
- Singh N, Gupta S, Pandey RM, Chauhan SS, Saraya A. High levels of cell-free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival. Cancer Invest. 2015;33(3):78–85. doi:10.3109/07357907.2014.1001894.
- Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665.
- Bachem MG, Schunemann M, Ramadani M, Siech M, Beger H, Buck A, Zhou S, Schmid-Kotsas A, Adler G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–921.
- Nielsen MF, Mortensen MB, Detlefsen S. Key players in pancreatic cancer-stroma interaction: cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol. 2016;22(9):2678–2700. doi:10.3748/wjg.v22.i9.2678.
- Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541.
- Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29:179–187.
- Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;152(6):1492–1506.e24. doi:10.1053/j.gastro.2017.01.010.
- Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121–1132. doi:10.1053/j.gastro.2013.07.025.
- Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, Ban Y, Lavania S, Dawra R, Banerjee S, et al. NFκB in pancreatic stellate cells reduces infiltration of tumors by cytotoxic t cells and killing of cancer cells, via upregulationof CXCL12. Gastroenterology. 2018;155:880–891.e8. doi:10.1053/j.gastro.2018.05.051.
- Xia C, Braunstein Z, Toomey AC, Zhong J, Rao X. S100 proteins as an important regulator of macrophage inflammation. Front Immunol. 2017;8:1908. doi:10.3389/fimmu.2017.01908.
- Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, Jin X, Liu J, Mo S, Zhang T, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. 2018;9(1):873. doi:10.1038/s41467-018-03225-9.
- Rojas A, Delgado-Lopez F, Perez-Castro R, Gonzalez I, Romero J, Rojas I, Araya P, Añazco C, Morales E, Llanos J. HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumour Biol. 2016;37(3):3321–3329. doi:10.1007/s13277-015-3940-y.
- Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–1299. doi:10.1084/jem.20180139.
- Iurescia S, Fioretti D, Rinaldi M. Targeting cytosolic nucleic acid-sensing pathways for cancer immunotherapies. Front Immunol. 2018;9:711. doi:10.3389/fimmu.2018.00711.
- Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, Akira S, Matsuda T, Kawai T. DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J Immunol. 2017;198(4):1649–1659. doi:10.4049/jimmunol.1601694.
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi:10.1016/j.immuni.2014.10.017.
- Baird JR, Feng Z, Xiao HD, Friedman D, Cottam B, Fox BA, Kramer G, Leidner RS, Bell RB, Young KH, et al. STING expression and response to treatment with STING ligands in premalignant and malignant disease. PLoS One. 2017;12(11):e0187532. doi:10.1371/journal.pone.0187532.
- Baird JR, Friedman D, Cottam B, Dubensky TW Jr., Kanne DB, Bambina S, Bahjat K, Crittenden MR, Gough MJ. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res. 2016;76(1):50–61. doi:10.1158/0008-5472.CAN-14-3619.
- Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA, Coonrod SA. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One. 2011;6(7):e22043. doi:10.1371/journal.pone.0022043.
- Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne H-J, Kurschus FC, Schmidt AM, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113(11):1641–1650. doi:10.1172/JCI18704.
- Brinkmann V, Laube B, Abu Abed U, Goosmann C, Zychlinsky A. Neutrophil extracellular traps: how to generate and visualize them. J Vis Exp. 2010;36:1724.
- Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp. 2013;77:e50586.
- Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Gröne H-J, Brinkmann V, Jenne DE. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15(6):623–625. doi:10.1038/nm.1959.