
ABSTRACT
HPV16 infections are associated with a variety of cancers and there is compelling evidence that the transforming activity of HPV16 critically depends on the expression of the viral oncoproteins E6 and E7. Therapeutic cancer vaccines capable of generating durable and specific immunity against these HPV16 antigens hold great promise to achieve long-term disease control. Here we show in mice that HPV16 E7 RNA-LPX, an intravenously administered cancer vaccine based on immuno-pharmacologically optimized antigen-encoding mRNA, efficiently primes and expands antigen-specific effector and memory CD8+ T cells. HPV-positive TC-1 and C3 tumors of immunized mice are heavily infiltrated with activated immune cells and HPV16-specific T cells and are polarized towards a proinflammatory, cytotoxic and less immune-suppressive contexture. E7 RNA-LPX immunization mediates complete and durable remission of progressing tumors. Circulating memory T cells are highly cytotoxic and protect from tumor rechallenge. Moreover, E7 RNA-LPX immunization sensitizes anti-PD-L1 refractory tumors to checkpoint blockade. In conclusion, our data highlight the potential of HPV16 RNA-LPX for the treatment of HPV-driven cancers.
Introduction
In the past decade, infections with oncogenic human papillomavirus (HPV) types have been established as being causative for a variety of cancers including head and neck squamous cell carcinoma (HNSCC), cervical and anogenital cancers.Citation1
The most prevalent HPV subtype in HPV-positive HNSCC is HPV16. HPV-positive HNSCC is clinically, histo-pathologically and molecularly distinctCitation2 with increasing incidence and a significantly better prognosis than HPV-negative HNSCC, independent of the treatment modality.Citation3,Citation4 In HNSCC the standard of care includes surgery, radiotherapy and chemotherapy and is associated with long-term physical, functional and psychosocial impairments, strongly affecting the patient’s quality of life. Despite aggressive treatment, about 50% of patients die of their disease.Citation5,Citation6 Alternative therapies are needed to improve survival while reducing treatment-associated morbidity.
Interest in exploring immunological approaches has increased since the recent approval of immune checkpoint blockade therapy targeting the PD-1/PD-L1 axis for the treatment of patients with recurrent or metastatic HNSCC.7 The HPV oncoproteins E6 and E7, which drive malignant transformation and are constitutively expressed by the cancer cells,Citation8 are ideal targets for immunotherapy. This is further supported by the observation that spontaneously occurring T cells against HPV16 oncoproteins are critical for viral clearance and regression of HPV-positive premalignant lesions,Citation9 and that the density of tumor-infiltrating lymphocytes (TIL) is a strong predictor for the outcome of HPV-positive HNSCC.Citation10
In clinical trials, therapeutic HPV16 vaccines have demonstrated the feasibility of inducing a systemic CD8+ T cell response against E6 and E7 through immunization.Citation9,Citation11,Citation12
The ultimate goal of cancer vaccines is the induction of potent, durable and clinically relevant immune responses by delivering the antigen to antigen-presenting cells preferentially in the lymph node for its presentation under optimal immune-stimulatory conditions.Citation13
Synthetic mRNA is emerging as an attractive vaccine format due to its advantageous characteristics: mRNA is non-integrating and therefore considered as safe. It delivers the encoded antigen in an HLA-independent manner, is a natural adjuvant due to its TLR7/8 ligand activity and its production is time- and cost-efficient. We have developed mRNA that is pharmacologically optimized for stability and efficient translationCitation14 by the design of its cap element, 5ʹ- and 3ʹ-untranslated region and its poly(A)-tail. Further, the encoded antigen is fused to the MHC class I signal sequence and transmembrane and cytoplasmic domains for routing to the endoplasmic reticulum, resulting in increased presentation efficacy of MHC class I and II epitopes.Citation15 The mRNA is encapsulated in a liposomal formulation, RNA-lipoplex (RNA-LPX), which can be administered intravenously (i.v.) and is selectively taken up by dendritic cells (DC) residing in lymphoid compartments.Citation16 We have previously shown that the body-wide uptake of antigen-encoding RNA by APCs in mice is spatiotemporally aligned with a type I IFN response, orchestrating innate and adaptive immune mechanisms and resulting in strong anti-tumor immunity.Citation16,Citation17 Our first clinical data support the preclinically described mode of action.Citation16–Citation19
Here we present the characterization of HPV16 RNA-LPX, the first intravenously administered RNA-based therapeutic HPV16 vaccine, in mouse models. As there are no naturally processed and presented CD8+ T cell epitopes derived from the E6 oncoprotein in mice, our study focuses on the E7 oncoprotein to conceptually evaluate our HPV16 RNA-LPX vaccine.
We show the induction of strong T cell immunity by E7 RNA-LPX, which results in durable remission of murine HPV16-positive TC-1 and C3 tumors, prevents relapse and synergizes with checkpoint blockade therapy. Our data warrant further exploration of therapeutic HPV16 RNA-LPX vaccination.
Results
E7 RNA-LPX immunization induces a strong antigen-specific effector and memory CD8+ T cell response in mice
To assess the immunogenicity of HPV16 E7 RNA-LPX, naïve C57BL/6 mice were immunized twice with vaccine or saline control. An increase of type I and II IFN, IL6 and TNFα plasma levels were detected 3 hours after the first immunization, indicating the intended systemic immune stimulation ((a)). The percentage of T cells stained with multimers featuring E749-57, the only known murine H2-Db restricted CD8+ epitope,Citation20 was quantified six days after each immunization. In E7 RNA-LPX vaccinated mice a significant increase of E7 antigen-specific T cells was observed in the blood after the first immunization and further expanded upon secondary immunization, reaching up to 35 % of antigen-specific CD8+ T cells ((b)). Whereas the vast majority of cells in the multimer-negative population of repeatedly HPV16 E7 RNA-LPX immunized mice were naïve, multimer-positive cells were found to have entered various stages of differentiation, including robustly expanded CD127− KLRG1+ short-lived effector cells (SLEC) and CD127+ KLRG1− memory precursor cell (MPEC) phenotypes ((c)). The major fraction of E7 antigen-specific MPECs were CD62L negative early effector memory T cells (Tem,), a smaller fraction was CD62L positive, a phenotype associated with commitment towards early central memory T cells (Tcm) ((d)).
Figure 1. E7 RNA-LPX immunization induces a strong antigen-specific effector and memory CD8+ T cell response in mice (a-d) Mice (n=5/group) were immunized twice with E7 RNA-LPX or NaCl (control) (d0, d7) and blood samples were harvested six days after each immunization (d6, d13). (a) Serum cytokines 3 h after first immunization were determined by multiplex immunoassay. (b-d) Flow cytometric characterization of de novo primed E749-57 MHC-class I multimer positive CD8+ T cells in the blood with regard to (b) their frequency and (c, d) their phenotype on d13. (c) Differential expression of CD127 and KLRG1 on E749-57 MHC-class I multimer positive (pos) and negative (neg) CD8+ T cells of E7 RNA-LPX immunized mice. (d) Differential expression of CD62L within KLRG1−CD127+ (MPEC) E749-57-specific CD8+ T cells. (b, c) Representative pseudocolor plots are shown. Significance was determined using (a, b) unpaired, two-tailed Student’s t-test and (c) two-way ANOVA, Bonferroni post-test. Mean±SD. SLEC: Short-lived effector cells; MPEC: Memory precursor effector cells; Tem: T effector memory cells; Tcm: T central memory cells.
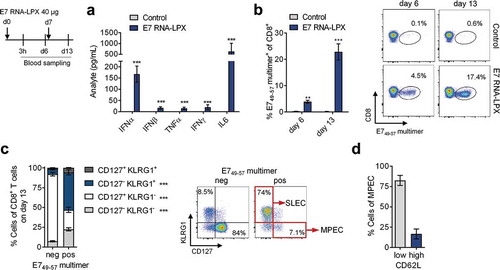
Together, these data indicate that HPV16 E7 RNA-LPX efficiently primes and expands endogenous E749-57 specific CD8+ T cells of both the effector as well as memory phenotype.
E7 RNA-LPX immunization mediates complete remission of progressing HPV16-positive tumors and establishes protective T cell memory
We next assessed the anti-tumor efficacy of E7 RNA-LPX induced T cells in two HPV16 antigen-positive syngeneic mouse tumor models – TC-1 and C3 – of which TC-1 has higher E7 expression levels than C3 (Supplementary Figure 1).
HPV-positive malignancies typically arise at sites of infection in oral or genital regions in the mucosa. As this poses a peculiar challenge for T cell trafficking,Citation21,Citation22 we first explored the HPV16 E7 vaccine in an orthotopic tumor setting. TC-1 luc tumors were implanted submucosally at the base of the tongue. A few days after a single E7 RNA-LPX immunization dramatic tumor regression was observed in all mice ((a)). Nine days after immunization, orthotopic tumor lesions of RNA-LPX treated mice were found to be heavily infiltrated with leukocytes ((b)). Up to 42 % of the infiltrating CD8+ T cells were HPV 16 E749-57 specific ((c)). The majority of HPV16 E749-57 specific T cells in orthotopic tumors were positive for the homing-associated integrin CD49α, whereas all antigen-specific CD8+ T cells circulating in blood were CD49α negative ((d)), further supporting effective trafficking of primed T cells to tumors located at mucosal sites.
Figure 2. E7 RNA-LPX immunization mediates complete remission of progressing HPV16-positive tumors and establishes protective T cell memory (a-d) TC-1 luc tumor cells were grafted into the submucosal lining of the tongue. Five days later mice were immunized with E7 RNA-LPX (n=13) or irrelevant (OVA257-264) RNA-LPX (n=12). (a) TC-1 luc tumor growth kinetics as measured by in vivo bioluminescence. (b-d) Mice were sacrificed 10 days after tumor-challenge and tumor tissues harvested (n=3/group). The proportion of (b) CD45+ and (c) E749-57 multimer+ CD8+ T cells in mice treated either with E7 RNA-LPX or irrelevant (OVA257-264) RNA-LPX was measured by flow cytometry. (d) CD49α expression of E749-57 multimer+ CD8+ T cells of E7 RNA-LPX treated mice in blood and tumor. (e, f) TC-1 tumor growth (e, left) and survival (e, right) in mice (n=10/group) immunized three times with E7 RNA-LPX or irrelevant (eGFP) RNA-LPX. Average tumor size of ~6 mm3 at start of treatment. (f) Survival of E7 RNA-LPX treated mice (n=10) rechallenged with TC-1 after initial TC-1 tumor challenge. Treatment-naive mice (n=10) served as control group. (g, h) C3 tumor growth (g, left) and survival (g, right) in mice immunized four times with E7 RNA-LPX (n=9) or irrelevant (OVA257-264) RNA-LPX (n=10). Average tumor size of ~25 mm3 at start of treatment. (h) Survival of E7 RNA-LPX treated mice rechallenged with TC-1 after C3 tumor challenge (n=12/group). Treatment-naive mice (n=10) served as control group. (i, j) Mice (n=10) bearing C3 tumors (average size of 25 mm3 at start of treatment) were immunized four times with E7 RNA-LPX and left without further treatment after complete tumor rejection. (i) CD8+ T cell phenotype in blood was analyzed before (pre-boost) and 5 days after receiving a booster dose (post-boost) of E7 RNA-LPX or NaCl (n=5/group). (j) Specific lysis of target cells in vivo was determined 18 h after adoptive transfer of CFSE-labeled splenocytes pulsed with E749-57 or OVA257-264 (control) peptide. Significance was determined using (e-h) log-rank test, (a, i) two-way ANOVA, Sidak’s multiple comparison test, and (j) one-way ANOVA, Tukey’s multiple comparison test. Ratios depict the frequency of mice with complete tumor responses (e, g). Mean±SD.
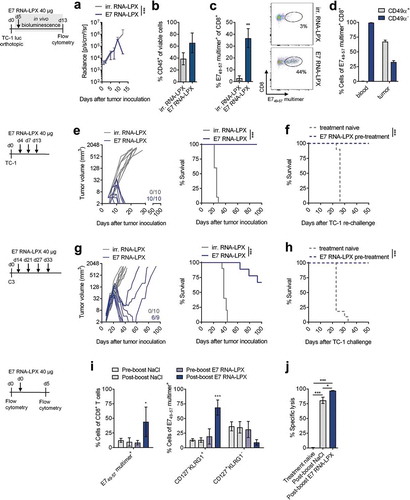
Next, tumor cells were implanted subcutaneously (s.c.) into the flank of mice and mice were dosed repeatedly i.v. with RNA-LPX encoding either HPV16 E7 or an irrelevant (irr.) antigen. All control-treated mice experienced progressive tumor growth and had to be sacrificed within 30 to 40 days after tumor challenge ((e, g)). RNA-LPX immunization was associated with a rapid and complete rejection of tumors in all TC-1 tumor-bearing mice ((e)) and in 6 of 9 C3 tumor-bearing mice ((g)). The remaining three C3 tumor-bearing mice initially responded with tumor shrinkage but tumor growth eventually progressed ((g)). All mice with a complete anti-tumoral response to E7 RNA-LPX immunization remained tumor-free for the entire observation period. Mice rechallenged with TC-1 cells remained tumor-free, indicating the prevalence of a functional memory T cell response against the HPV16 E7 antigen ( (f and h)).
This finding, together with our observation that a fraction of primed T cells retaining CD127 expression, prompted us to study the memory response in greater detail. A boosting immunization with E7 RNA-LPX induced expansion of the E749-57 multimer positive cells, predominantly of the CD127− KLRG1+ short-lived effector cell (SLEC) phenotype ()). Most importantly, when CFSE-labeled splenocytes pulsed with E749-57 peptide were injected after this boost, they were found to be similarly well lysed by NaCl-boosted mice as compared to E7 RNA-LPX-boosted mice ( (j)).
Collectively, these data indicate that E7 RNA-LPX efficiently primes T cells which not only execute anti-tumor activity and reject progressing tumors, but develop a durable memory response functionally capable of exerting immediate cytotoxic effector activity upon re-encountering the antigen without prior boost.
Tumors of E7 RNA-LPX immunized mice have brisk immune infiltrates, E7-specific CD8 + T cells and a proinflammatory, cytotoxic and less immune-suppressive contexture
Tumor-infiltrating cell populations from both TC-1 and C3 tumor-bearing mice were analyzed by flow cytometry 11 days after a single E7 RNA-LPX immunization or control treatment with irrelevant (OVA257-264) RNA-LPX. In both tumor models, immunization was associated with early leukocyte infiltration ((a)) and strong expansion of E7-specific CD8+ T cells ((b)). Tumor-infiltrating CD8+ T cells of E7 RNA-LPX immunized, but not of control-treated mice, expressed high levels of IFNγ and granzyme B (GzmB) when stimulated ex vivo with E749-57 peptide (Supplementary Figure 2). TC-1 tumors of E7 RNA-LPX vaccinated mice had higher frequencies of tumor-infiltrating CD8+ and CD4+ T cells ((c)), which were also highly proliferative as measured by Ki-67 staining ((d)). In C3 tumors, this finding was confirmed for the CD8+ T cell population, whereas the frequency and proliferative capacity of CD4+ T cells were not affected by the E7 vaccine ((f and g)). CD8+ T cells evenly infiltrated TC-1 and C3 tumors, displaying features of non-excluded, CD8+ T cell inflamed tumors ((i)). In both tumor models, E7 RNA-LPX was associated with a significant increase of NK cells and tumor-associated macrophages (TAM) ((c and f)). TAMs were slightly skewed towards the proinflammatory iNOS-secreting M1-like phenotype, whereas the frequency of suppressive, CD206+ M2-like macrophages were lower compared to control vaccinated mice ((e and h)). Immunization had no effect on Tregs in either model ((c and f)).
Figure 3. Tumors of E7 RNA-LPX immunized mice have brisk immune infiltrates, E7-specific CD8+ T cells and a proinflammatory, cytotoxic and less immune-suppressive contexture Analysis of TIL in TC-1 (a, b, c-e) and C3 tumors (a, b, f-h) resected from mice (n=5/group) 11 days after one immunization with E7 RNA-LPX or irrelevant (OVA257-264) RNA-LPX (TC-1: ~63 mm3 and C3: ~65 mm3 at therapy start). (a) Percentage of CD45+ cells and (b) frequency of E749-57-specific CD8+ T cells in TC-1 and C3 tumors as measured by flow cytometry. (b, right) Representative E749-57 multimer staining in TC-1 tumors. (c, f) Frequency of leukocyte subsets in (c) TC-1 and (f) C3 tumors. (d, g) Ki67+ fraction of tumor-infiltrating CD4+ and CD8+ T cells in (d) TC-1 and (g) C3 tumors. (e, h) Expression of iNOS and CD206 in TAM of (e) TC-1 and (h) C3 tumors. (i) Immunofluorescent staining of CD8+ T cell infiltration (green) in TC-1 (top) and C3 (bottom) tumors (nuclear staining: Hoechst (blue), scale bar = 50 µm). (j) Gene expression analysis of selected genes in TC-1 (top) and C3 tumors (bottom) resected after two immunizations with E7 RNA-LPX or irrelevant (OVA257-264) RNA-LPX determined by qRT-PCR. Tumor tissues were harvested two days after the last immunization. Heatmaps display log2-fold changes calculated and normalized to irrelevant (OVA257-264) RNA-LPX treated mice. (a-h) Significance was determined using unpaired, two-tailed Student’s t-test and (j) Holm-Šídák multiple comparison test. Mean±SD. TIL: Tumor-infiltrating lymphocytes; TAM: Tumor-associated macrophages; iNOS: Inducible nitric oxide synthase.
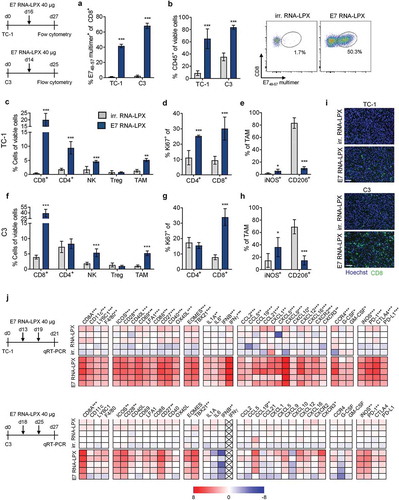
Gene expression analysis of bulk tumor samples of immunized mice confirmed and further extended the observations we made by flow cytometry on cellular level ((j)). E7 RNA-LPX immunization was associated with the upregulation of markers for CD8+ T cell function, attraction and Th1 development,Citation23 including T cell transcription factors EOMES and TBX21 (T-bet), T cell co-stimulatory molecules CD28, CD27, ICOS and chemokines such as CCL5, CXCL9 and CXCL12 together with their co-receptor CXCR3 known to be expressed on CD8+ T cellsCitation24 and NK cells.Citation25 Proinflammatory molecules such as IL-1, IL-6, interferons and CCL2 were increased, as were PD-1 and iNOS. Most, but not all these markers were upregulated in both tumors models. Moreover, TC-1 tumors but not C3 tumors of vaccinated mice displayed a higher expression of DC activation markers such as CD86, CD40, markers for monocyte/macrophage recruitment, such as F4/80, CCL5 and GM-CSF, and the immune checkpoint molecules PD-L1 and CTLA-4.
In summary, vaccination with HPV16 E7 RNA-LPX was markedly associated with polarization towards a proinflammatory, cytotoxic and less immune-suppressive contexture, which was more pronounced in TC-1 compared to C3 tumors.
E7 RNA-LPX immunization synergizes with checkpoint-blockade by rendering anti-PD-L1 refractory tumors responsive
C3 and TC-1 tumors are known to be resistant to PD-L1/PD-1 immune checkpoint inhibitor (CPI) monotherapy.Citation26–Citation28 E7 RNA-LPX vaccination appears to be associated with an array of immunologically favorable alterations in the tumor microenvironment including upregulation of PD-1 and of PD-L1, which correlates with IFNɣ induction observed in HPV-antigen expressing TC-1 tumors (, Supplementary Figure 3). Hypothesizing that increase in PD-L1 expression in conjunction with a stronger inflamed, non-excluded and less suppressed local milieu could convert a tumor refractory to PD-L1/PD-1 blockade to a susceptible one, we tested combination treatment of vaccine and anti-PD-L1 (aPD-L1) in mice. To allow a therapeutic window despite the efficacy of E7 RNA-LPX monotherapy, tumors were grown to a well advanced stage (TC1 model: treatment start at day 16, tumor average size of 48 mm3; C3 model: treatment start at day 17, tumor average size of 157 mm3) and only a single dose of vaccine was administered followed by aPD-L1 treatment every 3–4 days.
In the TC-1 model, the majority of mice treated with aPD-L1 alone had to be sacrificed within 40 days, whereas with E7 RNA-LPX monotherapy, complete remission in 4 of 15 mice and a significant survival benefit was observed ((a–b)). The combination regimen further augmented the rate of complete remissions (10 of 15 mice) and improved overall survival (70% of mice surviving compared to 30% with vaccine alone). As early tumor growth kinetics were largely comparable between E7 RNA-LPX vaccine and aPD-L1 combination therapy groups, the addition of aPD-L1 to the vaccine appeared to prevent the late relapse of tumors.
Figure 4. E7 RNA-LPX immunization synergizes with checkpoint-blockade by rendering anti-PD-L1 refractory tumors responsive (a, c) Survival and (b, d) tumor growth kinetics of (a, b) TC-1 or (c, d) C3-bearing mice (n=14–15/group) immunized with E7 RNA-LPX or irrelevant (OVA257-264) RNA-LPX on day 16 or day 17 respectively and treated with anti-PD-L1 (aPD-L1) antibody or isotype control from day 19 or day 20 onwards every 3 to 4 days. The average size of TC-1 tumors was ~48 mm3 and of C3 tumors at ~157 mm3 at start of treatment. Representative data is shown for two (each TC-1 and C3) similar, but independent experiments. Significance was determined using (a, c) log-rank test. (b, d) Ratios depict frequency of mice with complete tumor responses.
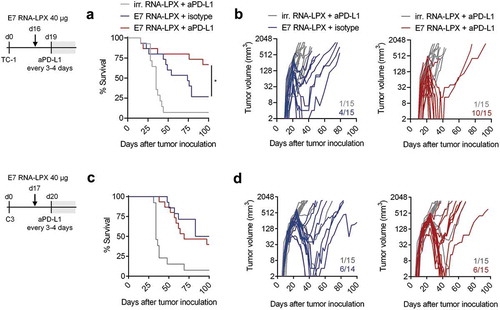
In the C3 model, the addition of aPD-L1 to the vaccine did not further improve the response rate or overall survival of C3 tumor-bearing mice compared to E7 RNA-LPX vaccine alone ((c–d)).
Discussion
Our study introduces a novel HPV16 RNA-LPX vaccine that is capable of mediating rejection of both HPV16-positive TC-1 and C3 tumors and improving the survival of tumor-bearing mice without the addition of an extrinsic adjuvant or another active treatment modality. We demonstrate that HPV16 RNA-LPX primes and expands E7 antigen-specific CD8+ T cells with an effector and effector memory phenotype, which acquire homing characteristics that support the penetration of subcutaneous and submucosal tumors.
Vaccines based on various formats including DCs,Citation29 DNACitation30,Citation31 and peptidesCitation20,Citation32–Citation34 are being studied for induction of HPV16-specific immunity in the same mouse models we used here. The efficacy of E744-62 peptide in incomplete Freud’s adjuvants (IFA) vaccine as well as E7-antigen expressing DC vaccine was shown in a prophylactic setting, protecting mice from s.c. C3Citation20 and TC-1Citation29 tumor challenge. For the treatment of prevalent HPV16-positive tumors, different DNA-based and synthetic-long peptide (SLP)-based vaccines were used in combination with other treatment modalities or proper immune-stimulatory adjuvants. Efficacious treatment of subcutaneous TC-1 tumors was reported with a DNA vaccine encoding E7 antigen fused to calreticulin but required the combination with cisplatin chemotherapy or radiotherapyCitation30,Citation31 to achieve full survival benefits. SLP-based HPV vaccines have been shown to induce anti-tumoral effects in subcutaneous TC-1 tumor models, when co-administered with ODN-CpG-ODN (CpG-oligodeoxynucleotide) or incomplete Freud’s adjuvants (IFA).Citation34 Prime-boost vaccinations with E743-77 SLP and the DC-activating adjuvant CpG-ODN led to the complete eradication of 80% of TC-1 tumors, whereas E743-77 SLP co-administered with IFA resulted in an initial response followed by later relapse. Furthermore, E743-77 SLP vaccine emulsified in Montanide ISA 51 has been reported to synergize with cisplatin chemotherapy in TC-1 tumor-bearing mice, where 80% survival could be achieved over 20% survival with E743-77 SLP peptide vaccine alone.Citation35 In another study, TriVAX HPV vaccine,Citation33 was shown to reject subcutanous TC-1 and C3.43 tumors by combining E749-57 peptides, poly-IC adjuvant and an costimulatory anti-CD40 antibody.
HPV16 RNA-LPX induced anti-tumoral responses were accompanied by a profound reshaping of the tumor-microenvironment in TC-1 and C3 tumors towards an inflammatory and cytotoxic signature with increased expression of T cell- and monocyte-attracting chemokines and a strong infiltration of E749-57 specific CD8+ T cells as well as NK cells and macrophages. Macrophages were skewed moderately towards an iNOS+CD206− M1-like proinflammatory phenotype, which is a favorable prognostic factor and associated with anti-tumor activity.Citation36,Citation37 This appears to be a common feature of successful vaccine-induced regression of HPV-positive tumors. In mouse studies exploring HPV16 E743-77 SLP vaccination as well, regression of TC-1 tumors depended on attraction and polarization of tumor-infiltrating macrophages.Citation32
Another key observation is that HPV16 RNA-LPX vaccination builds up strong and sustainable T cell memory. Following complete tumor rejection, circulating E7 antigen-specific T cells were capable of efficiently lysing E749-57 peptide-pulsed target cells in vivo without being boosted. This may contribute to the observed strong protection against rechallenge and relapse we have observed.
The PD-1 checkpoint inhibitors pembrolizumab and nivolumab were approved in 2016 for patients with advanced HNSCC and the aPD-L1 antibody durvalumab has shown clinical activity in a phase 2 trial.Citation38 Only a fraction of patients responds to these checkpoint blockers, whereas others progress under treatment. The HPV16-positive TC-1 and C3 cell lines represent murine models for PD-1/PD-L1 blockade refractory cancers. In the TC-1 model, the combination of aPD-L1 with the HPV16 E7 RNA-LPX vaccine resulted in synergistic inhibition of tumor growth and significant survival benefit. This supports the notion that in later lines of treatment, patients – even if non-responsive to CPI alone – may profit from the combination of a CPI with an HPV16-directed vaccine. In contrast, in the C3 model, the combination of aPD-L1 with HPV16 RNA-LPX vaccine was not beneficial over vaccine alone. Others have also reported that C3 is more difficult to treat by vaccine approaches,Citation32 to which our observations of a lower HPV antigen expression and only a modest reshaping of the immune contexture by the vaccine, also in addition to intrinsic resistance mechanisms shown by others such as the Qa-1/NKG2A axis,Citation39 may contribute.
In conclusion, our study demonstrates the potency of the HPV16 RNA-LPX vaccine format and warrants further translational pursuit of this concept. In this regard, clinical testing of an HPV16 RNA-LPX targeting both of the viral oncogenes E6 and E7 has been initiated in patients with HPV-driven cancers including HNSCC, anogenital, cervical and penile cancers (EudraCT: 2014–002061-30).
Material and methods
Mice
C57BL/6 mice were purchased from Charles River and Envigo. Throughout all experiments, age-matched (6–12 weeks) female animals were used. Procedures and experimental group sizes were approved by the regulatory authorities for animal welfare. All mice were kept in accordance with federal and state policies on animal research at the University of Mainz and BioNTech AG.
Tumor cell lines
The TC-1 tumor cell line derived from primary lung cells by immortalization and retroviral transduction with HPV16 E6/E7Citation40 was obtained together with the luciferase-transfected variant TC-1 luc from T.-C. Wu (Johns Hopkins University). The C3 tumor cell line generated by immortalization and transfection of B6 mouse embryonic cells with the complete HPV16 genome,Citation20 was a kind gift from S.H. van der Burg (Leiden University Medical Center, The Netherlands). Re-authentication of cells and generation of master and working cell banks were performed immediately upon receipt. The third to ninth passages were used for tumor experiments.
RNA constructs and in vitro transcription
Plasmid templates for in vitro transcription of antigen-encoding RNAs were based on the pST1-A120 and pST1-MITD vectorCitation14 which feature 5` and 3`UTRs and poly(A) tails pharmacologically optimized for stability and protein translation. pST1-MITD features a signal sequence for routing to the endoplasmic reticulum and the major histocompatibility complex (MHC) class I transmembrane and cytoplasmic domains for improved presentation of MHC class I and II epitopes. pST1-eGFP-A120 (eGFP), pST1-OVA-MITD (OVA257-264) and pST1-E7-MITD vectors were described previously.Citation14–Citation16,Citation41 pST1-OVA-MITD encodes the H-2Kb-restricted, immunodominant epitope OVA257-264. pST1-E7-MITD encodes HPV16 full-length E7 protein. Templates for in vitro transcription of mRNA were generated by cloning target sequences into the pST1-MITD vector. In vitro transcription and capping with β-Santi-reverse cap analog (ARCA)Citation41 were performed as described previously.Citation14
Liposomes, RNA-LPX preparation, and immunization
Liposomes with cationic net charge comprised of the cationic lipid DOTMA (Merck & Cie) and the helper lipid DOPE (Avanti Polar Lipids or Corden Pharma) were used to complex RNA for the formation of RNA-LPX. Liposome manufacture and LPX formation were performed as previously described.Citation16 In brief, RNA was stored in HEPES-buffered solution at an RNA concentration of 1 mg mL.−1 RNA-LPX were prepared by diluting the RNA with H2O and 1.5 M NaCl solution followed by adding an appropriate amount of liposome dispersion to reach a charge ratio of (+):(-) 1.3:2 at a final NaCl concentration of 150 mM. At the indicated charge ratio, RNA-LPX preparations (E7, eGFP, and OVA257–264 RNA-LPX) had a particle size of 200–250 nm, a polydispersity index of ~0.25 and a zeta potential (mV) of −20–30 mV. If not mentioned otherwise, mice were immunized with 40 µg RNA-LPX. Control mice received RNA-LPX encoding for irrelevant protein (eGFP or OVA257–264) or NaCl. Arrows in vaccination schemes indicate immunization.
Tumor models and treatment
For therapeutic tumor experiments, C57BL/6 mice were injected s.c. with 1 × 105 TC-1 tumor cells or 5 × 105 C3 tumor cells. Tumor sizes were measured unblinded with a caliper every three to four days for calculating tumor volumes using the equation (a2 × b)/2 (a, width; b, length). For orthotopic tumor experiments, 5 × 104 TC-1 luc tumor cells were inoculated into the submucosa of the tongue. In some experiments, 200 µg of PD-L1 antibody (10F.9G2, BioXCell) or IgG2b isotype control (LF-2, BioXCell) were administered every three to four days. Animals were euthanized when predefined endpoints were reached.
Bioluminescence imaging
Orthotopic TC-1 luc tumor growth was evaluated by in vivo bioluminescence imaging using the Xenogen IVIS Spectrum imaging system (Perkin Elmer, Waltham, MA, USA). An aqueous solution of L-luciferin (1.3 mg/mouse; BD Biosciences) was injected intraperitoneally. Emitted photons from live animals were quantified 5 min after luciferin administration. Regions of interest (ROI) were quantified as average radiance (photons s−1 cm−2 sr−1) using IVIS Living Image 4.0 software.
Tissue preparation
For the generation of single cell suspensions, tumors were excised, cut into pieces and subsequently incubated in digestion buffer containing 5 µg/mL collagenase (Roche Diagnostics), 500 units hyaluronidase (Sigma-Aldrich) and 50 µg/mL DNAse I (Roche Diagnostics) in PBS at 37°C for 15 min. Tumor samples were forced through a 70 µm cell strainer (BD Falcon) using a plunger end of a syringe while rinsing with PBS. Cells were centrifuged at 460 g for 6 min and resuspended in fresh PBS. Erythrocytes were lysed with a hypotonic electrolyte solution for 5 min. Similarly, spleens were forced through a 70 µm cell strainer while rinsing with PBS and erythrocytes lysed with a hypotonic electrolyte solution. Peripheral blood (50 µl) was collected from the orbital sinus to obtain serum or PBMCs for FACS staining.
In vivo cytotoxicity assay
Splenocytes from C57BL/6 mice were labeled with 0.5 µM (low) or 5 µM (high) CFSE (Thermo Scientific) as previously described.Citation42 CFSEhigh cells were pulsed with E749-57 peptide (4 µg/mL) and CFSElow cells pulsed with OVA257-264 peptide (4 µg/mL) (Jerini Peptide Technologies). Cell suspensions were washed with PBS and combined at a ratio of 1:1. Mice received 2 × 107 labeled, peptide-pulsed splenocytes in 200 µL PBS. After 18 h, recipients were sacrificed and splenocytes analyzed by flow cytometry. Antigen-specific lysis was calculated by the equation: Specific lysis (%) = (1-(% CFSElow/% CFSEhigh))×100.
Flow cytometry
Flow cytometry staining was conducted on full blood, tumor, and spleen single cell suspension. Monoclonal antibodies for extracellular staining included CD45, CD8α, CD4, CD44, CD86, PD-1, NK1.1, CD11b, PD-L1, I-A/I-E (BD Pharmingen), CD3, F4/80, Gr-1, CD127 (Biolegend), CD25 and KLRG1 (eBioscience). For intracellular staining, antibodies against IFNγ, Ki-67 (eBioscience), TNFα, Foxp3 (BD Pharmingen), granzyme B, iNOS (Invitrogen) and CD206 (BioLegend) were used. Flow cytometry staining was performed as follows: Live cells were stained with viability dyes (eBioscience) according to manufacturer’s instructions. E7-specific CD8+ T cells were stained with E749-57 H2-Db-restricted dextramers (Immudex) for 10 min at 4°C in the dark. Extracellular targets were stained for 30 min at 4°C in the dark. PBS containing 5% FCS and 5 mM EDTA was used as washing and staining buffer. For the staining of IFNγ, TNFα and granzyme B, samples were fixed and permeabilized with Cytofix/Cytoperm (BD Pharmingen), whereas for iNOS, CD206, Ki67 and Foxp3 staining, samples were fixed and permeabilized using Foxp3 Fixation Kit (eBioscience) according to manufacturer’s instructions. Full blood was stained with MHC-dextramers prior to erythrocyte lysis using BD FACS lysing solution (BD Pharmingen). Intracellular cytokine staining was performed as earlier described,Citation43 stimulating tumor single cell suspensions for 5 h at 37°C in the presence of 10 µg/mL Brefeldin A (Sigma) and 2 µg/mL E749-57 (RAHYNIVTF) or irrelevant VSV-NP52-59 (RGYVYQGL) peptide (Jerini Peptide Technologies). Immune cell populations were defined by pre-gating on viable cells and singlets and determined as follows: NK cells (CD45+ CD3− NK1.1+), CD8+ T cells (CD45+ CD8+), E749-57 specific CD8+ T cells (CD45+ CD8+ E749-57 multimer+), CD4+ T cells (CD45+ CD4+), Treg (CD45+ CD4+ Foxp3+ CD25+), tumor-associated macrophages (CD45+ CD11b+ F4/80+). Flow cytometric data was acquired on a FACS Canto II or LSR Fortessa flow cytometer (both BD Biosciences, Franklin Lakes, USA) and analyzed with FlowJo 7.6.5 or FlowJo 10.4 software (Tree Star).
Immunofluorescence staining
8-µm sections of cryconserved tumors were fixed in 4 % paraformaldehyde (PFA) for 10 min and blocked in PBS supplemented with 1 % BSA, 5 % mouse serum, 5 % rat serum and 0.02 % Nonident for 1 h, in the dark at room temperature. Fluorescence-labeled antibody against CD8α (BD Pharmingen) and PD-L1 (Biolegend) were used to stain sections overnight at 4°C, followed by nuclear staining with Hoechst (Sigma-Aldrich). Immunofluorescence images were acquired using an epifluorescence microscope (Axio Scan.Z1, Zeiss, Oberkochen, Germany).
Fluidigm qPCR expression analysis
Total RNA was extracted from cryo-conserved tumors using Tissue Lyser II (Quiagen). RNA was isolated by classical phenol/chloroform extraction following an initial RNA-cleanup on QIAcube using RNeasy mini spin columns. RNA was reverse-transcribed to cDNA using TAKARA PrimeScript™ RT Reagent Kit (Perfect Real Time). The cDNA was subjected to the Fluidigm BioMark HD system, following Fluidigm® Advanced Development Protocol 28 – Fast Gene Expression Analysis using TaqMan® Gene Expression Assays (PN 100–2594 A2). Detection of 6-carboxyfluorescein signal, linear baseline correction, and the auto Ct threshold method were used for Ct-value determination. For data analysis, the R software package “qpcrPANEL” developed in-house was used. Raw Ct-values were read in and undetected Cts were set to the 0.975 quantiles of all Ct. ΔΔCt-values were calculated as described.Citation44 Hypoxanthine-Guanine phosphoribosyltransferase (HPRT) was used as the reference gene to compute the ΔCt for each data point (ΔCti,j = Cti,j – CtHPRT,j) with i as gene and j as sample index. ΔCti,j were calibrated with the ΔCt-value of the means (χ) of the control sample for each gene versus the mean of control samples in HPRT (ΔCti,calib = χCti,control – χCtHPRT,control). Primer efficiencies were assumed to be equal and defined as ΔΔCt = 2-(- ΔCti,j – ΔCti,calib). Differences in log2-fold changes of ΔΔCt-values were plotted to generate heat maps (GraphPad PRISM 7).
Multiplex Immunoassay
Analysis of serum samples was performed by using the Th1/Th2 cytokine panel 6-plex (eBioscience) according to manufactures instruction. Data was acquired using the Magpix system (Luminex, TX, USA) and analyzed with xPONENT 4.2 software.
Statistical analysis
Individual treatment and corresponding control group means were compared by unpaired, two-tailed Student’s t test using corrected p values (Holm-Šídák approach) if appropriate. Two-way ANOVA was performed when both time and treatment were compared, and when significant (p < .05), multiple comparisons were performed using Bonferroni posthoc tests. Survival benefit was determined with the log-rank test (Mantel-Cox). *p ≤ .05, **p ≤ .01, ***p ≤ .001. If not mentioned otherwise, results are depicted as mean±SD. All statistical analyses were performed with GraphPad PRISM 7.
Disclosure of potential conflicts of interest
C.G., Ö.T., and U.S. are employees at BioNTech (Mainz, Germany). U.S., M.D. and S.K. are inventors on patents and patent applications related to this study. U.S. is cofounder; Ö.T. and U.S management board member of BioNTech and have an ownership interest in TRON and BioNTech. All other authors have no potential conflict of interest.
Author contributions
U.S., Ö.T., S.K., M.D. and F.V. conceived and guided the study. C.G., N.S., A.S., T.B. and F.V. performed and analyzed experiments. C.G., N.S., Ö.T. and U.S. interpreted the results and wrote the manuscript.
Supplemental Material
Download PDF (1.6 MB)Acknowledgments
We thank A. König, C. Ettingshausen, U. Schmitt, R. Roth, I. Beulshausen, E. Petscherskich, E. Daniel, M. Brkic and J. Neumaier for technical assistance; M. Suchan, B. Schrörs for qPCR gene expression analysis; S. Witzel and B. Tillmann for cloning of constructs; K. Tillmann and team for RNA production; J. Schumacher and A. Gerdts for the technical assistance in the measurement of RNA-LPX preparations. We are grateful to L.M. Kranz for conceptual and technical discussions and to L.M. Kranz, R. Rae and K. Chu for their diligent proofreading of this manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- IARC Working Group. Human Papillomaviruses. IARC Monographs on the evaluation of carcinogenic risks to humans; 2007. p. 90. doi:10.1094/PDIS-91-4-0467B.
- Lawrence, M. S. M. S. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi:10.1038/nature14129.
- Ang KK, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapía M, Guanira-Carranza JV, Ramirez-Cardich ME, et al. Human papillomavirus and survival of patients with oropharyngeal cancer — NEJM. N Engl J Med. 2010;363:24–35. doi:10.1056/NEJMoa1011205.
- Dayyani F, Etzel CJ, Liu M, Ho C-H, Lippman SM, Tsao AS. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC). Head Neck Oncol. 2010;2. doi:10.1186/1758-3284-2-15.
- Argiris A, Karamouzis MV, Raben D, Ferris RL. Seminar: head and neck cancer. Lancet. 2008;371:1695–1709. doi:10.1016/S0140-6736(08)60728-X.
- Razzaghi H, Saraiya M, Thompson TD, Henley SJ, Viens L, Wilson R. Five-year relative survival for human papillomavirus-associated cancer sites. Cancer. 2018;124:203–211. doi:10.1002/cncr.v124.1.
- Soulieres, D., Cohen E, Le Tourneau C, Dinis J, Licitra L, Ahn MJ, Soria A, Machiels JP, Mach N, Mehra R, et al. Abstract CT115: updated survival results of the KEYNOTE-040 study of pembrolizumab vs standard-of-care chemotherapy for recurrent or metastatic head and neck squamous cell carcinoma. Cancer Res. 2018. doi:10.1158/1538-7445.AM2018-CT115.
- Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe. 2014;15:266–282. doi:10.1016/j.chom.2014.02.011.
- Kenter GG, Welters MJP, Valentijn ARPM, Lowik MJG, Berends-van der Meer DMA, Vloon APG, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–1847. doi:10.1056/NEJMoa0810097.
- Ward MJ, Thirdborough SM, Mellows T, Riley C, Harris S, Suchak K, Webb A, Hampton C, Patel NN, Randall CJ, et al. Tumour-infiltrating lymphocytes predict for outcome in HPV-positive oropharyngeal cancer. Br J Cancer. 2014;110:489–500. doi:10.1038/bjc.2013.639.
- Trimble CL, Veillard D, Laplaud DA, Hamonic S, Wardi R, Lebrun C, Zagnoli F, Wiertlewski S, Deburghgraeve V, Coustans M, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386:2078–2088. doi:10.1016/S0140-6736(15)61137-0.
- Welters MJP, Ma W, Santegoets SJAM, Goedemans R, Ehsan I, Jordanova ES, van Ham VJ, van Unen V, Koning F, van Egmond SI, et al. Intratumoral HPV16-specific T cells constitute a type I-oriented tumor microenvironment to improve survival in HPV16-driven oropharyngeal cancer. Clin Cancer Res. 2018;24:634–647. doi:10.1158/1078-0432.CCR-17-2140.
- Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509–524. doi:10.1038/nrclinonc.2014.111.
- Holtkamp S, Bledi Y, Atallah M, Trahtemberg U, Verbovetski I, Nahari E, Zelig O, Linial M, Mevorach D. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108:4009–4017. doi:10.1182/blood-2006-03-013334.
- Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, Huber C, Tureci O, Sahin U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2007;180:309–318. doi:10.4049/jimmunol.180.1.309.
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi:10.1038/nature18300.
- Jabulowsky, R. A., Loquai, C., Diken, M., Kranz, L.M., Haas, H., Attig, S., Bidmon, N., Buck, J., Derhovanessian, E., Diekmann, J., et al. Abstract CT032: A first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles for potent cancer immunotherapy in patients with malignant melanoma. Cancer Res. 2016;76:CT032–CT032.
- Heesen L, Barrios CH, Kim TM, Cosgriff T, Srimuninnimit V, Pittman K, Sabbatini R, Rha SY, Flaig TW, Page RD, et al. First-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles encoding shared tumor antigens for potent melanoma immunotherapy. Ann Oncol. 2017;28. doi:10.1093/annonc/mdx075.
- Jabulowsky RA, Barrios CH, Kim TM, Cosgriff T, Srimuninnimit V, Pittman K, Sabbatini R, Rha SY, Flaig TW, Page RD, et al. A first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles encoding shared tumor antigens for immunotherapy of malignant melanoma. Ann Oncol. 2018;29. doi:10.1093/annonc/mdx807.
- Feltkamp MCW, Smits HL, Vierboom MPM, Minnaar RP, De Jongh BM, Drijfhout JW, Schegget JT, Melief CJM, Kast WM. Vaccination with cytotoxic T lymphocyte epitope containing peptide protects against a tumor induced by human papillomavirus type 16 transformed cells. Eur J Immunol. 1993;23:2242–2249. doi:10.1002/(ISSN)1521-4141.
- Sandoval, F., Terme, M., Nizard, M., Badoual, C., Bureau, M.F., Freyburger, L., Clement, O., Marcheteau, E., Gey, A., Fraisse, G., et al. Mucosal imprinting of vaccine induced-CD8+ T cells is crucial to inhibit mucosal tumors. Sci Transl Med. 2013 Mar 27;5(178):178er2.
- Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun. 2017;8:15221. doi:10.1038/ncomms15221.
- Hertweck A, Evans C, Eskandarpour M, Lau JH, Oleinika K, Jackson I, Kelly A, Ambrose J, Adamson P, Cousins D, et al. T-bet activates Th1 genes through mediator and the super elongation complex. Cell Rep. 2016;15:2756–2770. doi:10.1016/j.celrep.2016.05.054.
- Hickman HD, Reynoso G, Ngudiankama B, Cush S, Gibbs J, Bennink J, Yewdell J. CXCR3 chemokine receptor enables local CD8+ T cell migration for the destruction of virus-infected cells. Immunity. 2015;42:524–537. doi:10.1016/j.immuni.2015.02.009.
- Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008;68:8437–8445. doi:10.1158/0008-5472.CAN-08-1440.
- Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N, Levionnois E, Nizard M, Si-Mohamed A, Besnier N, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013;73:128–138. doi:10.1158/0008-5472.CAN-12-2606.
- Rice AE, Latchman YE, Balint JP, Lee JH, Gabitzsch ES, Jones FR. An HPV-E6/E7 immunotherapy plus PD-1 checkpoint inhibition results in tumor regression and reduction in PD-L1 expression. Cancer Gene Ther. 2015;22:454–462. doi:10.1038/cgt.2015.40.
- Weir GM, Hrytsenko O, Quinton T, Berinstein NL, Stanford MM, Mansour M. Anti-PD-1 increases the clonality and activity of tumor infiltrating antigen specific T cells induced by a potent immune therapy consisting of vaccine and metronomic cyclophosphamide. J Immunother Cancer. 2016:4. doi:10.1186/s40425-016-0169-2.
- Wang T-L, Ling M, Shih I-M, Pham T, Pai SI, Lu Z, Kurman RJ, Pardoll DM, Wu T-C. Intramuscular administration of E7-transfected dendritic cells generates the most potent E7-specific anti-tumor immunity. Gene Ther. 2000;7:726–733. doi:10.1038/sj.gt.3301160.
- Tseng CW, Hung C-F, Alvarez RD, Trimble C, Huh WK, Kim D, Chuang C-M, Lin C-T, Tsai Y-C, He L, et al. Pretreatment with cisplatin enhances E7-specific CD8+T-cell -mediated antitumor immunity induced by DNA vaccination. Clin Cancer Res. 2008;14:3185–3192. doi:10.1158/1078-0432.CCR-08-0037.
- Tseng C-W, Trimble C, Zeng Q, Monie A, Alvarez RD, Huh WK, Hoory T, Wang M-C, Hung C-F, Wu T-C. Low-dose radiation enhances therapeutic HPV DNA vaccination in tumor-bearing hosts. Cancer Immunol Immunother. 2009;58:737–748. doi:10.1007/s00262-008-0596-0.
- van der Sluis TC, Sluijter M, van Duikeren S, West BL, Melief CJM, Arens R, van der Burg SH, van Hall T. Therapeutic peptide vaccine-induced CD8 T cells strongly modulate intratumoral macrophages required for tumor regression. Cancer Immunol Res. 2015;3:1042–1051. doi:10.1158/2326-6066.CIR-15-0052.
- Barrios K, Celis E. TriVax-HPV: an improved peptide-based therapeutic vaccination strategy against human papillomavirus-induced cancers. Cancer Immunol Immunother. 2012;61:1307–1317. doi:10.1007/s00262-012-1259-8.
- Zwaveling S, Gilbert D, Hubert M, Drouot L, Machour N, Lange C, Charlionet R, Tron F. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002;169:350–358. doi:10.4049/jimmunol.169.7.4046.
- Van Der Sluis TC, van Duikeren S, Huppelschoten S, Jordanova ES, Beyranvand Nejad E, Sloots A, Boon L, Smit VTHBM, Welters MJP, Ossendorp F, et al. Vaccine-Induced tumor necrosis factor- Producing T cells synergize with cisplatin to promote tumor cell death. Clin Cancer Res. 2015;21:781–794. doi:10.1158/1078-0432.CCR-14-2142.
- Lisi L, Ciotti GMP, Braun D, Kalinin S, Currò D, Dello Russo C, Coli A, Mangiola A, Anile C, Feinstein DL, et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci Lett. 2017;645:106–112. doi:10.1016/j.neulet.2017.02.076.
- Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi:10.1038/ni.1937.
- Gilbert J, Le Tourneau C, Mehanna H, Fayette J, Goswami T, Emeribe U, Jarkowski A III, Melillo G, Siu LL. Phase II, randomized, open-label study of durvalumab (MEDI4736) or tremelimumab monotherapy, or durvalumab + tremelimumab, in patients with recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN): CONDOR. J Immunother Cancer. 2015;3(Suppl 2):P152. Published 2015 Nov 4. doi:10.1186/2051-1426-3-S2-P152.
- Van Montfoort N, Pantelyushin S, Kreutzfeldt M, Page N, Musardo S, Coras R, Steinbach K, Vincenti I, Klimek B, Lingner T, et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell. 2018;175:1744–1755.e15. doi:10.1016/j.cell.2018.07.049.
- Lin, K. Y. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26.
- Kuhn AN, Diken M, Kreiter S, Selmi A, Kowalska J, Jemielity J, Darzynkiewicz E, Huber C, Türeci Ö, Sahin U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010;17:961–971. doi:10.1038/gt.2010.52.
- Quah BJC, Warren HS. Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc. 2007;2:2049–2056. doi:10.1038/nprot.2007.296
- Diken, M.Vormehr M, Grunwitz C, Kreiter S, Türeci Ö, Sahin U. Discovery and subtyping of neo-epitope specific T-cell responses for cancer immunotherapy: addressing the mutanome. In: RNA vaccines. New York (NY): Humana Press; 2017. p. 223–236.
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:45e–45. doi:10.1093/nar/29.9.e45.